

Abstract
Pathogenesis of Mycoplasma pneumoniae infection is considered to be in part attributed to excessive immune responses. Mycoplasma pneumoniae shows strong cytoadherence to host cells and this cytoadherence is thought to be involved in the progression of pneumonia. However, the interaction between the cytoadherence and the immune responses is not known in detail. In this study, we demonstrated that the induction of pro-inflammatory cytokines in the human monocyte cell line THP-1 is dependent on the property of cytoadherence of M. pneumoniae. A wild-type strain of M. pneumoniae with cytoadherence ability induced pro-inflammatory cytokines such as tumour necrosis factor-α and interleukin-1β (IL-1β). Whereas, heat-killed M. pneumoniae and cytoadherence-deficient mutants of M. pneumoniae caused significantly less production of pro-inflammatory cytokines than the wild-type strain. The wild-type strain induced pro-inflammatory cytokines in an endocytosis-independent manners, but the induction by heat-killed M. pneumoniae and cytoadherence-deficient mutants was dependent on endocytosis. Moreover, the wild-type strain induced caspase-1 production and ATP efflux, promoting the maturation of IL-1β and release of the pro-IL-1β precursor, whereas heat-killed M. pneumoniae and the cytoadherence-deficient mutants failed to induce them. These data suggest that the cytoadherence ability of M. pneumoniae activates immune responses and is involved in the pathogenesis of M. pneumoniae infection.
Keywords: cytoadherence, inflammasome, mycoplasma, THP-1, toll-like receptors
Introduction
Mycoplasmas are wall-less parasitic bacteria, and the smallest organisms capable of self-replication.1Mycoplasma pneumoniae causes primary atypical pneumonia, tracheobronchitis, pharyngitis and asthma in humans.2–4 However, pathogenic agents such as endotoxins and exotoxins that cause such diseases have not been identified in M. pneumoniae. Cytoadherence of invading mycoplasma to the respiratory epithelium, localized host cell injury, and overaggressive inappropriate immune responses seem to contribute to the pathogenesis of M. pneumoniae infection.2,5
We previously identified three lipoproteins responsible for nuclear factor-κB (NF-κB) activation.6 One was MPN602, a subunit b of F0F1-type ATPase (F0F1-ATPase),7 a diacylated lipoprotein. The activation of NF-κB by F0F1-ATPase was dependent on Toll-like receptor 1 (TLR1), TLR2 and TLR6. We identified two other lipoproteins, MPN611 and MPN162, and designated them NF-κB-activating lipoprotein 1 (N-ALP1) and N-ALP2, respectively. N-ALP1 and N-ALP2 activated TLR signalling through TLR1 and TLR2. Both N-ALP1 and N-ALP2 were assumed to be triacylated lipoproteins based on their functional analysis.8
Toll-like receptors with the function of pattern-recognition receptors play critical roles in early innate recognition and inflammatory responses by the host against invading microbes.9,10 Among 10 TLR family members reported, TLR2, TLR4, TLR5 and TLR9 have been implicated in the recognition of different bacterial components. Peptidoglycan, lipoarabinomannan, zymosan and lipoproteins from various microorganisms are recognized by TLR2.11–17 On the other hand, lipopolysaccharide, bacterial flagellin and bacterial DNA are recognized by TLR4, TLR5 and TLR9, respectively.18–21 These TLR family members are shown to activate NF-κB via interleukin-1 receptor (IL-1R) -associated signal molecules, including myeloid differentiation protein, IL-1R-activated kinase, tumour necrosis factor receptor-associated factor 6 and NF-κB-inducing kinase.22
Interleukin-1β is an important pro-inflammatory cytokine and mature IL-1β is produced by cleavage of the pro-IL-1β precursor by caspase-1.23 Caspase-1 is activated within a large protein complex called the inflammasome.24 The inflammasome is composed of nucleotide-binding domain, leucine-rich repeat-containing (NLR) proteins, apoptosis-associated speck-like protein containing a C-terminal caspase recruitment domain, and caspase-1. The inflammasome responds to a variety of signals. Intracellular bacterial toxins,25 pathogen-associated molecular patterns (PAMPs) such as muramyl dipeptide26 or flagellin,27 the stress-associated danger signals (DAMPs) such as ATP28–30 or monosodium urate28 activate the inflammasome. The ATP activates the inflammasome through binding to the P2X7 receptor followed by potassium efflux31 and recruitment of pore-forming hemi-channel, pannexin-1.32 Phagocytosis-dependent production of reactive oxygen species is also reported to stimulate the inflammasome.33,34
Cytoadherence by M. pneumoniae in the respiratory tract is the initial event of infection. The cytoadherence is mediated by P1 adhesin and other additional proteins such as P30 or high molecular weight (HMW) proteins.35–38 Although the cytoadherence of M. pneumoniae is thought to be responsible for the pathogenesis of M. pneumoniae infection,39,40 it is not known precisely how the cytoadherence is involved in inflammatory responses.
In this study, we examined the interaction between the cytoadherence and inflammatory responses. A wild-type strain of M. pneumoniae with cytoadherence ability induced pro-inflammatory cytokines, such as tumour necrosis factor-α (TNF-α) and IL-1β, whereas heat-killed M. pneumoniae or cytoadherence-deficient mutants induced significantly fewer pro-inflammatory cytokines than the wild-type strain. The wild-type strain of M. pneumoniae induced TNF-α and the precursor of IL-1β in an endocytosis-independent manner, and also activated caspase-1 production and ATP efflux, promoting the maturation of IL-1β. These data suggest that the cytoadherence capability of M. pneumoniae activates inflammatory responses and is involved in the pathogenesis of M. pneumoniae infection.
Materials and methods
Mycoplasma pneumoniae strains
Cytoadherence-deficient mutants of M. pneumoniae, M5, M6, class I, class II, class III and class IV were obtained from Dr Miyata.41 These mutants originated from wild-type strain M129.42–45 The M. pneumoniae wild-type strain (M129) and cytoadherence-deficient mutants were cultured in Pleuropneumonia-like organisms (PPLO) broth (Difco, Franklin Lakes, NJ, USA) containing 10% horse serum, 0·25% glucose, 0·25% yeast extract, 0·002% phenol red, at pH 7·6. Each strain was cultured to the beginning of a stationary phase (the colour of medium turned slightly orange). The amount of each strain was adjusted by optical density (OD595) in PBS. The DNA of M. pneumoniae was purified using a FastPure DNA kit (Takara, Tokyo, Japan). The amounts of DNA were measured by optical density (OD280).
Cells
Cells of a human monocytic cell line, THP-1 and a human lung squamous cell carcinoma cell line, EBC-1 were cultured in RPMI-1640 medium containing 10% fetal calf serum (Mitsubishi Chemical, Tokyo, Japan) and 2 mm l-glutamine. Cells of a human kidney cell line, 293T were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal calf serum and 2 mm l-glutamine. To exclude the possibility of mycoplasma infections in the cell cultures, these cell suspensions were inoculated onto PPLO agar medium (Difco) once a month. Cells were also stained with 4′,6-diamidino-2-phenylindole and contaminated mycoplasmas were checked once a week. Peripheral blood mononuclear cells (PBMC) were separated using Lymphoprep (Axis-Shield PoC, Oslo, Norway) and cultured in RPMI-1640 medium containing 10% fetal calf serum.
Cell treatment
A total of 2 × 105 cells/500 μl THP-1 or EBC-1 were cultured for 24 hr before infection in a 24-well plate. Cells were infected with 50 μl M. pneumoniae (OD595 = 0·1) for 6 hr and the cells were harvested by centrifugation for 10 min at 300 g. A total of 5 × 105 cells/500 μl PBMC were cultured for 3 hr before infection in a 24-well plate. Cells were infected with 50 μl M. pneumoniae (OD595 = 0·01) for 3 hr and the cells were harvested by centrifugation for 10 min at 300 g. Concentrations of pro-inflammatory cytokines in supernatants were measured by ELISA. The TLR2 was blocked by 10 μg/ml mouse anti-human TLR2 monoclonal antibody, IMG-416 (Imgenex, San Diego, CA, USA). Endocytosis was examined in the presence of 1·0 μm cytochalasin D or 100 nm bafilomycin A1 (Wako, Osaka, Japan). P2X7 receptor was blocked using 300 μm oxidized ATP (Sigma, St Louis, MO, USA). Inhibition of caspase-1 was performed with 100 μm z-Val-Ala-Asp(oMe)-CH2F (z-VAD-FMK) or Ac-Trp-Glu-His-Asp-H (Ac-WEHD-CHO) (Bachem, Bubendorf, Switzerland).
ELISA
Amounts of TNF-α in the supernatants of cell culture were measured using ELISA development kits (R&D Systems, Minneapolis, MN, USA) according to the manufacturer's instructions. Amounts of IL-1β in the supernatants of cell culture were measured using ELISA development kits (R&D Systems). This kit detects both precursor and mature forms of IL-1β. Amounts of mature caspase-1 were measured using a Quantikine ELISA kit (R&D Systems).
ATP concentration
THP-1 cells were cultured in the presence of 200 μm ecto-ATPase inhibitor ARL 1 hr before infection. The cells were infected with M. pneumoniae for 6 hr and the levels of ATP in the supernatant were determined using ENLITEN luciferase reagent (Promega, Madison, WI, USA).
Western blot analysis
A total of 2 × 105 cells/500 μl THP-1 cells were cultured for 24 hr before infection in a 24-well plate. The cells were infected with 50 μl M. pneumoniae (OD595 = 0·1) for 6 hr. Cells and supernatants were collected by centrifugation for 10 min at 3000 g, and then mixed with lithium dodecyl sulphate sample buffer (Invitrogen, Carlsbad, CA, USA) and heated at 70° for 10 min. The proteins were resolved by SDS–PAGE and transferred to PVDF membranes. The membranes were probed with goat anti-human IL-1β (R&D Systems) and horseradish peroxidase-conjugated donkey anti-goat IgG (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Protein bands were visualized using ECL Advance (GE Healthcare, Amersham, UK).
Lipopeptides
A synthetic lipopeptide containing the N-terminal 20 amino acids of F0F1-ATPase derived from M. pneumoniae (FAM20) was synthesized by Bio Synthesis (Lewisville, TX, USA).7
The (S)-[2,3-Bis(palmitoyloxy)-(2-RS)-propyl]-N-palmitoyl-(R)-Cys-(S)-Ser-(S)-Lys4-OH, 3HCl (P3CSK4)11 was purchased from Calbiochem (Darmstadt, Germany).
Expression vectors
To prepare TLR2 expression vectors (pFLAG-TLR2), the coding regions of TLR2 minus the respective N-terminal signal sequences were amplified by PCR from a cDNA of THP-1 and cloned into the expression vector pFLAG-CMV1 (Sigma), in which a preprotrypsin leader precedes an N-terminal FLAG epitope. An NF-κB Cis-Reporting System containing pNF-kB-luc was purchased from Stratagene (La Jolla, CA, USA).
Transfection and luciferase assay
Transient transfection was performed using FuGENE6 (Roche, Basel, Switzerland) according to the manufacturer's instructions. A total of 1 × 105 293T cells were transfected with 0·1 μg pFLAG-TLR2, 0·1 μg pNF-kB-luc and 0·01 μg pRL-TK internal control plasmid (Promega) in 24-well plates. After 48 hr, the transfected cells were infected with M. pneumoniae or stimulated with lipopeptides. After a further 6 hr of incubation, the cells were lysed and assayed for luciferase activity using a Dual-Luciferase Reporter Assay System (Promega). Both firefly and Renilla luciferase activity were monitored with a Lumat LB9507 luminometer (Berthold, Wildbad, Germany). Normalized reporter activity is expressed as the firefly luciferase value divided by the Renilla luciferase value. Relative fold induction is calculated as the normalized reporter activity of the test samples divided by the unstimulated samples.
Statistical analysis
Results expressed as means and standard deviations were compared using one-way analysis of variance. The differences between each group were compared by multiple comparisons (Bonferroni t-test). Differences were considered significant at P < 0·01.
Results
Pro-inflammatory cytokine induction by M. pneumoniae strains and heat-killed M. pneumoniae
To examine the interaction between cytoadherence and inflammatory responses, wild-type strain (M129), cytoadherence-deficient mutants (M5 and M6) originating from M12942–45 or heat-killed M. pneumoniae were infected on THP-1 cells and the levels of pro-inflammatory cytokines induced by infection were measured by ELISA. As M. pneumoniae strongly adheres to itself, it is difficult to determine a precise colony-forming unit count. Therefore, the amounts of mycoplasma cells were adjusted using the OD595 in PBS. Table 1 shows the DNA amounts of wild-type (M129) and cytoadherence-deficient mutants (M5 and M6) of M. pneumoniae in 100 μl PBS at 0·1 OD595. M129 showed the same DNA amount as M5 and M6 at the same optical density.
Table 1.
Strains of Mycoplasma pneumoniae used in this study
| Strain | Missing proteins | Mutated gene | DNA1 (μg) | Reference |
|---|---|---|---|---|
| M129 | – | – | 4·57 ± 0·67 | – |
| M5 | P90, P40 | NR2 | 4·39 ± 0·62 | 42 |
| M6 | HMW1, P30 | hmw1, p30 | 4·42 ± 0·71 | 43–45 |
DNA, the amounts of M. pneumoniae in 100 μl PBS were adjusted to OD595 = 0·1 in 96-well plates. DNA was purified as described in the Materials and methods. The amounts of DNA were measured by optical density at 280 nm. All values represent the means and SD of three assays.
NR, not reported.
When THP-1 cells were infected with M129, TNF-α and IL-1β were strikingly induced (Fig. 1a,b). In contrast, heat-killed M129 induced the production of TNF-α at lower levels, but the induction of IL-1β was completely decreased to the control level. Next, THP-1 cells were infected with M129 and cytoadherence-deficient mutants (M5 and M6) at graded concentrations (Fig. 2a,b). M129 induced TNF-α and IL-1β in a dose-dependent manner. Although M5 at high doses (OD595 = 0·2) induced TNF-α as much as M129, M5 at low doses (OD595 = 0·1 and 0·05) and M6 at all doses induced lower levels of TNF-α than M129. In contrast, M5 and M6 at all doses induced lower levels of IL-1β than M129. We also tested four other cytoadherence-deficient mutants, class I, class II, class III and class IV (data not shown). Similar to M5 and M6, the induction levels of TNF-α and IL-1β by these mutants were also decreased compared with M129. To examine the time kinetics of pro-inflammatory cytokines induced by M. pneumoniae, THP-1 cells were infected with M129, M5 and M6. The induced TNF-α and IL-1β were measured by ELISA at 0, 1, 3, 6, 12 and 24 hr post-infection (Fig. 2c,d). The induction levels of TNF-α were maximal at 3 and 6 hr whereas the maximal induction levels of IL-1β were observed at 6 and 12 hr. When THP-1 cells were infected with M5 and M6, the induction levels of TNF-α and IL-1β were strikingly low compared with M129 at all time-points. To confirm the importance of cytoadherence in cytokine production of different types of cells, human PBMC and a human lung squamous cell carcinoma cell line, EBC-1 cells were infected with M129, M5 and M6. CD14+ cells in PBMC were 12·8% by flow cytometry analysis. When PBMC were infected with M129, high levels of TNF-α and IL-1β were induced. In contrast, M5 and M6 induced low levels of these cytokines (Supplementary materials, Fig. S1a,b). Likewise, in EBC-1 cells, M129 induced high levels of IL-8, but M5 and M6 failed to induce IL-8 (Fig. S1c). These results indicate that the adherent M. pneumoniae possesses the ability to induce potent inflammatory responses compared with the cytoadherence-deficient mutants.
Figure 1.
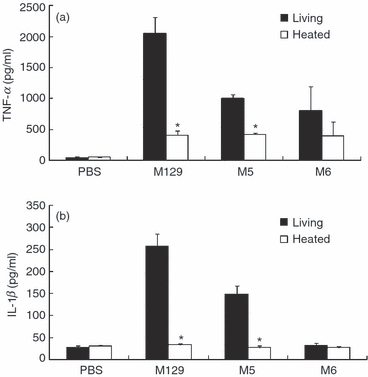
Induction of tumour necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) by heat-killed Mycoplasma pneumoniae. The M. pneumoniae was killed by heating at 60° for 30 min. THP-1 cells (2 × 105 cells/500 μl) were treated with 50 μl living or heat-killed M. pneumoniae (OD595 = 0.1). After 6 hr incubation, amounts of TNF-α (a) and IL-1β (b) in culture medium were measured by ELISA. All values represent the means and SD of three assays. An asterisk indicates that the P-value is < 0.01 for a comparison with living M. pneumoniae by multiple comparison.
Figure 2.
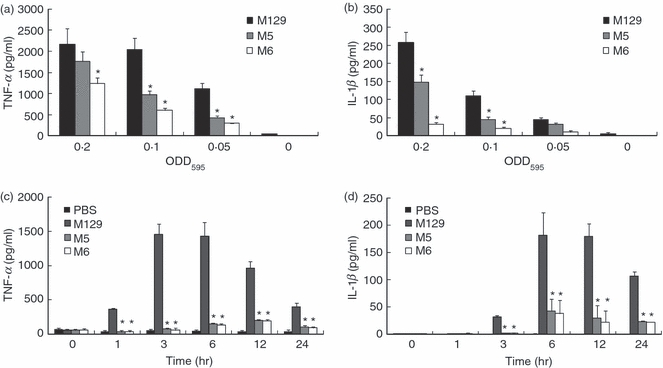
Induction of tumour necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) by cytoadherence-deficient mutants of Mycoplasma pneumoniae. THP-1 cells (2 × 105 cells/500 μl) were infected with 50 μl of the indicated amounts of wild-type or mutants of M. pneumoniae. After 6 hr incubation, amounts of TNF-α (a) and IL-1β (b) in culture medium were measured by ELISA. THP-1 cells (2 × 105 cells/500 ml) were infected with 50 ml of M. pneumoniae (OD595 = 0.1). Amounts of TNF-α (c) and IL-1β (d) in culture medium were measured by ELISA at the indicated time post-infection. All values represent the means and SD of three assays. An asterisk indicates that the P-value is < 0.01 for a comparison with wild-type of M. pneumoniae by multiple comparison.
Induction of TLR2 signalling
We previously demonstrated that lipoproteins of M. pneumoniae induced inflammatory responses through TLR2.7,8 To examine whether M129 augments TLR2 signalling, the levels of NF-κB induction through TLR2 were measured using NF-κB reporter assay system. A human kidney cell line, 293T cells, transfected with TLR2 expression vector (pFLAG-TLR2) and NF-κB reporter vector (pNF-kB-luc) was infected with M. pneumoniae and the induced luciferase activity was measured (Fig. 3a). The M. pneumoniae failed to induce NF-κB activation in 293T cells transfected with pNF-kB-luc alone, indicating that TLR2 is necessary for M. pneumoniae to induce the NF-κB activation. Surprisingly, M5 and M6 as well as M129 induced comparable levels of NF-κB activation through TLR2. Heat-killed M. pneumoniae seemed to slightly increase the induction levels of NF-κB compared with living mycoplasmas. Anti-TLR2 monoclonal antibody decreased the levels of TNF-α and IL-1β induction by M5 and M6 down to the control levels. In contrast, the induction levels of TNF-α and IL-1β by M129 were partially blocked by the antibody (Fig. 3b,c).
Figure 3.
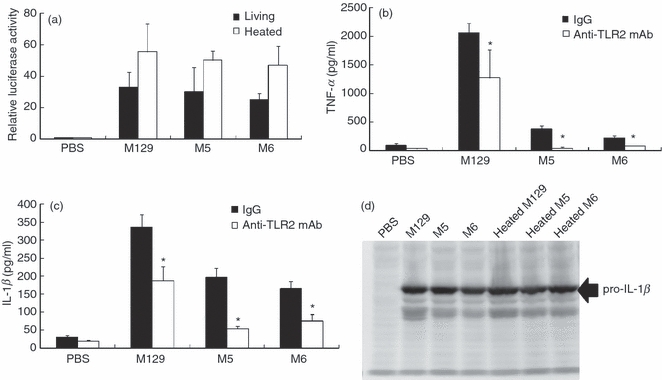
Induction of Toll-like receptor 2 (TLR2) signalling. (a) 293T cells (2 × 105 cells/500 μl) were transfected with the 0.1 μg/ml pFLAG-TLR2, 0.01 μg/ml pNF-kB-luc and 0.01 μg/ml pRL-TK. After 24 hr incubation, the cells were infected with 50 μl wild-type or mutants of Mycoplasma pneumoniae (OD595 = 0.1). After 6 hr incubation, the activity of luciferase was measured as described in the Materials and methods. All values represent the means and SD of three assays. (b, c) THP-1 cells (2 × 105 cells/500 μl) were treated with 10 μg/ml anti-TLR2 monoclonal antibody and infected with 50 μl wild-type or mutants of M. pneumoniae (OD595 = 0.1). After 6 hr incubation, amounts of tumour necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) in culture medium were measured by ELISA. All values represent the means and SD of three assays. An asterisk indicates that the P-value is < 0.01 for a comparison with control IgG by multiple comparison (d) THP-1 cells (2 × 105 cells/500 μl) were treated with 50 μl living or heat-killed M. pneumoniae (OD595 = 0.1). After 6 hr incubation, THP-1 cells were harvested and the induction of pro-IL-1β precursor was investigated by Western blotting.
The wild-type strain of M. pneumoniae strongly induced IL-1β production compared with cytoadherence-deficient mutants (Figs 1b and 2b). The induction of pro-IL-1β precursor in THP-1 cells was therefore examined by Western blotting (Fig. 3d). Similar to the results of TLR 2 signalling, the pro-IL-1β precursor was induced at the same levels by treatment with M129, M5, M6 and heat-killed mycoplasmas. These results suggest that the enhancement of inflammatory responses by cytoadherent mycoplasma is mostly TLR2-independent.
Endocytosis-independent induction of pro-inflammatory cytokines
To elucidate the mechanism by which cytoadherent mycoplasma enhances pro-inflammatory cytokine induction, we examined the involvement of endocytosis. THP-1 cells were treated with cytochalasin D, which disrupts actin filaments and inhibits the initiation of endocytosis.46 Cytochalasin D completely inhibited TNF-α production by M5, M6 and heat-killed mycoplasma strains, whereas TNF-α production by M129 was not affected (Fig. 4a). Interleukin-1β production by M129 was slightly decreased by cytochalasin D, but the production by M5, M6 and heat-killed mycoplasma strains was diminished (Fig. 4b). Subsequently, induction of pro-IL-1β precursor was examined by Western blotting. The induction levels of M5, M6 and heat-killed mycoplasma strains were lowered in the presence of cytochalasin D, but the induction by M129 was unaffected (Fig. 4c and data not shown). These results suggest that cytoadherent mycoplasma activates TNF-α and pro-IL-1β precursor production in an endocytosis-independent manner, whereas endocytosis is important for cytoadherence-deficient mycoplasma strains to induce TNF-α and IL-1β.
Figure 4.
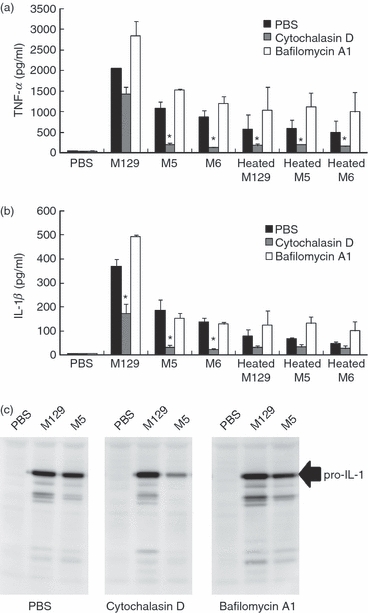
Endocytosis-independent induction of pro-inflammatory cytokines. THP-1 cells (2 × 105 cells/500 μl) were cultured in the presence of 1.0 μm cytochalasin D or 100 nm bafilomycin A1 for 1 hr, and treated with 50 μl of living or heat-killed Mycoplasma pneumoniae (OD595 = 0.1). After 6 hr incubation, amounts of tumour necrosis factor-α (TNF-α) (a) and interleukin-1β (IL-1β) (b) in culture medium were measured by ELISA. Cells were harvested and the induction levels of pro-IL-1β precursor were determined by Western blotting (c). All values represent the means and SD of three assays. An asterisk indicates that the P-value is < 0.01 for a comparison with PBS treatment by multiple comparison.
To further examine whether the maturation of endosome participates in the induction of pro-inflammatory cytokines, THP-1 cells were treated with bafilomycin A1, which inhibits the maturation of early endosome via inhibition of the vacuolar H+ ATPase system.47 In the presence of bafilomycin A1, the induction of TNF-α by mycoplasma infection was slightly enhanced (Fig. 4a). Interleukin-1β induction was slightly increased by bafilomycin A1 when THP-1 cells were treated with M129 and heat-killed mycoplasmas, but the induction was unaffected in the case of M5 and M6 infection (Fig. 4b). Taken together, these results indicate that cytoadherent M. pneumoniae induced pro-inflammatory cytokines in an endocytosis-independent manner, and that the early endosome was important for the induction of pro-inflammatory cytokines by heat-killed and cytoadherence-deficient M. pneumoniae.
Involvement of the inflammasome
Despite the same induction levels of pro-IL-1β precursor, cytoadherent M. pneumoniae induced a higher level of IL-1β than cytoadherence-deficient mutants (Figs 1b and 3b). Mature IL-1β is produced by cleavage of the pro-IL-1β precursor by caspase-1.23 Caspase-1 is activated within a large protein complex, called the inflammasome.24 The inflammasome plays a central role in sensing various intracellular bacterial components. To examine the involvement of the inflammasome, we transfected diacylated lipopeptide derived from M. pneumoniae (FAM20) and triacylated lipopeptide (P3CSK4) into the cytoplasm of THP-1 cells by lipofection. The induction levels of TNF-α and IL-1β remained low when THP-1 cells were treated with either FAM20 or P3CSK4 alone, but the induction levels were augmented when the lipopeptides were transfected using lipofectamin (Fig. 5b). These results suggest that PAMPs like mycoplasmal lipopeptides are recognized by an intracellular receptor such as the inflammasome, leading to the release of IL-1β.
Figure 5.
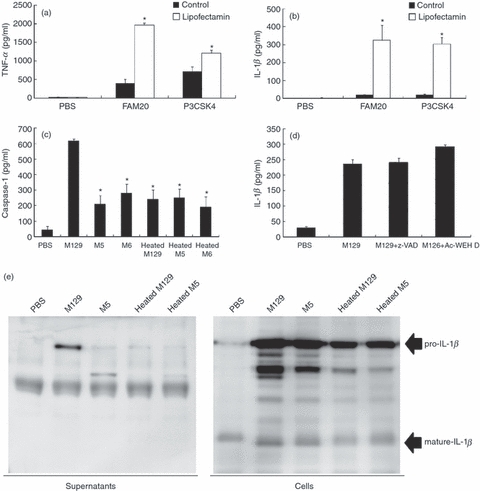
Involvement of the inflammasome. (a, b) 1000 ng/ml of FAM20 was incubated with 1% of lipofectamin. THP-1 cells (2 × 105 cells/500 μl) were transfected with 100 ng/ml FAM20. After 6 hr incubation, the amount of tumour necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) in the culture medium was measured by ELISA. All values represent the means and SD of three assays. An asterisk indicates that the P-value is < 0.01 for a comparison with control by multiple comparison. (c) THP-1 cells (2 × 105 cells/500 μl) were infected with 50 μl of indicated Mycoplasma pneumoniae (OD595 = 0.1). After 6 hr incubation, levels of caspase-1 in culture medium were measured by ELISA. All values represent the means and SD of three assays. An asterisk indicates that the P-value is < 0.01 for a comparison with M129 by multiple comparison. (d) THP-1 cells (2 × 105 cells/500 μl) were cultured in the presence of 100 μm z-Val-Ala-Asp(oMe)-CH2F (z-VAD-FMK) or Ac-Trp-Glu-His-Asp-H (Ac-WEHD-CHO) for 1 hr, and infected with 50 μl M129 (OD595 = 0.1). After 6 hr incubation, amounts of IL-1β in culture medium were measured using ELISA. All values represent the means and SD of three assays. (e) THP-1 cells (2 × 105 cells/500 μl) were infected with 50 μl of the indicated M. pneumoniae (OD595 = 0.1). After 6 hr incubation, the IL-1β levels in the supernatants or infected cells were determined by Western blotting.
Next, we examined the activation of caspase-1. Active caspase-1 is composed of 20 000 and 10 000 molecular weight polypeptides (p20 and p10) derived from the processing of a cytosolic 45 000 MW precursor protein (p45).48 After caspase-1 is activated within the inflammasome, both p20 and p10 are released from cytoplasm to outside the cells.49 We measured the amount of caspase-1 p20 in the culture medium of THP-1 cells infected with M. pneumoniae (Fig. 5c). Infection of M129 released high concentrations of active caspase-1 p20, but the release of the p20 was depressed when M5, M6 and heat-killed M. pneumoniae were used for infection or treatment. To further confirm the involvement of caspase-1, we used several caspase inhibitors in the assay. Interestingly, the release of IL-1β was unaffected when THP-1 cells were treated with a pan-caspase inhibitor, z-VAD or a caspase-1 inhibitor, ac-WEHD (Fig. 5d). To check whether caspase-1 cleaves the pro-IL-1β precursor, the levels of IL-1β precursor in the cells and supernatants of THP-1 cells were examined by Western blotting (Fig. 5e). Surprisingly, pro-IL-1β precursor was cleaved and the mature form of IL-1β was detected in the cells, but only pro-IL-1β precursor was released into the supernatants when the cells were infected with M129. When the cells were treated with M6 or heat-killed M. pneumoniae, lower amounts of mature IL-1β were induced and pro-IL-1β precursor was not released into the supernatant. These results indicate that cytoadherent M. pneumoniae induces caspase-1 activation and maturation of IL-1β, but only pro-IL-1β precursor was released from the cell.
Extracellular ATP serves as a danger signal that alerts the immune system by binding to the receptor P2X7, thereby activating the inflammasome and caspase-1.28–31 Binding of ATP to P2X7 receptor also augments the release of pro-IL-1β precursor.50 It was reported that M. fermentans and M. salivarium induced the extracellular ATP and regulated cell death through the P2X7 receptor in lymphocytes and monocytes.51 Then, we investigated the involvement of extracellular ATP and the P2X7 receptor. Initially, THP-1 cells were infected with M. pneumoniae, and the amounts of extracellular ATP were measured. The extracellular ATP concentration reflects the balance between ATP release and ATP hydrolysis by ectonucleotidase.52 Therefore, to determine the levels of ATP release, THP-1 cells were pre-treated with the ecto-ATPase inhibitor ARL and in turn infected with M. pneumoniae. The levels of ATP release by M129 were increased about threefold compared with M5, M6 and heat-killed M. pneumoniae strains (Fig. 6a). To further examine the role of the P2X7 receptor, THP-1 cells were treated with an inhibitor of the P2X7 receptor, oxidized ATP (oATP). Although the induction of pro-IL-1β precursor by M129 was unaffected by oATP (Fig. 6b), the release of IL-1β was strikingly inhibited by oATP (Fig. 6c). Taken together, these results suggest that cytoadherent M. pneumoniae activates the inflammasome via ‘an ATP autocrine system,’ leading to the release of IL-1β.
Figure 6.
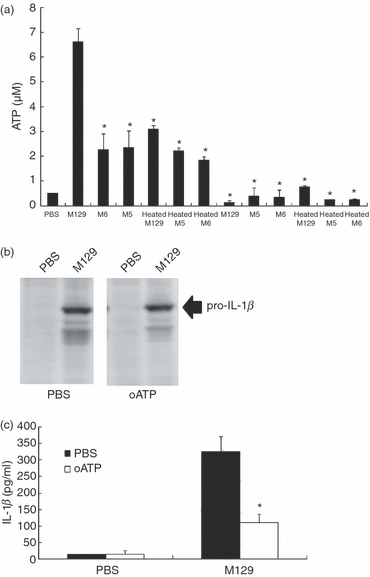
Involvement of ATP in the interleukin-1β (IL-1β) induction. (a) THP-1 cells (2 × 105 cells/500 μl) were cultured in the presence of 200 μm ecto-ATPase inhibitor, ARL for 1 hr, and infected with 50 μl of the indicated Mycoplasma pneumoniae sOD595 = 0.1). After 6 hr incubation, concentrations of ATP were measured as described in the Materials and methods. All values represent the means and SD of three assays. An asterisk indicates that the P-value is < 0.05 for a comparison with M129 by multiple comparison. (b) THP-1 cells (2 × 105 cells/500 μl) were cultured in the presence of 300 μm oxidized ATP for 1 hr and infected with 50 μl M129 (OD595 = 0.1). After 6 hr incubation, THP-1 cells were harvested and the induction levels of pro-IL-1β precursor were determined by Western blotting. (c) THP-1 cells were treated with oxidized ATP and infected with M129 as described above. Release levels of IL-1β were measured by ELISA. All values represent the means and SD of three assays. An asterisk indicates that the P-value is < 0.01 for a comparison with PBS treatment by multiple comparison.
Discussion
The cytoadherence of M. pneumoniae is mediated by attachment organelle, including P1 adhesin and other additional proteins such as P30 or HMW proteins.35–38 These proteins are unique in mycoplasma species, and their homologues are not identified in any other bacteria.53 In this study, we found that cytoadherent M. pneumoniae enhanced the induction of pro-inflammatory cytokines TNF-α and IL-1β compared with cytoadherence-deficient mutants and heat-killed M. pneumoniae (Figs 1 and 2). These results indicate that the property of cytoadherence is critical for the progression of diseases caused by M. pneumoniae. These results are consistent with the earlier reports that a protease treatment decreases the induction of pro-inflammatory cytokines by M. pneumoniae,40 and that M. pneumoniae cultured in non-adherent condition fails to induce IL-4 from rodent mast cells.39 Although the cytoadherence of M. pneumoniae is suggested to be involved in the induction of pro-inflammatory cytokines, the mechanism whereby cytoadherence of M. pneumoniae activates the induction of pro-inflammatory responses has not been clarified.
We previously reported that M. pneumoniae induced pro-inflammatory cytokines through TLR2.6 Interestingly, cytoadherence-deficient M. pneumoniae as well as cytoadherent M. pneumoniae induced TLR2 signalling and pro-IL-1β precursor (Fig. 3a,d). Anti-TLR 2 antibody inhibited the TNF-α and IL-1β production by cytoadherence-deficient mutants, but only partially inhibited the production by cytoadherent M. pneumoniae (Fig. 3b). In addition, wild-type M. pneumoniae had TNF-α inducing activity in macrophages of TLR 2 knockout mice (data not shown). These findings suggest that augmentation of pro-inflammatory cytokine induction by cytoadherent M. pneumoniae is independent of TLR 2 signalling. The NLR proteins have been reported as recognizing intracellular components of microbes and endogenous molecules associated with inflammation, known as PAMPs and DAMPs, respectively.54 Some of the NLR proteins such as NOD1, NOD2, NLRX1 and NLRP12 induce NF-κB and mitogen-activated protein kinase signalling leading to TNF-α induction.55 In this study, we found that lipofection of lipopeptides derived from M. pneumoniae enhances TNF-α production (Fig. 5a). Recognition of intracellular components of M. pneumoniae by intracellular receptors, such as NLR proteins, may explain TLR2-independent pro-inflammatory cytokine induction by cytoadherent M. pneumoniae. The mechanisms of the injection of mycoplasmal components into the cytoplasm of host cells remain unclear. The secretion system in mycoplasma species has not been found. Although it is believed that mycoplasmas remain attached to the surface of epithelial cells, some mycoplasmas including M. pneumoniae can reside within non-phagocytic cells under certain circumstances.56 Moreover, several reports suggest that mycoplasma species are wall-less bacteria and the cell membranes of mycoplasma fuse with the membranes of host cells.57,58 Invasion and fusion may result in the direct delivery of mycoplasmal components into host cells. Alternatively, a pore-forming hemi-channel, pannexin-1 induced by ATP may participate in the process. Pannexin-1-mediated infiltration of bacterial components was reported to activate the production of pro-inflammatory cytokines.32
We also found that an inhibitor of initiation of endocytosis, cytochalasin D inhibited the induction of TNF-α and IL-1β by cytoadherence-deficient mutants, demonstrating that the induction of TNF-α and IL-1β by cytoadherence-deficient M. pneumoniae was completely dependent on endocytosis. In contrast, cytochalasin D partially inhibited the induction by cytoadherent M. pneumoniae, indicating that cytoadherent M. pneumoniae induces pro-inflammatory cytokines in an endocytosis-independent as well as a dependent manner (Fig. 4). In our experimental system, an inhibitor of maturation of early endosome, bafilomycin A1, failed to affect the induction levels of TNF-α and IL-1β. The results indicate that M. pneumoniae induces the production of pro-inflammatory cytokines through the early endosome rather than the late endosome. This result is inconsistent with the recent report that late endosome inducing reactive oxygen species are necessary for IL-1β induction by influenza virus or silica.33,34
Mature IL-1β is produced by cleavage of the pro-IL-1β precursor by caspase-1.23 Caspase-1 is activated within a large protein complex, called the inflammasome.24 Activation of the inflammasome is also mediated by ATP through the P2X7 receptor.28–31 Our results suggest that the inflammasome is involved in the release of IL-1β by M. pneumoniae. Lipofection with the lipopeptide FAM20 derived from M. pneumoniae augmented the release levels of IL-1β (Fig. 5b). Cytoadherent M. pneumoniae strongly induced caspase-1 compared with cytoadherence-deficient mutants (Fig. 5c). Cytoadherent M. pneumoniae efficiently induced efflux of ATP from THP-1 cells (Fig. 6a,b). The oATP, an inhibitor of the P2X7 receptor, decreased the release levels of IL-1β by M. pneumoniae (Fig. 6c). In the present study, however, caspase inhibitors such as z-VAD and ac-WEHD failed to affect the release of IL-1β (Fig. 5d). Pro-IL-1β was cleaved and mature IL-1β was produced in the cells infected with cytoadherent M. pneumoniae, but only pro-IL-1β was released into the supernatants of infected cells (Fig. 5e). The findings suggest that cytoadherent M. pneumoniae induced caspase-1 activation followed by maturation of IL-1β, but only pro-IL-1β precursor was released in an ATP- and P2X7 receptor-dependent manner. These results are consistent with the earlier report that binding of ATP to P2X7 receptor augments the release of pro-IL-1β precursor.50 Although a recent report demonstrated that ATP is released by monocytes stimulated with pathogen-sensing receptor ligands followed by induction of IL-1β,59 the mechanisms by which such ligands release ATP from host cells are still unknown. To our knowledge, this is the first report that the cytoadherence of bacteria enhances the release of ATP from the host cells. Released pro-IL-1β precursor was reported to be cleaved by neutrophil- and macrophage-derived serine proteases such as protease3, elastase and cathepsin-G.60,61 In particular, the crucial role of neutrophil-dependent activation of pro-IL-1β precursor has been validated by the experiments using neutrophil-derived protease inhibitors.62 In M. pneumoniae infection, a large amount of neutrophils are present in human broncho-alveolar lavage fluid,2 and lipoproteins derived from M. pneumoniae induce many neutrophils in mouse broncho-alveolar lavage fluid.6 These evidences indicate that such released pro-IL-1β precursor would be cleaved into mature IL-1β in the extracellular milieu.
In conclusion, our results suggest that cytoadherence of M. pneumoniae is one of the key factors to induce inflammatory responses leading to pneumonia by M. pneumoniae. Hence, the proteins such as P1 and the HMW proteins involved in cytoadherence might be molecules for use in the development of alternative strategies for the prevention and treatment of M. pneumoniae infection.
Acknowledgments
This work was supported in part by a grant from the Ishibashi Foundation, and Grant-in-aid from the Ministry of Education, Science, Sports and Culture of Japan.
Acknowledgments
The authors declare no financial conflicts of interest.
Glossary
Abbreviations:
- Ac-WEHD-CHO
Ac-Trp-Glu-His-Asp-H
- DAMPs
stress associated danger signals
- F0F1-ATPase
F0F1-type ATPase
- HMW
high molecular weight
- IL
interleukin
- NF-κB
nuclear factor-κB
- NLR
nucleotide-binding domain, leucinerich repeat-containing
- oATP
oxidized ATP
- PAMPs
pathogen-associated molecular patterns
- PBMC
peripheral blood mononuclear cells
- TLR
toll-like receptor
- TNF
tumor necrosis factor
- z-VAD-FMK
z-Val-Ala-Asp(oMe)-CH2F
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. Induction of cytokines in peripheral blood mononuclear cells and EBC-1 cells by cytoadherence-deficient mutants of Mycoplasma pneumoniae.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than about missing material) should be directed to the corresponding author for the article.
References
- 1.Razin S. Peculiar properties of mycoplasmas: the smallest self-replicating prokaryotes. FEMS Microbiol Lett. 1992;79:423–31. doi: 10.1111/j.1574-6968.1992.tb14072.x. [DOI] [PubMed] [Google Scholar]
- 2.Waites KB, Talkington DF. Mycoplasma pneumoniae and its role as a human pathogen. Clin Microbiol Rev. 2004;17:697–728. doi: 10.1128/CMR.17.4.697-728.2004. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gil JC, Cedillo RL, Mayagoitia BG, Paz MD. Isolation of Mycoplasma pneumoniae from asthmatic patients. Ann Allergy. 1993;70:23–5. [PubMed] [Google Scholar]
- 4.Kraft M, Cassell GH, Henson JE, Watson H, Williamson J, Marmion BP, Gaydos CA, Martin RJ. Detection of Mycoplasma pneumoniae in the airways of adults with chronic asthma. Am J Respir Crit Care Med. 1998;158:998–1001. doi: 10.1164/ajrccm.158.3.9711092. [DOI] [PubMed] [Google Scholar]
- 5.Tryon VV, Baseman JB. Pathgenic determinant and mechanisms. In: Maniloff J, McElhaney RN, Finch LR, Baseman JB, editors. Mycoplasmas – Molecular Biology and Pathogenesis. Washington, D.C: American Society for Microbiology; 1992. pp. 457–89. [Google Scholar]
- 6.Shimizu T, Kida Y, Kuwano K. Mycoplasma pneumoniae-derived lipopeptides induce acute inflammatory responses in the lungs of mice. Infect Immun. 2008;76:270–7. doi: 10.1128/IAI.00955-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shimizu T, Kida Y, Kuwano K. A dipalmitoylated lipoprotein from Mycoplasma pneumoniae activates NF-kappa B through TLR1, TLR2, and TLR6. J Immunol. 2005;175:4641–6. doi: 10.4049/jimmunol.175.7.4641. [DOI] [PubMed] [Google Scholar]
- 8.Shimizu T, Kida Y, Kuwano K. Triacylated lipoproteins derived from Mycoplasma pneumoniae activate nuclear factor-kappaB through toll-like receptors 1 and 2. Immunology. 2007;121:473–83. doi: 10.1111/j.1365-2567.2007.02594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Akira S, Takeda K. Toll-like receptor signaling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 10.Kopp EB, Medzhitov R. The toll-receptor family and control of innate immunity. Curr Opin Immunol. 1999;11:13–8. doi: 10.1016/s0952-7915(99)80003-x. [DOI] [PubMed] [Google Scholar]
- 11.Aliprantis AO, Yang RB, Mark MR, et al. Cell activation and apoptosis by bacterial lipoproteins through toll-like receptor-2. Science. 1999;285:736–9. doi: 10.1126/science.285.5428.736. [DOI] [PubMed] [Google Scholar]
- 12.Brightbill HD, Libraty DH, Krutzik SR, et al. Host defense mechanisms triggered by microbial lipoproteins through toll-like receptors. Science. 1999;285:732–6. doi: 10.1126/science.285.5428.732. [DOI] [PubMed] [Google Scholar]
- 13.Lien E, Sellati TJ, Yoshimura A, et al. Toll-like receptor 2 functions as a pattern recognition receptor for diverse bacterial products. J Biol Chem. 1999;274:33419–25. doi: 10.1074/jbc.274.47.33419. [DOI] [PubMed] [Google Scholar]
- 14.Means TK, Lien E, Yoshimura A, Wang S, Golenbock DT, Fenton MJ. The CD14 ligands lipoarabinomannan and lipopolysaccharide differ in their requirement for toll-like receptors. J Immunol. 1999;163:6748–55. [PubMed] [Google Scholar]
- 15.Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, Akira S. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 1999;11:443–51. doi: 10.1016/s1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]
- 16.Takeuchi O, Kaufmann A, Grote K, Kawai T, Hoshino K, Morr M, Muhlradt PF, Akira S. Preferentially the R-stereoisomer of the mycoplasmal lipopeptide macrophage-activating lipopeptide-2 activates immune cells through a toll-like receptor 2- and MyD88-dependent signaling pathway. J Immunol. 2000;164:554–7. doi: 10.4049/jimmunol.164.2.554. [DOI] [PubMed] [Google Scholar]
- 17.Underhill DM, Ozinsky A, Hajjar AM, Stevens A, Wilson CB, Bassetti M, Aderem A. The toll-like receptor 2 is recruited to macrophage phagosomes and discriminates between pathogens. Nature. 1999;401:811–5. doi: 10.1038/44605. [DOI] [PubMed] [Google Scholar]
- 18.Hayashi F, Smith KD, Ozinsky A, et al. The innate immune response to bacterial flagellin is mediated by toll-like receptor 5. Nature. 2001;410:1099–103. doi: 10.1038/35074106. [DOI] [PubMed] [Google Scholar]
- 19.Hemmi H, Takeuchi O, Kawai T, et al. A toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–5. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 20.Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, Akira S. Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol. 1999;162:3749–52. [PubMed] [Google Scholar]
- 21.Poltorak A, He X, Smirnova I, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–8. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 22.Medzhitov R, Preston-Hurlburt P, Kopp E, Stadlen A, Chen C, Ghosh S, Janeway CA., Jr MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol Cell. 1998;2:253–8. doi: 10.1016/s1097-2765(00)80136-7. [DOI] [PubMed] [Google Scholar]
- 23.Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10:417–26. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 24.Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol. 2009;27:229–65. doi: 10.1146/annurev.immunol.021908.132715. [DOI] [PubMed] [Google Scholar]
- 25.Boyden ED, Dietrich WF. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat Genet. 2006;38:240–4. doi: 10.1038/ng1724. [DOI] [PubMed] [Google Scholar]
- 26.Martinon F, Agostini L, Meylan E, Tschopp J. Identification of bacterial muramyl dipeptide as activator of the NALP3/cryopyrin inflammasome. Curr Biol. 2004;14:1929–34. doi: 10.1016/j.cub.2004.10.027. [DOI] [PubMed] [Google Scholar]
- 27.Miao EA, Andersen-Nissen E, Warren SE, Aderem A. TLR5 and Ipaf: dual sensors of bacterial flagellin in the innate immune system. Semin Immunopathol. 2007;29:275–88. doi: 10.1007/s00281-007-0078-z. [DOI] [PubMed] [Google Scholar]
- 28.Mariathasan S, Weiss DS, Newton K, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–32. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 29.Sutterwala FS, Ogura Y, Szczepanik M, et al. Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity. 2006;24:317–27. doi: 10.1016/j.immuni.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 30.Kanneganti TD, Ozoren N, Body-Malapel M, et al. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature. 2006;440:233–6. doi: 10.1038/nature04517. [DOI] [PubMed] [Google Scholar]
- 31.Franchi L, Kanneganti TD, Dubyak GR, Nunez G. Differential requirement of P2X7 receptor and intracellular K+ for caspase-1 activation induced by intracellular and extracellular bacteria. J Biol Chem. 2007;282:18810–8. doi: 10.1074/jbc.M610762200. [DOI] [PubMed] [Google Scholar]
- 32.Kanneganti TD, Lamkanfi M, Kim YG, Chen G, Park JH, Franchi L, Vandenabeele P, Nunez G. Pannexin-1-mediated recognition of bacterial molecules activates the cryopyrin inflammasome independent of toll-like receptor signaling. Immunity. 2007;26:433–43. doi: 10.1016/j.immuni.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 33.Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320:674–7. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Allen IC, Scull MA, Moore CB, et al. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity. 2009;30:556–65. doi: 10.1016/j.immuni.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krause DC, Balish MF. Structure, function, and assembly of the terminal organelle of Mycoplasma pneumoniae. FEMS Microbiol Lett. 2001;198:1–7. doi: 10.1111/j.1574-6968.2001.tb10610.x. [DOI] [PubMed] [Google Scholar]
- 36.Balish MF, Krause DC. Cytoadherence and the cytoskeleton. In: Razin S, Herrmann R, editors. Molecular Biology and Pathogenicity of Mycoplasmas. New York: Kluwer Academic/Plenum Publishers; 2002. pp. 491–518. [Google Scholar]
- 37.Miyata M. Centipede and inchworm models to explain Mycoplasma gliding. Trends Microbiol. 2008;16:6–12. doi: 10.1016/j.tim.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 38.Miyata M. Molecular mechanism of mycoplasma gliding – a novel cell motility system. In: Lenz P, editor. Cell Motility. New York: Springer Science; 2008. pp. 137–75. [Google Scholar]
- 39.Hoek KL, Duffy LB, Cassell GH, Dai Y, Atkinson TP. A role for the Mycoplasma pneumoniae adhesin P1 in interleukin (IL)-4 synthesis and release from rodent mast cells. Microb Pathog. 2005;39:149–58. doi: 10.1016/j.micpath.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 40.Yang J, Hooper WC, Phillips DJ, Talkington DF. Regulation of proinflammatory cytokines in human lung epithelial cells infected with Mycoplasma pneumoniae. Infect Immun. 2002;70:3649–55. doi: 10.1128/IAI.70.7.3649-3655.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seto S, Miyata M. Attachment organelle formation represented by localization of cytoadherence proteins and formation of the electron-dense core in wild-type and mutant strains of Mycoplasma pneumoniae. J Bacteriol. 2003;185:1082–91. doi: 10.1128/JB.185.3.1082-1091.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Layh-Schmitt G, Harkenthal M. The 40- and 90-kDa membrane proteins (ORF6 gene product) of Mycoplasma pneumoniae are responsible for the tip structure formation and P1 (adhesin) association with the Triton shell. FEMS Microbiol Lett. 1999;174:143–9. doi: 10.1111/j.1574-6968.1999.tb13561.x. [DOI] [PubMed] [Google Scholar]
- 43.Layh-Schmitt G, Hilbert H, Pirkl E. A spontaneous hemadsorption-negative mutant of Mycoplasma pneumoniae exhibits a truncated adhesin-related 30-kilodalton protein and lacks the cytadherence-accessory protein HMW1. J Bacteriol. 1995;177:843–6. doi: 10.1128/jb.177.3.843-846.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Romero-Arroyo CE, Jordan J, Peacock SJ, Willby MJ, Farmer MA, Krause DC. Mycoplasma pneumoniae protein P30 is required for cytadherence and associated with proper cell development. J Bacteriol. 1999;181:1079–87. doi: 10.1128/jb.181.4.1079-1087.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jordan JL, Berry KM, Balish MF, Krause DC. Stability and subcellular localization of cytadherence-associated protein P65 in Mycoplasma pneumoniae. J Bacteriol. 2001;183:7387–91. doi: 10.1128/JB.183.24.7387-7391.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mimura N, Asano A. Synergistic effect of colchicine and cytochalasin D on phagocytosis by peritoneal macrophages. Nature. 1976;261:319–21. doi: 10.1038/261319a0. [DOI] [PubMed] [Google Scholar]
- 47.Bowman EJ, Siebers A, Altendorf K. Bafilomycins: a class of inhibitors of membrane ATPases from microorganisms, animal cells, and plant cells. Proc Natl Acad Sci USA. 1988;85:7972–6. doi: 10.1073/pnas.85.21.7972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yamin TT, Ayala JM, Miller DK. Activation of the native 45-kDa precursor form of interleukin-1-converting enzyme. J Biol Chem. 1996;271:13273–82. doi: 10.1074/jbc.271.22.13273. [DOI] [PubMed] [Google Scholar]
- 49.Ferrari D, Pizzirani C, Adinolfi E, Lemoli RM, Curti A, Idzko M, Panther E, Di Virgilio F. The P2X7 receptor: a key player in IL-1 processing and release. J Immunol. 2006;176:3877–83. doi: 10.4049/jimmunol.176.7.3877. [DOI] [PubMed] [Google Scholar]
- 50.Mehta VB, Hart J, Wewers MD. ATP-stimulated release of interleukin (IL)-1beta and IL-18 requires priming by lipopolysaccharide and is independent of caspase-1 cleavage. J Biol Chem. 2001;276:3820–6. doi: 10.1074/jbc.M006814200. [DOI] [PubMed] [Google Scholar]
- 51.Into T, Okada K, Inoue N, Yasuda M, Shibata K. Extracellular ATP regulates cell death of lymphocytes and monocytes induced by membrane-bound lipoproteins of Mycoplasma fermentans and Mycoplasma salivarium. Microbiol Immunol. 2002;46:667–75. doi: 10.1111/j.1348-0421.2002.tb02750.x. [DOI] [PubMed] [Google Scholar]
- 52.Joseph SM, Buchakjian MR, Dubyak GR. Colocalization of ATP release sites and ecto-ATPase activity at the extracellular surface of human astrocytes. J Biol Chem. 2003;278:23331–42. doi: 10.1074/jbc.M302680200. [DOI] [PubMed] [Google Scholar]
- 53.Himmelreich R, Hilbert H, Plagens H, Pirkl E, Li BC, Herrmann R. Complete sequence analysis of the genome of the bacterium Mycoplasma pneumoniae. Nucleic Acids Res. 1996;24:4420–49. doi: 10.1093/nar/24.22.4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bryant C, Fitzgerald KA. Molecular mechanisms involved in inflammasome activation. Trends Cell Biol. 2009;19:455–64. doi: 10.1016/j.tcb.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 55.Ting JP, Duncan JA, Lei Y. How the noninflammasome NLRs function in the innate immune system. Science. 2010;327:286–90. doi: 10.1126/science.1184004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Baseman JB, Lange M, Criscimagna NL, Giron JA, Thomas CA. Interplay between mycoplasmas and host target cells. Microb Pathog. 1995;19:105–16. doi: 10.1006/mpat.1995.0050. [DOI] [PubMed] [Google Scholar]
- 57.Dimitrov DS, Franzoso G, Salman M, Blumenthal R, Tarshis M, Barile MF, Rottem S. Mycoplasma fermentans (incognitus strain) cells are able to fuse with T lymphocytes. Clin Infect Dis. 1993;17(Suppl. 1):S305–8. doi: 10.1093/clinids/17.supplement_1.s305. [DOI] [PubMed] [Google Scholar]
- 58.Franzoso G, Dimitrov DS, Blumenthal R, Barile MF, Rottem S. Fusion of Mycoplasma fermentans strain incognitus with T-lymphocytes. FEBS Lett. 1992;3:251–4. doi: 10.1016/0014-5793(92)80531-k. [DOI] [PubMed] [Google Scholar]
- 59.Piccini A, Carta S, Tassi S, Lasiglie D, Fossati G, Rubartelli A. ATP is released by monocytes stimulated with pathogen-sensing receptor ligands and induces IL-1beta and IL-18 secretion in an autocrine way. Proc Natl Acad Sci USA. 2008;105:8067–72. doi: 10.1073/pnas.0709684105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dinarello CA. Biologic basis for interleukin-1 in disease. Blood. 1996;87:2095–147. [PubMed] [Google Scholar]
- 61.Coeshott C, Ohnemus C, Pilyavskaya A, Ross S, Wieczorek M, Kroona H, Leimer AH, Cheronis J. Converting enzyme-independent release of tumor necrosis factor alpha and IL-1beta from a stimulated human monocytic cell line in the presence of activated neutrophils or purified proteinase 3. Proc Natl Acad Sci USA. 1999;96:6261–6. doi: 10.1073/pnas.96.11.6261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Greten FR, Arkan MC, Bollrath J, et al. NF-kappaB is a negative regulator of IL-1beta secretion as revealed by genetic and pharmacological inhibition of IKKbeta. Cell. 2007;130:918–31. doi: 10.1016/j.cell.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.