

Abstract
The biochemical properties of the HLA-C antigen differ substantially from those of HLA-A and -B molecules. For this reason, HLA-C diversity and expression at the cell surface are much lower than its counterparts and in consequence HLA-C-restricted responses have been infrequently detected and described. In this review we summarise the key differences between HLA-C and other class I molecules and provide an update on natural killer and T-cell responses restricted by HLA-C. We also discuss the different clinical settings associated with HLA-C alleles which mainly consist of autoimmune disorders, cancers and chronic infections.
Keywords: epitopes, HIV, HLA-C, killer immunoglobulin receptors, T cells
Introduction
For many years HLA-C has been the poor relation of the HLA class I molecular family, largely neglected by cellular immunologists because of its relatively low level of cell-surface expression, leading to difficulties in serological HLA-C typing, and its apparently minor role in mediating antigen-specific T-cell responses. The major role of HLA-C has been assumed to be in acting as a ligand for killer immunoglobulin receptors (KIRs) expressed on natural killer (NK cells). However, the seminal observation that HIV-1 Nef has the ability to selectively down-regulate HLA class I A and B molecules to minimize cytotoxic T lymphocyte (CTL) surveillance, while maintaining HLA-C expression (presumably to maintain NK cell inhibition), has led to an appreciation of the role of HLA-C as a T-cell restriction element, particularly in HIV-1 infection.1 This role in HIV-1 infection has been highlighted by the recent description of a single nucleotide polymorphism (SNP) in the HLA-C promoter region as an important factor linked with virus control in genome-wide association studies,2 although the mechanism remains unknown. Here we review our current understanding of the relative importance of HLA-C in NK and T-cell recognition.
Distinctive features of the HLA-C molecule
Over the last 20 years, molecular analysis of HLA-C alleles has been impeded by limitations such as a lack of suitable antisera and technical difficulties in protein isolation, purification and characterization. In the 1990s, application of single specific primer PCR to specific cloning of HLA-C alleles helped to circumvent this problem and revealed a 37% discrepancy between single specific primer PCR and serology results.3 However, analysis of responses restricted by HLA-C is still arduous and dubious as HLA-C alleles are often in strong linkage disequilibrium with HLA-B alleles, making it difficult to distinguish HLA-C from HLA-B-restricted responses. This raises the possibility that HLA-C epitopes might mistakenly have been identified as restricted by HLA-A or HLA-B (for example, some B14 epitopes in HIV-1 p24 are now thought to be Cw8-restricted, Los Alamos Immunology Database). HLA-C shares sequence homology with other classical human class I HLA-A and HLA-B molecules (its α2 helix is particularly similar to that of HLA-B) but it also differs from other HLA antigens on several levels. HLA-C alleles are closely related to each other and their α1 domain is unusually conserved and so most characteristic of HLA-C (Fig. 1a). The KYRV motif of residues 66, 67, 69 and 76 in the α1 helix highlights this, as it is conserved in all HLA-C alleles and absent in HLA-A and HLA-B molecules except B46.4 Furthermore a conserved glycine at amino acid 45 (α1 helix), the presence of four HLA-C-unique residues in the α2 domain and the reduced diversity at the B pocket of the antigen recognition site are particularly striking of HLA-C and correlate with binding of a restricted set of self-peptides in comparison with HLA-A and HLA-B antigens, emphasizing the distinctive character of this antigen.5,6
Figure 1.
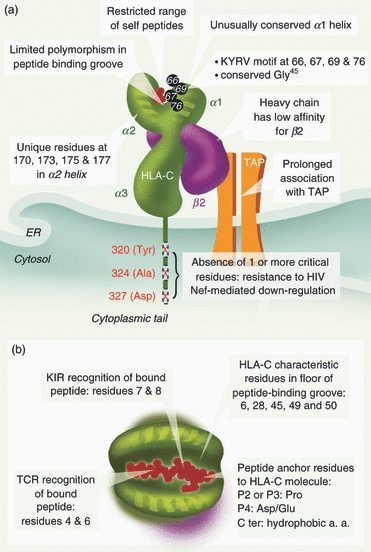
Schematic representation illustrating the distinctive features of the HLA-C molecule. (a) Conserved residues are found in α1 and α2 domains, which results in binding to a limited range of self-peptides. HLA-C alleles are resistant to HIV Nef-mediated down-regulation because of the absence of any one of the three key amino acids in the cytoplasmic tail. Prolonged association with transporters associated with antigen processing (TAP) and low affinity for the light chain (β) are also characteristic of HLA-C. (b) Residues important for binding of peptide (red) to HLA-C molecule (green) and for T-cell receptor and killer immunoglobulin receptor (KIR) recognition are highlighted.
The main feature distinguishing HLA-C is its low expression level at the cell surface, which is only ∼ 10% of that of HLA-A and HLA-B molecules. Several explanations have been offered to account for low surface expression of HLA-C: low levels of mRNA, poor assembly with β2-microglobulin (β2m) and restricted peptide binding leading to the accumulation of un/misfolded intermediates in the endoplasmic reticulum (ER). There is conflicting evidence on whether or not levels of HLA-C mRNA are lower than those of HLA-A and HLA-B. McCutcheon et al.7 showed that reduced protein expression of HLA-C in B cells correlated with low mRNA levels resulting from faster degradation of the HLA-C message than HLA-A or HLA-B transcripts. On the other hand, at least two other independent groups have detected similar levels of HLA-A/B/C mRNA in Epstein—Barr virus-transformed B-cell lines.8,9 Consistent with this, Neisig et al.5 found that intracellular protein levels of HLA-Cw04 heavy chains in human B lymphoblastoid cells were comparable to those of HLA-A and HLA-B. However, a recent study using real-time PCR on laser-assisted microdissected tissues has shown that the gene expression profile of HLA-A, HLA-B and HLA-C varies according to the tissues examined. The authors looked at mRNA levels of HLA-ABC in peripheral blood lymphocytes, colon mucosa and larynx mucosa and found that HLA-C mRNA expression was lowest in larynx mucosa but comparable with that of HLA-A in peripheral blood lymphocytes.10 These data are in agreement with both previous reports, as they indicate that levels of HLA-C transcripts could be similar or different from those of HLA-A and HLA-B depending on the type and function of the cells under study (hence their location in the body) and the type of immune response they are involved in (mucosal innate or specific T-cellular). Nevertheless, in settings in which HLA-C mRNA levels are similar to those of HLA-A and HLA-B, expression of HLA-C molecules at the cell surface is consistently 15–35% lower than that of its counterparts, suggesting that a mechanism at the post-transcriptional level prevents HLA-C molecules from being properly exported at the cell surface.7
Association of Class I heavy chain with β2m is a prerequisite for surface expression. Several groups have reported that HLA-C associates inefficiently with β2m, leading to an accumulation of folding intermediates free of β2m (free heavy chains) in the ER5 (Fig. 1a). As a result of this, HLA-C heavy chains remain associated with calnexin even in the absence of impaired antigen-processing/peptide-loading machinery, hence leading to their degradation in the ER.9,11,12 However, some HLA-C heavy chains do bind β2m and become properly assembled. An explanation for their poor expression at the cell surface is their prolonged association with transporters associated with antigen processing (TAP) in the ER, resulting in slower exocytosis. Neisig et al. have elegantly shown in an in vitro peptide-binding assay that HLA-Cw04 and HLA-Cw02 alleles require a 10-fold higher concentration of a degenerated 9-mer peptide mix to induce release from TAP than HLA-A and HLA-B.5 This stable association with TAP resulting in a reduced formation of HLA-C–peptide complexes is a consequence of HLA-C molecules being more selective in their peptide binding than HLA-A and HLA-B, which is likely to be a result of their limited polymorphism in the peptide-binding groove. In agreement with this, it was recently shown that the retention of TAP depends on the KYRV motif in the α1 helix of HLA-C (Fig. 1a). This motif might restrict the range of acceptable self-ligands for HLA-C heavy chains by reducing the plasticity of the antigen recognition groove, hence prolonging the interaction with TAP while waiting for the ‘perfect’ peptide.11 Self peptides containing the optimal peptide-binding motif for most HLA-C alleles (Pro at position 2 or 3 and a hydrophobic C-terminal anchor residue) are present in very small amounts in the ER and it has been suggested that HLA-C molecules have a specialized function in the immune system because they are not saturated with endogenous proteins and are therefore free to bind and present a set of viral peptides that are not efficiently presented by HLA-A and HLA-B. Consistent with this idea, it was also proposed that low HLA-C expression could also play a role in shaping the T-cell repertoire during positive selection in the thymus.4,5
How do the interactions of HLA-C with its NK and T-cell ligands differ?
The crystal structures of the inhibitory KIR, KIR2DL2 in complex with HLA-Cw3,13 together with that of KIR2DL1–HLA-Cw4,14 revealed that positions 7 and 8 at the C-terminus of HLA-C-binding peptides are involved in direct contact with the KIR molecule, and showed that any residue at p8 larger than valine would not be tolerated for KIR2DL2–Cw3–peptide binding (Fig. 1b). The other residues in the peptide do not contribute significantly to KIR recognition. These crystal structures represent the two groups of HLA-C ligands, namely C1 (including HLA-Cw2, -4, -5, -6 and -15), which interact with KIR2DL1 and KIR2DS1, and C2 (HLA-Cw1, -3, -7 and -8), which bind to KIR2DL2 and KIR2DL3. These two groups of molecules differ at position 80, which is lysine in C1 and asparagine in the C2 group.15 The crystal structures revealed how this position mediates the distinct specificities of HLA-C/KIR binding, showing that the nature of residue 44 in the KIR molecule (methionine in KIR2DL1/S1 and lysine in KIR2DL2/3) is critical for the interaction with residue 80 of the HLA-C molecule.
Although KIR binding to HLA-C peptide complexes is analogous to that of the T-cell receptor, the precise contact regions differ, with the T-cell receptor showing interaction with the central region of the bound peptide at residues 4–6 (Fig. 1b).16 The footprints of KIR and HLA lead to burying of a similar amount of the accessible surface area of the peptide–HLA complex, and they also overlap, meaning that it would be impossible for an HLA-C molecule to be simultaneously in contact with both a T-cell and an NK cell (reviewed in ref. 17).
HLA-C associations with human disease
Because expression of HLA-C antigens is characteristically low, their physiological relevance has been questioned, particularly with regards to antigen presentation to CD8+ T cells, as only a minority of immunodominant cytotoxic CD8+ T-cell responses (CTL) are restricted by HLA-C.18 The HLA-C molecules clearly play a role in NK cell activation through binding of KIRs, but their involvement in T-cell-mediated responses is still poorly defined. This is not surprising considering that in 1993, it was still not clear whether or not HLA-C molecules ‘are regularly associated with peptide’,19 despite the fact that HLA-C-restricted CTL responses specific for influenza and Sendai viruses had been reported 5 years before in a mouse system where human Cw03 was over-expressed.19,20 Over the last 15 years, the biological significance of HLA-C-restricted responses was confirmed as the involvement of HLA-C antigens in alloreactivity was established and more HLA-C-restricted CTL epitopes have been identified, particularly in chronic infection settings such as Epstein–Barr virus and HIV infections (Table 1).21,22
Table 1.
Characterized HLA-C CTL epitopes
| Specificity | HLA-C allele | Antigen | References |
|---|---|---|---|
| Epstein–Barr virus | Cw*03 | EBNA1 | Ito et al., 2007 |
| Cw*04 | Rta | Pepperl et al., 1998 | |
| Cw*06 | Zta | Schendel et al., 1992 | |
| Cw*07 | – | ||
| Cytomegalovirus virus | Cw* | pp65 | Bihl et al., 2005 |
| 01Cw*04 | pp65 | ||
| Cw*08 | pp65 | ||
| Cw*12 | pp65 | ||
| Human immunodeficiency virus | Cw*01 | p24 gag | Bihl et al., 2005 |
| Cw*03 | gp41 | Bihl et al., 2005 | |
| Cw*04 | p24 gag/gp l20 | ||
| Cw*05 | rev | ||
| Cw*07 | nef | ||
| Cw*08 | p24 gag | ||
| Cw*12 | tat | ||
| Hepatitis C virus (HCV) | Cw*07 | P7 | Bihl et al., 2005 |
| Tumours | Cw*07 | MAGE-A12 | Nagata et al., 2005 |
| Cw*08 | MAGE-3 | Breckpot et al., 2004 | |
| Melan-A/MARTl | Larrieu et al., 2008 | ||
| Wilm's tumour antigen 1 | Wolfl et al., 2007 | ||
| Influenza virus + Sendai viruses | Cw*03 | – | Dill et al.20 |
The HLA-C alleles have been implicated in a number of human diseases, but it is not always clear whether the relationship is the result of the function of HLA-C as a T-cell restriction element or as a consequence of its interaction with KIR on NK cells (Table 2). Some associations are clearly attributable to HLA-C/KIR interactions, as they involve both a KIR as well as an HLA-C component. The first description of an HLA-C/KIR association with clinical outcome was made in hepatitis C virus infection, which leads to a chronic viral infection in approximately 80% of infected people. In a large study of hepatitis C virus-exposed individuals, Khakoo et al.23 found a strong association between the presence of KIR2DL3, along with homozygosity for the C1 group of HLA-C molecules that bind to KIR2DL2 and 2DL3, and clearance of infection. The interaction between KIR2DL3 and C1 molecules is notably weaker than that with KIR2DL2, and these two KIR variants act as alleles of one another, so the protective effect is thought to be mediated by NK cells that are relatively less inhibited than they would have been by KIR2DL2 interacting with C1 (or indeed by the more common KIR2DL1 molecule interacting with HLA-C2 group molecules).24
Table 2.
Clinical settings associated with HLA-C alleles
| Disease | Type of disease | HLA-C alleles | Type of association | References |
|---|---|---|---|---|
| Genital herpes simplex type 2 | Infection | Cw*02 | Severity of infection | Lekstrom-Himes et al., 1999 |
| Cw*04 | Susceptibility | Lekstrom-Himes et al., 1999 | ||
| Nasopharyngeal carcinoma | Cancer | Cw*0401 | Protection | Butsch Kovacic et al., 2005 |
| Human immunodeficiency virus | Infection | Cw*0401 | Protection (with B*8101) | Leslie et al. 2010 |
| B35 Cw4 haplotype | Rapid disease progression | Carrington, 1999 | ||
| Cw*12 | Protection | Thomas et al., 2009 | ||
| HLTV-1 | Infection | Cw*08 | Protection | Jeffery et al., 2000 |
| Cervical neoplasia | Cancer | HLA-C2 | Protection | Carrington et al., 2005 |
| Homozygosity | Invasive disease | Martin et al.35 | ||
| HLA-C1 | ||||
| Hepatitis C virus | Infection | HLA-C1 homozygosity | Protection in presence of KIR2DL3 | Khakoo et al.23 |
| Multinodular goitre | Autoimmunity | Cw*0401 | Protection | Rios et al., 2006 |
| Graves’ disease | Autoimmunity | Cw*03 | Protection | Simmonds et al., 2007 |
| Cw*07 | Susceptibility | Simmonds et al., 2007 | ||
| Cw*16 | Protection | Simmonds et al., 2007 | ||
| Psoriasis vulgaris | Autoimmunity | Cw*0501 | Protection when with HLA-A*3002 and B*1801 | Contu et al., 2004 |
| Cw*06 | Susceptibility | Szczerkowska-Dobosz et al., 2004 | ||
| Type I autoimmune hepatitis | Autoimmunity | Cw*0701 | Susceptibility | Strettell et al., 1997 |
| Psoriatic arthritis | Autoimmunity | Cw*0602 | Susceptibility | Gladman et al., 1999 |
| Cw*12 | Susceptibility | Liao and colleagues43 | ||
| Idiopathic inflammatory myopathies | Autoimmunity | Cw*0702 | Protection | O'Hanlon et al., 2005 |
| Primary sclerosing cholangitis | Autoimmunity | HLA-C1homozygosity | Susceptibility | Hov et al.22 |
A number of autoimmune diseases show HLA-C associations. The best known of these is the association of psoriasis with HLA-Cw6, which was first shown in candidate gene studies and subsequently confirmed in genome-wide association studies:25,26 people expressing this HLA-C allele not only have an earlier onset, but also more extensive and severe skin disease. HLA-Cw6 has also been linked with increased susceptibility to psoriaritic arthritis,27 but the association is less strong than with skin disease. It is thought that the disease may be mediated by CD8+ T-cell recognition of self-peptides derived from the affected tissues, presented by HLA-Cw6, a hypothesis supported by the recent finding of a genetic interaction between HLA-C and polymorphisms in ERAP1, a gene involved in the processing of antigenic peptides for T-cell recognition.28 However, HLA-C-restricted T cells have not so far been demonstrated in psoriaritic or any other HLA-C-associated autoimmune disease.
The interaction between fetal HLA-C molecules and KIRs expressed on uterine NK cells is thought to play a key role in the development of the placenta and the regulation of fetal growth.29 The main points of contact between the maternal immune system and the fetal placenta are the villous trophoblast (which expresses no MHC molecules and is therefore regarded as immunologically inert) and the extravillous trophoblast, which infiltrates the uterine wall and spiral arteries. The latter selectively expresses HLA-C and HLA-E molecules with the addition of trophoblast-specific HLA-G: the absence of class I A and B molecules is thought to prevent strong allogenic reactions that could damage the trophoblast (reviewed in ref. 29). Intriguingly, HLA-C molecules, the only polymorphic molecules on trophoblast, are found there largely in their fully assembled form, complexed with β2m (in contrast to the mixture of folded and unfolded HLA-C molecules characteristically found on somatic tissues):30 further evidence of the importance of HLA-C/KIR interactions at this site comes from the observation that uterine NK cells show preferential expression of HLA-C-interacting KIRs.31 Many pregnancy-related disorders, such as pre-eclampsia or recurrent miscarriage, can be attributed to inadequate trophoblast infiltration and such conditions frequently show HLA-C/KIR associations. Increased susceptibility to pre-eclampsia is associated with the combination of fetal HLA-C2 and predominantly inhibitory maternal KIRs,32 whereas reproductive failure mediated by fetal HLA-C233 can be overcome by the presence of maternal activating KIRs.34
HLA-C alleles, often in association with their interacting KIR molecules, have been linked with protection from or susceptibility to several malignancies, including cervical cancer:35 many of these studies suffer from relatively small patient numbers and the underlying mechanisms are not known.
Physiological importance of HLA-C-restricted T-cell responses
It has been suggested that the role of HLA-C in the context of T-cell priming differs from that of HLA-A and HLA-B molecules, as it is weakly expressed at the cell surface and displays relatively poor diversity at the antigen recognition site, which might hamper T-cell interaction with HLA molecules. However, when immunodominant HIV-specific T-cell responses are compared, HLA-C-restricted T cells are similar to their HLA-A and HLA-B-restricted counterparts, confirming that potent cytotoxic responses can be mediated by HLA-C as (i) they display the same activation markers; (ii) they display similar antiviral functions in vitro; (iii) they do not differ in polyfunctionality (five simultaneous functions such as interferon-γ, tumour necrosis factor-α, interleukin-2, macrophage inflammatory protein-1β and CD107) and (iv) they may represent the immunodominant HIV-1-specific T-cell response in some individuals.36–38 Moreover, HLA-Cw*03-restricted CD8+ T cells have recently been to shown to induce escape mutations in HIV as a result of immune pressure, demonstrating that HLA-C-restricted T cells are involved in defence against viral infections.39,40
Interestingly, two SNPs associated with increased susceptibility to psoriasis were also found to be associated with slow disease progression following HIV infection in two independent genome-wide association studies.2,41,42 One SNP lies in the HLA complex P5 (HCP5), which encodes a retroviral element, and the other is found ∼ 35 kb upstream of the transcriptional start site of HLA-C.43 Although the genotypes at – 35 kb associated with psoriasis and HIV are different (T correlates with increased susceptibility to psoriasis whereas C is associated with slow disease progression after HIV infection), this observation is quite interesting because psoriasis can be triggered by viral infections and HLA-C levels are directly affected by the SNP.43 It is not yet clear how the – 35 SNP variant slows HIV disease progression and whether the immune mechanisms of protection involve NK or T cells. Nevertheless, individuals with the protective genotype show increased expression of both HLA-C transcripts and surface protein, which correlates with better clinical status (CD4 counts and viral load) and delayed disease progression.42
The fact that HLA-C expression is associated with viral control is of interest considering the distinctive ability of HIV to selectively down-regulate HLA-A and HLA-B from the surface of infected cells without affecting HLA-C and HLA-E expression. Nef-mediated down-modulation of HLA-A and HLA-B, but not HLA-C and HLA-E,44 is based on amino acid differences in the cytoplasmic domain of these molecules.44,45 Briefly, Nef disrupts the MHC-1 endosomal trafficking through interactions with the cytoplasmic domain of MHC-I and clathrin adaptor protein.46,47 Three key amino acids (Tyr320, Ala324, and Asp327) in the cytoplasmic tails of class I HLAs are thought to be responsible for Nef down-regulation; the absence of one of these residues in HLA-C and HLA-E results in them being resistant to the Nef effect. Nef-mediated down-regulation of HLA-A and HLA-B has been thought to enable HIV to escape CTL recognition while maintaining resistance to NK recognition (Fig. 1a).
On the other hand, it is tempting to speculate that higher HLA-C expression results in a stronger HLA-C-restricted T-cell response, which might play a role in the control HIV replication in individuals with the protective variant. The fact that HLA-C molecules remain at the surface of infected cells in the absence of HLA-A and HLA-B during HIV infection may amplify the relative contribution of HLA-C-restricted CTLs in this particular situation. This raises the possibility that HLA-C-restricted T-cell responses also play a role in other chronic settings for which HLA-C-restricted epitopes have been identified, such as Epstein–Barr virus, cytomegalovirus and several malignancies, in which class I down-regulation is also observed as a probable immune evasion mechanism (Table 1). Although not selective like HIV Nef down-regulation of HLA-A and HLA-B and achieved through different mechanisms, Class I down-modulation is a common feature of all clinical settings associated with immunodominant T-cell responses restricted by HLA-C. Moreover, low HLA-C expression in the thymus might play an important role in shaping the T-cell repertoire, as the extent to which the TCR is engaged by peptide–MHC complexes in the thymus determines which clones will be positively and negatively selected.48 Low HLA-C expression on thymic dendritic cells could result in fewer clones being negatively selected in the thymus, which would result in an extremely diverse HLA-C-restricted T-cell repertoire. This would explain why HLA-C alleles are associated with several immunodominant responses in chronic infection (the most striking being those associated with control of HIV replication) as well as several autoimmune disorders. Therefore the extent to which these factors alter the specificity, the quality and the magnitude of the immune response merits further investigation.
Disclosures
The authors declare no financial conflicts of interest.
References
- 1.Collins KL, Chen BK, Kalams SA, Walker BD, Baltimore D. HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature. 1998;391:397–401. doi: 10.1038/34929. [DOI] [PubMed] [Google Scholar]
- 2.Fellay J, Shianna KV, Ge D, et al. A whole-genome association study of major determinants for host control of HIV-1. Science. 2007;317:944–7. doi: 10.1126/science.1143767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goulder PJ, Bunce M, Luzzi G, Phillips RE, McMichael AJ. Potential underestimation of HLA-C-restricted cytotoxic T-lymphocyte responses. AIDS. 1997;11:1884–6. doi: 10.1097/00002030-199715000-00016. [DOI] [PubMed] [Google Scholar]
- 4.Zemmour J, Parham P. Distinctive polymorphism at the HLA-C locus: implications for the expression of HLA-C. J Exp Med. 1992;176:937–50. doi: 10.1084/jem.176.4.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neisig A, Melief CJ, Neefjes J. Reduced cell surface expression of HLA-C molecules correlates with restricted peptide binding and stable TAP interaction. J Immunol. 1998;160:171–9. [PubMed] [Google Scholar]
- 6.Turner S, Ellexson ME, Hickman HD, Sidebottom DA, Fernandez-Vina M, Confer DL, Hildebrand WH. Sequence-based typing provides a new look at HLA-C diversity. J Immunol. 1998;161:1406–13. [PubMed] [Google Scholar]
- 7.McCutcheon JA, Gumperz J, Smith KD, Lutz CT, Parham P. Low HLA-C expression at cell surfaces correlates with increased turnover of heavy chain mRNA. J Exp Med. 1995;181:2085–95. doi: 10.1084/jem.181.6.2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tibensky D, Decary F, Delovitch TL. HLA-C genes are transcribed in HLA-C blank individuals. Immunogenetics. 1988;27:220–4. doi: 10.1007/BF00346590. [DOI] [PubMed] [Google Scholar]
- 9.Neefjes JJ, Ploegh HL. Allele and locus-specific differences in cell surface expression and the association of HLA class I heavy chain with beta 2-microglobulin: differential effects of inhibition of glycosylation on class I subunit association. Eur J Immunol. 1988;18:801–10. doi: 10.1002/eji.1830180522. [DOI] [PubMed] [Google Scholar]
- 10.Garcia-Ruano AB, Mendez R, Romero JM, Cabrera T, Ruiz-Cabello F, Garrido F. Analysis of HLA-ABC locus-specific transcription in normal tissues. Immunogenetics. 2010;62:711–9. doi: 10.1007/s00251-010-0470-z. [DOI] [PubMed] [Google Scholar]
- 11.Sibilio L, Martayan A, Setini A, Lo Monaco E, Tremante E, Butler RH, Giacomini P. A single bottleneck in HLA-C assembly. J Biol Chem. 2008;283:1267–74. doi: 10.1074/jbc.M708068200. [DOI] [PubMed] [Google Scholar]
- 12.Stam NJ, Spits H, Ploegh HL. Monoclonal antibodies raised against denatured HLA-B locus heavy chains permit biochemical characterization of certain HLA-C locus products. J Immunol. 1986;137:2299–306. [PubMed] [Google Scholar]
- 13.Boyington JC, Motyka SA, Schuck P, Brooks AG, Sun PD. Crystal structure of an NK cell immunoglobulin-like receptor in complex with its class I MHC ligand. Nature. 2000;405:537–43. doi: 10.1038/35014520. [DOI] [PubMed] [Google Scholar]
- 14.Fan QR, Long EO, Wiley DC. Crystal structure of the human natural killer cell inhibitory receptor KIR2DL1-HLA-Cw4 complex. Nat Immunol. 2001;2:452–60. doi: 10.1038/87766. [DOI] [PubMed] [Google Scholar]
- 15.Winter CC, Long EO. A single amino acid in the p58 killer cell inhibitory receptor controls the ability of natural killer cells to discriminate between the two groups of HLA-C allotypes. J Immunol. 1997;158:4026–8. [PubMed] [Google Scholar]
- 16.Garcia KC, Degano M, Stanfield RL, Brunmark A, Jackson MR, Peterson PA, Teyton L, Wilson IA. An alphabeta T cell receptor structure at 2.5 A and its orientation in the TCR–MHC complex. Science. 1996;274:209–19. doi: 10.1126/science.274.5285.209. [DOI] [PubMed] [Google Scholar]
- 17.Boyington JC, Sun PD. A structural perspective on MHC class I recognition by killer cell immunoglobulin-like receptors. Mol Immunol. 2002;38:1007–21. doi: 10.1016/s0161-5890(02)00030-5. [DOI] [PubMed] [Google Scholar]
- 18.Matthews PC, Prendergast A, Leslie A, et al. Central role of reverting mutations in HLA associations with human immunodeficiency virus set point. J Virol. 2008;82:8548–59. doi: 10.1128/JVI.00580-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Falk K, Rotzschke O, Grahovac B, et al. Allele-specific peptide ligand motifs of HLA-C molecules. Proc Natl Acad Sci U S A. 1993;90:12005–9. doi: 10.1073/pnas.90.24.12005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dill O, Kievits F, Koch S, Ivanyi P, Hammerling GJ. Immunological function of HLA-C antigens in HLA-Cw3 transgenic mice. Proc Natl Acad Sci U S A. 1988;85:5664–8. doi: 10.1073/pnas.85.15.5664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pei J, Akatsuka Y, Anasetti C, Lin MT, Petersdorf EW, Hansen JA, Martin PJ. Generation of HLA-C-specific cytotoxic T cells in association with marrow graft rejection: analysis of alloimmunity by T-cell cloning and testing of T-cell-receptor rearrangements. Biol Blood Marrow Transplant. 2001;7:378–83. doi: 10.1053/bbmt.2001.v7.pm11529487. [DOI] [PubMed] [Google Scholar]
- 22.Hov JR, Lleo A, Selmi C, Woldseth B, Fabris L, Strazzabosco M, Karlsen TH, Invernizzi P. Genetic associations in Italian primary sclerosing cholangitis: heterogeneity across Europe defines a critical role for HLA-C. J Hepatol. 2010;52:712–7. doi: 10.1016/j.jhep.2009.11.029. [DOI] [PubMed] [Google Scholar]
- 23.Khakoo SI, Thio CL, Martin MP, et al. HLA and NK cell inhibitory receptor genes in resolving hepatitis C virus infection. Science. 2004;305:872–4. doi: 10.1126/science.1097670. [DOI] [PubMed] [Google Scholar]
- 24.Parham P. Immunology. NK cells lose their inhibition. Science. 2004;305:786–7. doi: 10.1126/science.1102025. [DOI] [PubMed] [Google Scholar]
- 25.Cao J, McNevin J, McSweyn M, Liu Y, Mullins JI, McElrath MJ. Novel cytotoxic T-lymphocyte escape mutation by a three-amino-acid insertion in the human immunodeficiency virus type 1 p6Pol and p6Gag late domain associated with drug resistance. J Virol. 2008;82:495–502. doi: 10.1128/JVI.01096-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nair RP, Duffin KC, Helms C, et al. Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat Genet. 2009;41:199–204. doi: 10.1038/ng.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ho PY, Barton A, Worthington J, et al. Investigating the role of the HLA-Cw*06 and HLA-DRB1 genes in susceptibility to psoriatic arthritis: comparison with psoriasis and undifferentiated inflammatory arthritis. Ann Rheum Dis. 2008;67:677–82. doi: 10.1136/ard.2007.071399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Strange A, Capon F, Spencer CC, et al. A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat Genet. 2010;42:985–90. doi: 10.1038/ng.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Trowsdale J, Moffett A. NK receptor interactions with MHC class I molecules in pregnancy. Semin Immunol. 2008;20:317–20. doi: 10.1016/j.smim.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 30.Apps R, Gardner L, Hiby SE, Sharkey AM, Moffett A. Conformation of human leucocyte antigen-C molecules at the surface of human trophoblast cells. Immunology. 2008;124:322–8. doi: 10.1111/j.1365-2567.2007.02789.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sharkey AM, Gardner L, Hiby S, et al. Killer Ig-like receptor expression in uterine NK cells is biased toward recognition of HLA-C and alters with gestational age. J Immunol. 2008;181:39–46. doi: 10.4049/jimmunol.181.1.39. [DOI] [PubMed] [Google Scholar]
- 32.Hiby SE, Walker JJ, O'Shaughnessy KM, Redman CW, Carrington M, Trowsdale J, Moffett A. Combinations of maternal KIR and fetal HLA-C genes influence the risk of preeclampsia and reproductive success. J Exp Med. 2004;200:957–65. doi: 10.1084/jem.20041214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hiby SE, Regan L, Lo W, Farrell L, Carrington M, Moffett A. Association of maternal killer-cell immunoglobulin-like receptors and parental HLA-C genotypes with recurrent miscarriage. Hum Reprod. 2008;23:972–6. doi: 10.1093/humrep/den011. [DOI] [PubMed] [Google Scholar]
- 34.Hiby SE, Apps R, Sharkey AM, et al. Maternal activating KIRs protect against human reproductive failure mediated by fetal HLA-C2. J Clin Invest. 2010;120:4102–10. doi: 10.1172/JCI43998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martin MP, Borecki IB, Zhang Z, et al. HLA-Cw group 1 ligands for KIR increase susceptibility to invasive cervical cancer. Immunogenetics. 2010;62:761–5. doi: 10.1007/s00251-010-0477-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mkhwanazi N, Thobakgale CF, van der Stok M, et al. Immunodominant HIV-1-specific HLA-B- and HLA-C-restricted CD8+ T cells do not differ in polyfunctionality. Virology. 2010;405:483–91. doi: 10.1016/j.virol.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Makadzange AT, Gillespie G, Dong T, et al. Characterization of an HLA-C-restricted CTL response in chronic HIV infection. Eur J Immunol. 2010;40:1036–41. doi: 10.1002/eji.200939634. [DOI] [PubMed] [Google Scholar]
- 38.Adnan S, Balamurugan A, Trocha A, Bennett MS, Ng HL, Ali A, Brander C, Yang OO. Nef interference with HIV-1-specific CTL antiviral activity is epitope specific. Blood. 2006;108:3414–9. doi: 10.1182/blood-2006-06-030668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Honeyborne I, Codoner FM, Leslie A, et al. HLA-Cw*03-restricted CD8+ T-cell responses targeting the HIV-1 gag major homology region drive virus immune escape and fitness constraints compensated for by intracodon variation. J Virol. 2010;84:11279–88. doi: 10.1128/JVI.01144-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leslie A, Matthews PC, Listgarten J, et al. Additive contribution of HLA class I alleles in the immune control of HIV-1 infection. J Virol. 2010;84:9879–88. doi: 10.1128/JVI.00320-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fellay J, Ge D, Shianna KV, et al. Common genetic variation and the control of HIV-1 in humans. PLoS Genet. 2009;5:e1000791. doi: 10.1371/journal.pgen.1000791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thomas R, Apps R, Qi Y, et al. HLA-C cell surface expression and control of HIV/AIDS correlate with a variant upstream of HLA-C. Nat Genet. 2009;41:1290–4. doi: 10.1038/ng.486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu Y, Helms C, Liao W, et al. A genome-wide association study of psoriasis and psoriatic arthritis identifies new disease loci. PLoS Genet. 2008;4:e1000041. doi: 10.1371/journal.pgen.1000041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cohen GB, Gandhi RT, Davis DM, Mandelboim O, Chen BK, Strominger JL, Baltimore D. The selective downregulation of class I major histocompatibility complex proteins by HIV-1 protects HIV-infected cells from NK cells. Immunity. 1999;10:661–71. doi: 10.1016/s1074-7613(00)80065-5. [DOI] [PubMed] [Google Scholar]
- 45.Williams M, Roeth JF, Kasper MR, Fleis RI, Przybycin CG, Collins KL. Direct binding of human immunodeficiency virus type 1 Nef to the major histocompatibility complex class I (MHC-I) cytoplasmic tail disrupts MHC-I trafficking. J Virol. 2002;76:12173–84. doi: 10.1128/JVI.76.23.12173-12184.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Noviello CM, Benichou S, Guatelli JC. Cooperative binding of the class I major histocompatibility complex cytoplasmic domain and human immunodeficiency virus type 1 Nef to the endosomal AP-1 complex via its mu subunit. J Virol. 2008;82:1249–58. doi: 10.1128/JVI.00660-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wonderlich ER, Williams M, Collins KL. The tyrosine binding pocket in the adaptor protein 1 (AP-1) mu1 subunit is necessary for Nef to recruit AP-1 to the major histocompatibility complex class I cytoplasmic tail. J Biol Chem. 2008;283:3011–22. doi: 10.1074/jbc.M707760200. [DOI] [PubMed] [Google Scholar]
- 48.Potter TA, Hansen TH, Habbersett R, Ozato K, Ahmed A. Flow microfluorometric analysis of H-2L expression. J Immunol. 1981;127:580–4. [PubMed] [Google Scholar]