

Abstract
Cytoplasmic polyadenylation-induced translation controls germ cell development1,2, neuronal synaptic plasticity3-5, and cellular senescence6,7, a tumor-suppressor mechanism that limits the replicative lifespan of cells8,9 . The cytoplasmic polyadenylation element binding protein (CPEB) promotes polyadenylation by nucleating a group of factors including defective in germline development 2 (Gld2), a non-canonical poly(A) polymerase10,11, on specific mRNA 3’ untranslated regions (UTRs). Because CPEB regulation of p53 mRNA polyadenylation/translation is necessary for cellular senescence in primary human diploid fibroblasts6, we surmised that Gld2 would be the enzyme responsible for poly(A) addition. Here, we show that depletion of Gld2 surprisingly promotes rather than inhibits p53 mRNA polyadenylation/translation, induces premature senescence, and enhances the stability of CPEB mRNA. The CPEB 3’UTR contains two miR-122 binding sites, which when deleted, elevate mRNA translation, as does an antagomir of miR-122. Although miR-122 is thought to be liver-specific, it is present in primary fibroblasts and destabilized by Gld2 depletion. Gld4, a second non-canonical poly(A) polymerase, was found to regulate p53 mRNA polyadenylation/translation in a CPEB-dependent manner. Thus, translational regulation of p53 mRNA and cellular senescence is coordinated by Gld2/miR-122/CPEB/Gld4.
Keywords: CPEB, polyadenylation, translation, p53, Gld2, Gld4
Mouse embryo fibroblasts (MEFs) derived from CPEB knockout (KO) mice do not senesce as do MEFs derived from wild type (WT) mice, but instead are immortal. Senescence is rescued when ectopic CPEB is expressed in the KO MEFs and potentiated when expressed in WT MEFs7. Human foreskin fibroblasts depleted of CPEB also bypass senescence and divide ~270 days compared to WT cells, which senesce after about 90 days. As with the mouse cells, ectopic expression of CPEB rescues senescence in knockdown cells and potentiates senescence in WT cells. CPEB controls the polyadenylation-induced translation of p53 mRNA, and indeed CPEB-induced senescence requires p53. Depletion of CPEB also induces the “Warburg Effect” where mitochondrial respiration is reduced and cells produce ATP primarily through glycolysis6.
To investigate the possibility that CPEB control of p53 polyadenylation requires Gld2, human primary foreskin fibroblasts were stably transduced with lentiviruses expressing two different shRNAs against the Gld2 coding sequence. Surprisingly, Gld2 depletion (Fig. 1a, 1b) induced an increase in both p53 protein levels (Fig. 1c) and p53 mRNA polyadenylation (Fig. 1d; Supplemental Fig. 1). Also unexpectedly, depletion of Gld2 resulted in increased oxygen consumption (Fig. 1e) and entry into a senescence-like cell cycle arrest as evidenced by ß-galactosidase staining at acid pH (Fig. 1f). In comparison, CPEB depleted cells had decreased oxygen consumption, fewer cells staining with ß-galactosidase, increased lifespan, and most importantly, reduced poly(A) tail size on p53 mRNA and ~50% reduction in p53 protein levels6.
Figure 1.

Depletion of Gld2 enhances p53 expression. (a) RT-PCR of Gld2 and tubulin RNAs following infection of human foreskin fibroblasts with lentiviruses expressing shRNA against Gld2 or GFP (mock, same in all panels). (b) Knockdown of Gld2-HA in cells expressing ectopic Gld2-HA. Tubulin served as a loading control. (c) Western blot showing 2.5 fold enhanced expression of p53 relative to tubulin following Gld2 depletion. (d) Poly(A) tail analysis of p53 mRNA in WT and Gld2 depleted cells (two shRNAs targeting different regions of Gld2 were used). (e) Oxygen consumption in cells infected with shCPEB, shGld2, or empty vector (Mock). (f) Mock or shGld2 infected cells stained for ß-galactosidase, which denotes cellular senescence. Population doublings were determined in wild type or Gld2 depleted cells.
These paradoxical results prompted us to examine CPEB levels in Gld2 deficient cells since CPEB is required for normal p53 mRNA translation6. After comparing the amounts of CPEB nuclear pre-mRNA by intron-specific quantitative RT-PCR and mostly cytoplasmic mRNA by exon-specific quantitative RT-PCR, we found that the pre-mRNA levels, which generally reflect transcription, were nearly unchanged while cytoplasmic mRNA levels increased by about five-fold (Fig. 2a). Thus, in the absence of Gld2, CPEB mRNA unexpectedly was more stable.
Figure 2.
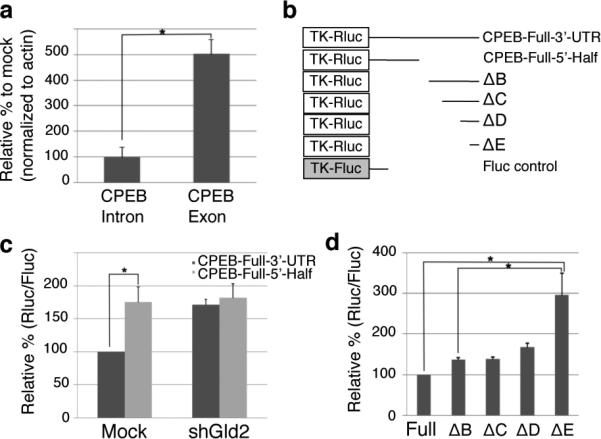
Gld2 knockdown increases CPEB reporter mRNA and translation by a post-transcriptional mechanism. (a) Fold change of nuclear (intron-containing) or predominantly cytoplasmic (exon-containing) CPEB RNA following Gld2 depletion (n=3, bars are s.e.m.) (b) Schematic of reporter constructs used in the following experiments (numbers refer to nucleotides of CPEB 3’UTR). (c and d) Cells expressing firefly luciferase (Fluc) as a control and Renilla luciferase (Rluc) as noted in panel b were depleted of Gld2; the amount of Renilla lucifease activity (relative to firefly) was derived from RNA containing the entire CPEB 3’UTR (full) and set at 100. In all panels, n = 3 and the bars refer to s.e.m.; one or two asterisks refer to statistical significance (Students t test) at the p<0.05 or p<0.01 levels, respectively.
Surmising that Gld2 might control p53 protein levels via CPEB, we next used a Renilla luciferase (Rluc) and firefly luciferase (Fluc) reporter system to investigate post-transcriptional regulation of CPEB by Gld2. As shown in Figs. 2b and 2c, the entire CPEB 3’ UTR was translated about 40% less efficiently compared to a reporter lacking the 3’ most 455 nucleotides (Mock). However, in Gld2-deficient cells, the two reporters were translated equally. Additional deletions (Δ) of the CPEB 3’ UTR suggested that there might be multiple regions that elicited increases in reporter translation following Gld2 knockdown (i.e., ΔE translation was about two fold greater than ΔB, ΔC, or ΔD translation) (Fig. 2d).
Analysis of the regions of the CPEB 3’ UTR that mediated translational repression by Gld2 revealed the presence of two potential miR-122 binding sites (Supplementary Fig. 2). Although miR-122 is thought to be liver-specific and account for ~70% of the total population of microRNAs in that tissue13, deletion of these specific sites, either individually or combined, alleviated translational repression in Gld2 depleted cells (Fig. 3a) and were nearly identical to that observed with the large deletions (Fig. 2d). These results suggest that miR-122 might repress CPEB mRNA translation in human skin fibroblasts and indicate that this miRNA is more widely distributed than originally thought. Indeed recent evidence shows that miR-122 is present in human skin14 and even HEK293 cells15.
Figure 3.

miR-122 activates p53 mRNA translation by repressing CPEB. (a) Gld2 depleted fibroblasts were transduced with firefly and Renilla luciferase with CPEB full length or deletion mutant 3’UTRs lacking putative miR-122 sites (Supplementary Fig. 2). The data are expressed as in Fig 2; in all panels, n = 3 and the bars refer to s.e.m. and one or two asterisks refer to statistical significance (Students t test) at the p<0.05 or p<0.01 levels, respectively. (b) Sequence of miR-122 from fibroblasts; a non-templated adenosine is shaded. (c) Fibroblasts expressing firefly and Renilla luciferase containing the CPEB 3’ UTR were electroporated with miR-122 (Anta-122), scrambled, (Anta-Scr), or no LNA antagomir (Mock); data are expressed as in Figure 2. (d) Quantitative RTPCR for miR-122 in cells expressing GFP (WT) or shGld2. (e and f) Immunoprecipitation of 35S-methonine-labeled p53 from MG132-treated cells transduced with no (Mock-GFP), miR-122 (Anta-122), or scrambled (Anta-Scram) LNA antagomirs. WCE refers to whole cell lysate. (g) Fibroblasts were treated as in panels e-g after first expressing either TET repressor (shTETR, a control) or CPEB shRNA.
To assess directly whether miR-122 might repress CPEB mRNA expression, we first cloned and sequenced it from human foreskin fibroblasts and found that it contained a nontemplated 3’ monoadenylate residue (Fig. 3b; see discussion). Next, cells were electroporated with a locked nucleic acid (LNA) antagomir for miR-122, or as a control, a scrambled LNA. The miR-122 antagomir enhanced reporter expression by about 3.25 fold relative to control (Fig. 3c), but had no stimulatory effect on a reporter whose 3’ UTR contained no miR-122 sites (Supplementary Fig. 3). Based on evidence from Katoh et al16, who demonstrated that in murine liver, Gld2 is essential for miR-122 stability, we suspected that Gld2 might influence CPEB expression and possibly p53 mRNA translation by controlling miR-122 steady-state levels. Indeed, Fig. 3d demonstrates that depletion of Gld2 from skin fibroblasts reduced the level of miR-122 by nearly 40-fold. Importantly, when miR-122 LNA antagomir-transduced cells were incubated with the proteasome inhibitor MG132 and pulsed-labeled with 35S-methionine for 15 min followed by p53 immunoprecipitation, there was a two-fold increase in the synthesis rate of p53 (Fig. 3e, 3f). Taken together, these data demonstrate that human primary skin fibroblasts contain miR-122 and that Gld2 controls its steady state levels or activity.
While consistent with the hypothesis that miR-122 mediates p53 mRNA translation via CPEB, these data do not eliminate the possibility that miR-122 could act via another molecule to regulate p53 synthesis (note that p53 mRNA has no miR-122 sites according to Targetscan.org or Microrna.org). Consequently, we infected cells with a lentivirus expressing shRNA for CPEB as well as the miR-122 antagomir followed by a 15 minute pulse of 35S-methionine and p53 immunoprecipitation. Fig. 3g shows that although miR-122 antagomir alone elicited an increase in p53 synthesis, the antagomir plus shRNA for CPEB induced no increase. Taken together, these data demonstrate that Gld2 activity stabilizes miR-122, which in turn reduces CPEB expression; CPEB then acts directly on p53 mRNA to control poly(A) tail length and translation.
If not Gld2, what poly(A) polymerase modifies p53 mRNA polyadenylation and translation? We surmised that an alternative non-canonical poly(A) polymerase, i.e., one that lacks an RNA binding domain and thus would require another factor such as CPEB to be tethered to the RNA, would most likely be involved. Two cytoplasmic enzymes have this characteristic: Gld4 (PAPD5)17 and MitoPAP (PAPD1)18. Both polymerases were depleted with shRNAs (Supplementary Fig. 4) but only the loss of Gld4 reduced p53 protein levels (Fig. 4a). To investigate whether Gld4 interacts with p53 mRNA in a CPEB-dependent manner, FLAG-Gld4 was expressed in cells (Supplementary Fig. 5) containing shRNA for tetracycline repressor (TETR, a control) or CPEB. Gld4 was then immunoprecipitated and the extracted RNA was examined for p53 and GAPDH (a control) RNAs by RT-PCR (Fig. 4b). p53 mRNA was detected only when CPEB was present, suggesting that Gld4 is anchored to p53 mRNA by CPEB, and indeed, CPEB co-immunoprecipitated Gld4 but not Mcl1, a nonspecific control (Fig. 4c). Finally, depletion of Gld4 reduced p53 mRNA polyadenylation (Fig. 4d), which probably then induced p53 mRNA destabilization (Fig. 4e; depletion of Gld4 reduced mostly cytoplasmic p53 mRNA as examined by RT-PCR using exon-specific primers but had no effect on p53 transcription as examined by RT-PCR using intron-specific primers).
Figure 4.
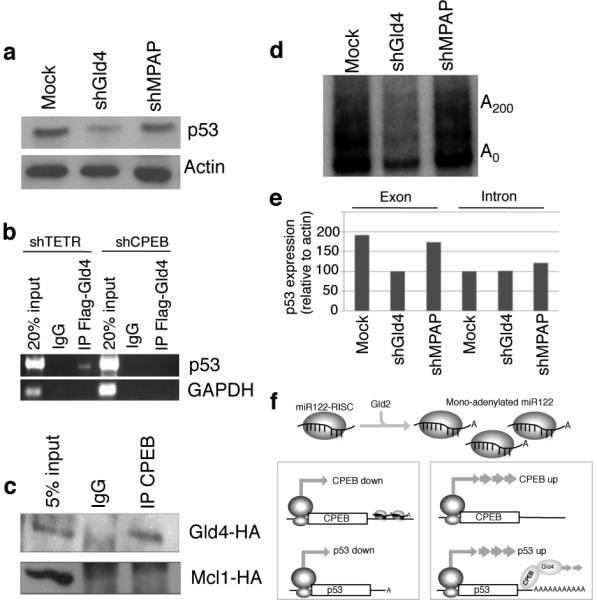
Gld4 controls p53 mRNA expression. (a) p53 and actin western blots from fibroblasts expressing GFP (mock), shGld4, or shMitoPAP (shMPAP). (b) Fibroblasts containing shTETR or CPEB were transfected with Gld4-FLAG followed by FLAG antibody or IgG immunoprecipitation of RNA complexes and RT-PCR for p53 or GAPDH (control) RNAs. (c) Protein from fibroblasts infected with Gld4-HA and CPEB-FLAG was FLAG or IgG immunoprecipitated and western blotted for HA. Other cells infected with CPEB-FLAG and MCl1-HA (a nonspecific control) were processed similarly. (d) Examination of p53 poly(A) tail6 from skin fibroblasts expressing GFP or (mock) Gld4 or mitoPAP shRNAs. (e) RT-PCR analysis of predominantly cytoplasmic p53 RNA (exon-specific primers), or nuclear p53 pre-mRNA (intron-specific primers) in cells expressing GFP (mock) or Gld4 or MitoPAP shRNAs. (f) Model for regulation of p53 translation; see text for explanation.
The results presented here and in Katoh et al. (ref. 16) suggest a model for homeostatic control of p53 synthesis in human skin fibroblasts (Fig. 4f). Gld2 stabilizes miR-122 by catalyzing the addition of a single adenylate residue to its 3’ end16. miR-122 then base-pairs to two regions of the CPEB 3’ UTR, causing instability and/or translational inhibition of the mRNA. CPEB, whose levels are modulated by these events, binds to the p53 3’UTR and recruits Gld4, which in turn maintains p53 mRNA polyadenylation and translation. We envision this hierarchical regulation of p53 to resemble a rheostat, where translation is turned up or down rather than a switch, where translation is turned on or off19, although p53 mRNA translation can also be controlled by a switch mechanism in response to DNA damage20,21. A 50% change in p53 synthesis can toggle a cell between growth and senescence6, demonstrating that drastic biological consequences result from a relatively modest change in protein level.
Although ectopically-expressed Gld2 immunoprecipitated from hepatocarcinoma cells adds a single adenosine to miR-122 in vitro16, Gld2 tethered to a small non-coding RNA by MS2 adds >300 adenylate residues in injected oocytes22, and about that same amount to mRNA when bound to CPEB10. How the enzyme can modulate its catalytic activity depending on the substrate is unknown, but we postulate that components of the RNA-induced silencing complex (RISC) might be responsible. In addition to our demonstration that miR-122 is 3’ mono-adenylated in skin fibroblasts, ~20% of all RNA deep sequencing reads from cloned neuroblastoma miRNAs contain a non-templated adenylate residue23, suggesting that miR-122 may be one of several miRNAs that are mono-adenylated by Gld2.
In conclusion, our results demonstrate that Gld2 control of miR-122 stability in human skin fibroblasts tunes CPEB expression, which in turn regulates p53 mRNA polyadenylation and translation by Gld4. The coordinated activities of these factors then gate entry into senescence. These studies also bring up two additional aspects of CPEB-related activities: how does Gld4, but not Gld2, associate with CPEB on p53 mRNA, and what molecular machinery is responsible for miR-122 destruction upon Gld2 depletion? Deciphering the mechanisms involved would likely require analysis of the combinatorial associations of factors on different RNA substrates.
METHODS SUMMARY
Molecular biology and cell culture
Primary human foreskin fibroblasts obtained from the Cell Culture Core Facility of the Yale University Skin Disease Research Center were cultured as described 24 in Dulbecco's Modified Eagle's Medium (DMEM) containing 10% fetal calf serum. Amphotropic retroviruses and shRNA-containing lentiviruses were produced by transient transfection of 293T cells with a transfer vector and amphotropic packaging plasmids encoding VSV-G and gag-pol using Lipofectamine 2000 (Invitrogen). Human cells at 50% confluency were infected for 8-12 hr with viral supernatants containing 7μg/ml polybrene. Typically 70-90% infection efficiency was achieved as assessed by a GFP-encoding viral gene or by immunostaining with HA antibody (Covance). After infection, fresh medium was added to the infected fibroblasts. Some cells were analyzed by western blotting for p53 (DO-1, Neomarkers) and Δ-actin (Abcam). Other cells were fixed with 0.2% glutaraldehyde and stained for Δ-galactosidase activity at acidic pH according to Dimri et al25.
Supplementary Material
Acknowledgements
We thank Victor Ambros, Joan Steitz, Timothy Kowalik, and Roger Davis for helpful comments. S.N. was supported by a fellowship of the Max Planck Society and by European Molecular Biology Organization fellowship ALTF 995-2004. This work was supported by NIH grant AG30323. Additional core support from the Diabetes Endocrinology Research Center (DK32520) is gratefully acknowledged.
Methods
Analysis of p53, Gld2, and Gld4
Lentiviruses expressing shRNAs for Gld2, Gld4, and mitoPAP were generated as described6. shRNA against the TET repressor has also been described6. A retrovirus expressing Gld2-HA was generated as described7. Control and shCPEB infected fibroblasts were cultured in methionine and cysteine-free media (Invitrogen) for 45 min and then cultured in media containing 140 mCi 35S-methionine and 35S-cysteine (ProMix, Amersham) for 30 min. The cells were then washed and cultured in fresh DMEM supplemented with 2 mM each of methionine and cysteine for the times indicated. The cells were then frozen and stored until they were used for p53 immunoprecipitation and analysis by SDS-PAGE and phosphorimaging. Cells were also cultured in methionine/cysteine free media in the presence of MG132, a proteasome inhibitor, for 30 min. followed by a 15 min culture in 100 µCi 35S-methionine and cysteine; p53 was then immunoprecipitated and analyzed as noted above.
Ligation-mediated polyadenylation test (LM-PAT) assays were used to detect the poly(A) tail of p53 mRNA in WT cells, shGld2 knockdown cells (two shRNAs targeting different regions of Gld2 were used), cells expressing ectopic CPEB, and cells expressing ectopic that lacked a zinc finger and hence was unable to bind RNA (CPEB∆ZF)6. Quantitative RT-PCR analyses were normalized against actin RNA.
Oxygen Consumption and cellular senescence
To measure oxygen consumption, ~4×105 cells were washed and suspended in 200 ml Krebs-Ringers solution plus HEPES (125 mM NaCl, 1.4 mM KCl, 20 mM HEPES, pH 7.4, 5 mM NaHCO3, 1.2 mM MgSO4, 1.2 mM KH2,PO4, 1 mM CaCl2) containing 1% BSA. Cells from each condition were aliquoted into a BD Oxygen Biosensor System plate (BD Biosciences) in triplicate. Plates were assayed on a SAFIRE multimode microplate spectrophotometer (Tecan) at 1-minute intervals for 60 minutes at an excitation wavelength of 485 nm and emission wavelength of 630 nm.
Mock or shGld2 infected cells were stained for ß-galactosidase at acidic pH, which denotes cellular senescence. Cell number was also determined with a haemocytometer; population doublings were plotted as growth curves of wild type cells or cells infected with shGld2.
miR-122 cloning and sequencing
Small RNAs from human foreskin fibroblasts were extracted with mirVANA PARIS kit (Ambion) and those corresponding in length to18-24 nucleotides were resolved by urea-PAGE, extracted, and ethanol precipitated. miRNA cloning llinker-1 (IDT) was ligated to the 3' ends and used to prime a reverse transcription reaction with Superscript III (Invitrogen) and the RT primer DP3 (5'-ATTGATGGTGCCTACAG-3’). cDNA was then PCR amplified with miR-122 specific primer 5'-AGGGGCGCCTGGAGTGTGACAATG-3' and DP3. The PCR product was cloned into pGEM-T (Promega) and sequenced. The chromatogram shown is adapted from 4peaks (http://mekentosj.com/science/4peaks/).
Antagomir depletion of miR-122
Locked nucleic acid (LNA) antagomir against miR-122, or a scrambled sequence LNA (Exiqon) were electroporated (Amaxa, Lonza) at a final concentration of 4 nM into ~106 human foreskin fibroblast cells together with 0.8 Δg firefly (pGL3, Promega) and 1.0 Δg Renilla (pRLTK) luciferase-encoding plasmid, the latter harboring the full length CPEB 3’ UTR or deletion mutations of the 3’UTR. Luciferase assays were performed 16-24 hours after electroporation according to methods described elsewhere26. The amount of Renilla luciferase activity derived from RNA containing the entire CPEB 3’UTR (full) was arbitrarily set at 100. When employed, the CPEB 3’UTR deletions (Δ) were (in CPEB nucleotide number): ΔA, 420-755; ΔB, 480-755; ΔC, 530-755; ΔD, 565-755; ΔE, 640-755.
CPEB-Gld4 Co-immunoprecipitation
Cells were transfected with plasmids encoding FLAG-CPEB and HA-Gld4 using Effectene (Qiagen). The cells were then harvested in PBS and lysed in lysis/wash buffer (50 mM Tris-HCl, pH7.4, 100 mM NaCl, 1 mM MgCl2, 0.1 mM CCl2, 0.1% SDS, and Complete Protease Inhibitor (Roche)). Extracts (0.5 mg protein) were incubated with M2-FLAG antibody (Sigma)-coated Dynabeads (InVitrogen) for 2 hours at 4°C. The beads were then washed 3 times with lysis/wash buffer and the bound proteins eluted by boiling in SDS sample buffer. Coimmunoprecipitates were detected by western blotting with HA antibody (HA.11 16B12, Covance). Control IPs were performed with generic mouse IgG-coated Dyanbeads.
Gld4-RNP Co-immunoprecipitation
Mock or CPEB-depleted human foreskin fibroblasts6 (Burns and Richter, 2008) were electroporated (Amaxa, Lonza) with a plasmid encoding FLAG-Gld4 according to the manufacturer's instructions. Immunoprecipitation with the FLAG antibody followed the procedure of Peritz et al27 with the following modifications: a.) M2 anti-FLAG (Sigma)-coated Dynabeads were used instead of agarose beads and b.) washes with buffer containing 1 M urea were omitted. p53 RNA was detected by RT-PCR as described6.
Footnotes
Author Information Reprints and permissions information is available at www.nature.com/reprints.
The authors declare no competing financial interests.
Full Methods and any associated references are available in the online version of the paper at www.nature.com/nature.
Supplementary information is linked to the online version of the paper at www.nature.com/nature.
References
- 1.Mendez R, et al. Phosphorylation of CPE binding factor by Eg2 regulates translation of c-mos mRNA. Nature. 2000;404:302–307. doi: 10.1038/35005126. [DOI] [PubMed] [Google Scholar]
- 2.Tay J, Richter JD. Germ cell differentiation and synaptonemal complex formation are disrupted in CPEB knockout mice. Dev Cell. 2001;1:201–213. doi: 10.1016/s1534-5807(01)00025-9. [DOI] [PubMed] [Google Scholar]
- 3.Alarcon JM, et al. Selective modulation of some forms of schaffer collateral-CA1 synaptic plasticity in mice with a disruption of the CPEB-1 gene. Learn Mem. 2004;11:318–327. doi: 10.1101/lm.72704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu L, et al. CPEB-mediated cytoplasmic polyadenylation and the regulation of experience-dependent translation of alpha-CaMKII mRNA at synapses. Neuron. 1998;21:1129–1139. doi: 10.1016/s0896-6273(00)80630-3. [DOI] [PubMed] [Google Scholar]
- 5.Zearfoss NR, Alarcon JM, Trifilieff P, Kandel E, Richter JD. A molecular circuit composed of CPEB-1 and c-Jun controls growth hormone-mediated synaptic plasticity in the mouse hippocampus. J Neurosci. 2008;28:8502–8509. doi: 10.1523/JNEUROSCI.1756-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burns DM, Richter JD. CPEB regulation of human cellular senescence, energy metabolism, and p53 mRNA translation. Genes Dev. 2008;22:3449–3460. doi: 10.1101/gad.1697808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Groisman I, et al. Control of cellular senescence by CPEB. Genes Dev. 2006;20:2701–2712. doi: 10.1101/gad.1438906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–740. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- 9.Stewart SA, Weinberg RA. Telomeres: cancer to human aging. Annu Rev Cell Dev Biol. 2006;22:531–557. doi: 10.1146/annurev.cellbio.22.010305.104518. [DOI] [PubMed] [Google Scholar]
- 10.Barnard DC, Ryan K, Manley JL, Richter JD. Symplekin and xGLD-2 are required for CPEB-mediated cytoplasmic polyadenylation. Cell. 2004;119:641–651. doi: 10.1016/j.cell.2004.10.029. [DOI] [PubMed] [Google Scholar]
- 11.Kim JH, Richter JD. Opposing polymerase-deadenylase activities regulate cytoplasmic polyadenylation. Mol Cell. 2006;24:173–183. doi: 10.1016/j.molcel.2006.08.016. [DOI] [PubMed] [Google Scholar]
- 12.Kim JH, Richter JD. RINGO/cdk1 and CPEB mediate poly(A) tail stabilization and translational regulation by ePAB. Genes Dev. 2007;21:2571–2579. doi: 10.1101/gad.1593007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T. Identification of tissue-specific microRNAs from mouse. Curr Biol. 2002;12:735–739. doi: 10.1016/s0960-9822(02)00809-6. [DOI] [PubMed] [Google Scholar]
- 14.Holst LM, Kaczkowski B, Gniadecki R. Reproducible pattern of microRNA in normal human skin. Exp Dermatol. 2010;19:e201–205. doi: 10.1111/j.1600-0625.2009.01049.x. [DOI] [PubMed] [Google Scholar]
- 15.Liao JY, et al. Deep sequencing of human nuclear and cytoplasmic small RNAs reveals an unexpectedly complex subcellular distribution of miRNAs and tRNA 3' trailers. PLoS One. 2010;5:e10563. doi: 10.1371/journal.pone.0010563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Katoh T, et al. Selective stabilization of mammalian microRNAs by 3' adenylation mediated by the cytoplasmic poly(A) polymerase GLD-2. Genes Dev. 2009;23:433–438. doi: 10.1101/gad.1761509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schmid M, Kuchler B, Eckmann CR. Two conserved regulatory cytoplasmic poly(A) polymerases, GLD-4 and GLD-2, regulate meiotic progression in C. elegans. Genes Dev. 2009;23:824–836. doi: 10.1101/gad.494009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mullen TE, Marzluff WF. Degradation of histone mRNA requires oligouridylation followed by decapping and simultaneous degradation of the mRNA both 5' to 3' and 3' to 5'. Genes Dev. 2008;22:50–65. doi: 10.1101/gad.1622708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Santos SD, Ferrell JE. Systems biology: On the cell cycle and its switches. Nature. 2008;454:288–289. doi: 10.1038/454288a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ofir-Rosenfeld Y, Boggs K, Michael D, Kastan MB, Oren M. Mdm2 regulates p53 mRNA translation through inhibitory interactions with ribosomal protein L26. Mol Cell. 2008;32:180–189. doi: 10.1016/j.molcel.2008.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takagi M, Absalon MJ, McLure KG, Kastan MB. Regulation of p53 translation and induction after DNA damage by ribosomal protein L26 and nucleolin. Cell. 2005;123:49–63. doi: 10.1016/j.cell.2005.07.034. [DOI] [PubMed] [Google Scholar]
- 22.Kwak JE, Wang L, Ballantyne S, Kimble J, Wickens M. Mammalian GLD-2 homologs are poly(A) polymerases. Proc Natl Acad Sci U S A. 2004;101:4407–4012. doi: 10.1073/pnas.0400779101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schulte JH, et al. MicroRNAs in the pathogenesis of neuroblastoma. Cancer Lett. 2009;274:10–15. doi: 10.1016/j.canlet.2008.06.010. [DOI] [PubMed] [Google Scholar]
- 24.Rangarajan A, Hong SJ, Gifford A, Weinberg RA. Species- and cell type-specific requirements for cellular transformation. Cancer Cell. 2004;6:171–183. doi: 10.1016/j.ccr.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 25.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nottrott S, Simard MJ, Richter JD. Human let-7a miRNA blocks protein production on actively translating polyribosomes. Nat Struct Mol Biol. 2006;13:1108–14. doi: 10.1038/nsmb1173. [DOI] [PubMed] [Google Scholar]
- 27.Peritz T, Zeng F, Kannanayakal TJ, Kilk K, Eiriksdottir E, Langel U, Eberwine J. Immunoprecipitation of mRNA-protein complexes. Nature Protocols. 2006;1:577–580. doi: 10.1038/nprot.2006.82. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.