

Abstract
To model inflammatory bowel disease, we assessed infection with Helicobacter hepaticus 3B1 (ATCC 51449) and a potential probiotic Lactobacillus reuteri (ATCC PTA-6475) in gnotobiotic B6.129P2-IL-10tm1Cgn (IL-10−/−) mice. No typhlocolitis developed in germ-free controls (n = 21) or in L. reuteri (n = 8) or H. hepaticus (n = 18) mono-associated mice for 20 weeks post-infection. As positive controls, three specific pathogen-free IL-10−/− mice dosed with H. hepaticus developed severe typhlocolitis within 11 weeks. Because L. reuteri PTA-6475 has anti-inflammatory properties in vitro, it was unexpected to observe significant typhlocolitis (P < 0·0001) in mice that had been infected with L. reuteri followed in 1 week by H. hepaticus (n = 16). The H. hepaticus colonization was not affected through 20 weeks post-infection but L. reuteri colonization was lower in co-infected compared with L. reuteri mono-associated mice at 8–11 weeks post-infection (P < 0·05). Typhlocolitis was associated with an increased T helper type 1 serum IgG2c response to H. hepaticus in co-infected mice compared with H. hepaticus mono-associated mice (P < 0·005) and similarly, mRNA expression in caecal–colonic tissue was elevated at least twofold for chemokine ligands and pro-inflammatory interleukin-1α (IL-1α), IL-1β, IL-12 receptor, tumour necrosis factor-α and inducible nitric oxide synthase. Anti-inflammatory transforming growth factor-β, lactotransferrin, peptidoglycan recognition proteins, Toll-like receptors 4, 6, 8 and particularly 9 gene expression, were also elevated only in co-infected mice (P < 0·05). These data support that the development of typhlocolitis in H. hepaticus-infected IL-10−/− mice required co-colonization with other microbiota and in this study, required only L. reuteri. Although the effects other microbiota may have on H. hepaticus virulence properties remain speculative, further investigations using this gnotobiotic model are now possible.
Keywords: bacteria/bacterial immunity, gut immunology/disease, inflammatory bowel diease, innate immunity, mucosal immunity
Introduction
Select human inflammatory bowel diseases such as Crohn's disease, are characterized by a genetically predisposed inflammatory response to commensal bacteria.1 The idiopathic nature of human inflammatory bowel diseases has promoted the use of interleukin-10-deficient (IL-10−/−) mice to fulfill Koch's postulates using potential bacterial pathogens,2,3 to examine the role of commensal organisms4 and to investigate the interplay between otherwise non-pathogenic bacterial species in causing or ameliorating disease.5,6Helicobacter hepaticus infection of IL-10−/− mice has been used to model mechanisms of human inflammatory bowel diseases ever since the association between enterohepatic helicobacter infections and chronic typhlocolitis in immunocompromised mice was first suspected and later experimentally established.7–10 Similar features between Crohn's enterocolitis and the H. hepaticus-infected IL-10−/− mouse model include a pro-inflammatory T helper type 1 (Th1) immune response to enteric microbiota.11 IL-10−/− mice develop a Th1-biased inflammatory response to H. hepaticus driven by macrophages, CD4+ T cells, IL-12 and interferon-γ (IFN-γ) in the absence of IL-10-secreting T regulatory cells.12,13 Comparable to Crohn's lesions, typhlocolitis in H. hepaticus-infected IL-10−/− mice is characterized by transmural inflammation, epithelial hyperplasia and dysplasia, primarily at the caecal–colonic junction with varying degrees of colitis and proctitis.14
Despite controlling for host background genetics, variable severity of disease in the H. hepaticus IL-10−/− mouse model has been reported.13,15 Genetic variation in a 70-kb genomic island (HHGI1), considered a putative pathogenicity island in the H. hepaticus strain 3B1 (ATCC 51449), was identified and shown to be important for the A/J mouse model of hepatitis and the IL-10−/− mouse model of colitis.2,16 Similarly, cytolethal distending toxin (CDT) present in H. hepaticus17,18 promoted typhlocolitis in the IL-10−/− mouse model.19 One study suggested that the commensal microbiota may be the primary promoter of colitis in the IL-10−/− model and that H. hepaticus does not induce or potentiate typhlocolitis in IL-10−/− mice.15
For these reasons, to further evaluate the virulence potential of H. hepaticus to cause colitis in the IL-10−/− mouse model under well-defined conditions, gnotobiotic IL-10−/− B6.129 mice were infected with H. hepaticus 3B1 (ATCC 51449). The H. hepaticus 3B1 was selected because it is the prototypic enterohepatic helicobacter that was first shown to cause chronic active hepatitis20 and hepatocellular carcinoma21 in male A/JCr mice and severe typhlocolitis in IL-10−/− mice.13 We further assessed co-infection with human-derived Lactobacillus reuteri (ATCC PTA-6475) because it was previously shown to have probiotic immunomodulatory activity in vitro22 and had not previously been evaluated in a mouse model of inflammatory bowel diseases. Our objectives were to determine if H. hepaticus mono-association would cause typhlocolitis and if inflammation developed, could it be ameliorated by a probiotic L. reuteri PTA-6475, as had been shown in H. hepaticus-infected B6.129 IL-10−/− male mice co-colonized with murine-derived Lactobacillus paracasei 1602 and L. reuteri 6798.6
Methods
Mice
B6.129P2-IL-10tm1Cgn (IL-10−/−) mice were originally obtained from Jackson Laboratories (Bar Harbor, ME) and were maintained specific pathogen-free (SPF) for murine viruses, pathogenic bacteria (including helicobacters) and parasites in an Association for Assessment and Accreditation of Laboratory Animal Care-accredited barrier facility. These IL-10−/− mice were re-derived by embryo transfer into the germ-free health status and subsequently bred to generate experimental groups. Germ-free IL-10−/− mice were housed in sterile plastic film isolators confirmed to be maintained free of all aerobic and anaerobic bacteria and fungi by weekly microbiological monitoring. After experimental infections, isolator interior surfaces, drinking water, food and faeces were sampled weekly for contaminants by culture. Within isolators segregated by infection status, mice were housed in sterile open-top polycarbonate cages on autoclaved hardwood bedding and fed autoclaved water and diet (ProLab 2000; Purina Mills, St Louis, MO) ad libitum. Macroenvironmental conditions included a 14 : 10-hr light/dark cycle and temperature maintenance at 20 ± 1°C. Control SPF IL-10−/− mice were housed in a separate facility in standard microisolator cages under similar environmental conditions except their food and water were not autoclaved. Experiments were approved by the MIT Committee on Animal Care.
Experimental infection
Lactobacillus reuteri (ATCC PTA-6475) was originally isolated from human milk and in vitro inhibition of tumour necrosis factor-α (TNF-α) activity has been demonstrated.22 The H. hepaticus strain 3B1 (ATCC 51449) was the original isolate from an outbreak of hepatitis and hepatocellular carcinoma in control A/J mice used in carcinogenesis assays.20,23 The 3B1 was sequenced24 and found to contain CDT and a putative pathogenicity island important for development of typhlocolitis in IL-10−/− mice.2 At 6–8 weeks of age, approximately equal numbers of male and female germ-free IL-10−/− mice were randomly assigned to separate isolators to be either unmanipulated as controls (n = 21) or to be orally gavaged every other day for three doses of 2 × 108 colony-forming units (CFU) of L. reuteri alone (n = 8), 2 × 108 CFU of H. hepaticus alone (n = 18), or L. reuteri followed in 1 week by H. hepaticus infection (n = 16). Three helicobacter-free SPF IL-10−/− mice were dosed with the same H. hepaticus inoculum as the gnotobiotic mice as a positive control to demonstrate its ability to cause typhlocolitis. Negative control SPF IL-10−/− mice (n = 3) were maintained helicobacter-free25 and then killed and examined post-mortem after 20 weeks.
Colonization levels of L. reuteri and H. hepaticus at the caecal–colonic junction
At 8–11 weeks post-infection (p.i.), tissue samples of the caecal–colonic junction from experimentally infected mice were obtained post-mortem and weighed using sterile technique for quantification of L. reuteri by limiting dilution culture and for H. hepaticus by real-time quantitative PCR, as previously described.26 In brief, samples for L. reuteri were 10-fold diluted in sterile saline and plated on blood agar under anaerobic conditions at 37°C and colony counts were performed after 24 hr of incubation. Because H. hepaticus does not grow in discrete colonies on agar, bacterial and host DNA were extracted from the caecum using the Boehringer-Mannheim High Pure PCR Template Preparation kit (Roche Molecular Biochemicals, Indianapolis, IN). Samples were probed with 18S rRNA-based primers for quantifying host DNA (Applied Biosystems, Foster City, CA) and with H. hepaticus DNA-specific primers and probe generated from the H. hepaticus cdtB gene as previously described26 using Primer Express software (Applied Biosystems). At 20 weeks p.i., the caecal–colonic junction of experimentally dosed mice was cultured for L. reuteri to confirm colonization but L. reuteri was not quantified.
Clinical assessment and post-mortem examination
Mice were assessed for evidence of diarrhoea, dehydration and deteriorating body condition27 with final body weights recorded at the post-mortem examination. Approximately 50% of randomly selected mice from each infection or control group were killed with carbon dioxide and examined post-mortem at 8–11 weeks p.i.; the remaining mice were killed and examined post-mortem at 20 weeks p.i. Serum and tissues were aseptically obtained for serology, microbiology, pathology and mRNA expression levels. Tissues for culture recovery of L. reuteri and H. hepaticus were stored in Brucella broth containing 30% glycerol at −70°C pending culture. Tissues for mRNA expression levels were flash frozen in liquid nitrogen and stored at −70°C pending extraction and PCR assays. Tissues for histology were fixed in 10% buffered formalin and processed routinely.
Histology
Formalin-fixed tissues were embedded in paraffin, sectioned at 4 μm, stained with haematoxylin and eosin and evaluated by a board-certified veterinary pathologist (S.M.) blinded to the sample identity. Lesion scores for inflammation, oedema, epithelial defects, hyperplasia and dysplasia of the caecal–colonic junction were evaluated on a scale of zero for normal to four for ascending severity and invasiveness of inflammatory lesions. Distinct from transmural inflammatory lesions, gut-associated lymphoid tissue (GALT) was semi-quantitatively assessed using a scale of zero (for minimal cellularity) to four for increased cellular expansion of organized lymphoid follicles, inclusive of Peyer's patches, and scattered aggregates of lymphoid cells that varied from small foci to isolated but organized follicles with germinal centres. To generate comparative scores, haematoxylin and eosin-stained longitudinal section of coiled small intestine, the entire caecum with adjacent terminal ileum and proximal colon as well as multiple representative linear sections from transverse and distal colon were examined under both low-power and high-power objective fields.
Immunohistochemistry
Immunohistochemistry was performed to phenotype B cells, T cells, macrophages and proliferating cells at the caecal–colonic junction of three germ-free and three gnotobiotic IL-10−/− mice infected with H. hepaticus alone, three infected with L. reuteri alone and three mice with H. hepaticus and L. reuteri infection at 20 weeks p.i. Formalin-fixed sections from the caecal–colonic junction were stained for the pan-B-cell marker CD45/B220 using monoclonal rat anti-mouse antibody (#550286; BD Pharmingen, San Diego, CA) and secondary goat anti-rat antibody (#559286; BD Pharmingen), pan-T-cell marker CD3 using rabbit anti-mouse CD3 antibody (#A0452; Dako, Carpinteria, CA), macrophage-restricted cell surface glycoprotein F4/80 using rat monoclonal antibody (#MF48015; Invitrogen, Carlsbad, CA) and secondary goat anti-rat antibody (#559286; BD Pharmingen), and nuclear cell proliferation marker Ki67 using mouse anti-human antibody (#550609; BD Pharmingen), developed using Dako Art kit (#K3954) per instructions. Methods for antigen retrieval, blocking steps and counterstaining were as previously described.28
ELISA for serum IgG2c and IgG1 and mucosal IgA responses to H. hepaticus
Serum Th1-associated IgG2c29 and Th2-associated IgG1 responses to outer membrane antigens of H. hepaticus were measured by ELISA as previously described.30 Antigen was coated on Immulon II plates (Thermo Fischer Scientific, Pittsburgh, PA) at a concentration of 10 μg/ml with sera diluted 1 : 100. Biotinylated secondary antibodies included monoclonal anti-mouse antibodies produced by clones A85-1 and 5.7 (BD Biosciences, San Jose, CA) for detecting IgG1 and IgG2c, respectively. Incubation with extravidin peroxidase (Sigma, St. Louis, MO) was followed by 2,2′-azinobis (3-ethylbenzthiazoline-6-sulphonic acid) diammonium salt (ABTS®) substrate (Kirkegaard and Perry Laboratories, Gaithersburg, MD) for colour development. Absorbance (or optical density) development at 405 nm was recorded by an ELISA plate reader (Dynatech MR7000; Dynatech Laboratories, Inc., Chantilly, VA).
To measure mucosal IgA responses to H. hepaticus, four fresh faecal pellets were emulsified in protease inhibitor (Sigma), centrifuged briefly to pellet debris and stored at −70°C pending ELISA. Assay conditions were as for serum IgG1 and IgG2c except the faecal sample was used neat and biotinylated goat anti-mouse IgA (Southern Biotechnology Associates, Birmingham, AL) diluted 1 : 2000 was used as the secondary antibody. Optical density values for H. hepaticus-specific IgA were normalized by optical densities measured for total IgA content of the faecal sample. Sheep anti-mouse IgA (Sigma M-1272) was used as a capture antibody at 5 μg/ml, followed by sample incubation and additional steps of extravidin peroxidase, ABTS® substrate as above.
PCR array for innate and adaptive immunity gene expression
Tissue samples of the caecal–colonic junction collected at post-mortem examination from three mice of each infection status at 8–11 weeks p.i. were snap frozen in liquid nitrogen. Total RNA was isolated using Trizol reagent following the supplier's instruction (Invitrogen) with further purification by removing DNA contamination using the RNeasy kit (Qiagen, Valencia, CA). Caecal–colonic mRNA (2 μg) was reverse-transcribed into cDNA using the High Capacity cDNA Archive kit following the supplier's instructions (Applied Biosystems). Expression of mRNA of mouse genes involved in innate and adaptive immunity was measured with an RT2 Profiler PCR array (SABioscience Corporation) using real-time quantitative PCR (qPCR) performed in the 7500 Fast Real-Time PCR System (Applied Biosystems). Transcript levels of the detected genes were normalized to the endogenous control GAPDH transcript and expressed as fold change in reference to germ-free control mice using the Comparative CT method (Applied Biosystems User Bulletin #2). Messenger RNA expression was considered significant if twofold or higher multiples of expression were measured compared with values obtained from the germ-free control mice.
Statistical analysis
Analysis of variance, regression and the Student's t-test were used to determine the influence of infection status on serum levels of antibodies, colonization levels of H. hepaticus and L. reuteri and gene expression levels in tissues collected from the caecal–colonic junction. Experimental groups were compared for severity of typhlocolitis using the Mann–Whitney non-parametric t-test or the Kruskal–Wallis one-way analysis of variance with the Dunns’ post-test (GraphPad Software Inc., San Diego, CA). Results were considered significant at P < 0·05.
Results
Co-infection of IL-10−/− mice with L. reuteri and H. hepaticus caused significant typhlocolitis
As positive controls for demonstrating the virulence of the H. hepaticus 3B1 strain, three SPF IL-10−/− mice experimentally infected with H. hepaticus 3B1 developed rectal prolapse with declining body condition by 11 weeks p.i. and were killed and examined post-mortem. There was gross thickening of the caecum and colon, moderately enlarged mesenteric lymph nodes and histologically, the caecal–colonic junction mucosa and submucosa had severe mononuclear inflammation, submucosal oedema, mild epithelial defects (glandular epithelial necrosis and luminal cell debris) and hyperplasia (Fig. 1c,d). Mild dysplasia was characterized by architectural distortion involving glandular elongation, gland splitting, loss of columnar orientation and mild cytological atypia. Two of three additional SPF IL-10−/− mice maintained under helicobacter-free husbandry conditions through 20 weeks p.i. were free of significant gross or histological lesions at post-mortem examination (Fig. 1a). One SPF IL-10−/− mouse had developed rectal prolapse and a thickened proximal colon but was PCR negative for H. hepaticus, as were the two clinically normal cage mates. Spontaneous colitis occurs in this helicobacter-free SPF IL-10−/− mouse colony with a very low incidence.
Figure 1.
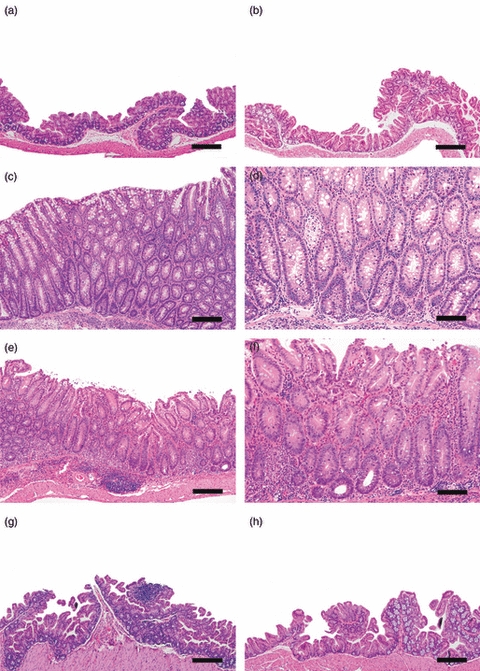
Germ-free B6.129P2-IL-10tm1Cgn (IL-10−/−) mice were colonized at 6–8 weeks of age with Lactobacillus reuteri, Helicobacter hepaticus or L. reuteri followed in 1 week by H. hepaticus and killed and examined post-mortem at 8–11 or 20 weeks post-infection (p.i.). (a) Caecal–colonic junction from helicobacter-free specific-pathogen-free (SPF) IL-10−/− mouse at 28 weeks of age that lacked gross or histological lesions at necropsy. (b) Caecal–colonic junction from IL-10−/− mouse mono-associated with L. reuteri for 20 weeks. Note absence of lymphoid development. (c, d) Representative low and high magnification images of the caecal–colonic junction from a positive control, 19-week-old SPF IL-10−/− mouse infected with H. hepaticus for 11 weeks characterized by mucosal and submucosal mononuclear inflammation, submucosal oedema, mild epithelial defects (glandular epithelial necrosis and luminal cell debris), hyperplasia and mild dysplasia characterized by architectural distortion involving glandular elongation, gland splitting, loss of columnar orientation and mild cytological atypia. (e, f) Representative low and high magnification images of the caecal–colonic junction of a gnotobiotic IL-10−/− mouse co-infected with L. reuteri and H. hepaticus for 20 weeks. Note mixed population of inflammatory cells in the lamina propria and distinct lymphoid aggregate in submucosa with associated mild oedema, epithelial defects (surface epithelial tethering, decrease in goblet cells, focal crypt necrosis and abcessation), mild hyperplasia and minimal to mild dysplasia (occasional loss of glandular columnar orientation and mild cytological atypia). (g) The H. hepaticus mono-association for 20 weeks did not result in significant inflammation and only mild lymphoid development in the caecal–colonic junction. (h) Caecal–colonic junction from germ-free IL-10−/− mouse at the 20 weeks p.i. time-point. Note lack of lymphoid development or histological lesions. Bars: a, b, c, e, g, h = 100 μm, d and f = 50 μm.
Germ-free IL-10−/− mice that were mono-associated with L. reuteri or H. hepaticus or co-infected with L. reuteri and H. hepaticus were killed and examined post-mortem at 8–11 and 20 weeks p.i. to confirm colonization with one or both bacteria and to assess potential pathology. The IL-10−/− mice mono-associated with L. reuteri or H. hepaticus were clinically normal except for gross dilation of the gallbladder and caecum, characteristic of germ-free mice, and lacked gross evidence of caecal–colonic inflammation. In L. reuteri mono-associated mice (Fig. 1b) and in H. hepaticus mono-associated mice (Fig. 1g), there was histological evidence of small numbers of neutrophils, lymphocytes and macrophages scattered in the lamina propria of the small intestine, caecal–colonic junction and colon, without qualitative differences in number or distribution of these cell types compared with the germ-free control mice (Fig. 1h). Notably, there was no significant inflammation at the caecal–colonic junction (typhlocolitis) unless mice were co-infected with L. reuteri and H. hepaticus (Fig. 1e,f). At post-mortem examination 8–11 and 20 weeks p.i., IL-10−/− mice co-infected with L. reuteri and H. hepaticus had gross dilation of the caecum comparable with the germ-free mice and the L. reuteri and H. hepaticus mono-associated mice but dual infection also produced gross enlargement of the mesenteric lymph nodes and thickening of the caecal–colonic junction. At both 8–11 and 20 weeks p.i., there was significant infiltration of inflammatory cells in the lamina propria of the caecal–colonic junction accompanied by distinct lymphoid aggregates in the submucosa with associated mild oedema, epithelial defects (surface epithelial tethering, decrease in goblet cells, focal crypt necrosis and abcessation), mild hyperplasia and minimal to mild dysplasia (occasional loss of glandular columnar orientation and mild cytological atypia). Compared with germ-free and mono-associated mice (Fig. 2a–c), there was significant inflammatory cell infiltration in co-infected mice at both 8–11 and 20 weeks p.i. (both P < 0·0001) accompanied by submucosal oedema (P < 0·03, P < 0·01), and epithelial defects (P < 0·0003, P < 0·0002). Scores for crypt atrophy, hyperplasia and dysplasia were only significant in co-infected mice (Fig. 3a; all P < 0·05) and did not differ in extent by time-point. Additionally, typhlocolitis in co-infected mice was similar in severity at 8–11 and 20 weeks p.i. with no gender differences in lesion severity among co-infected mice.
Figure 2.
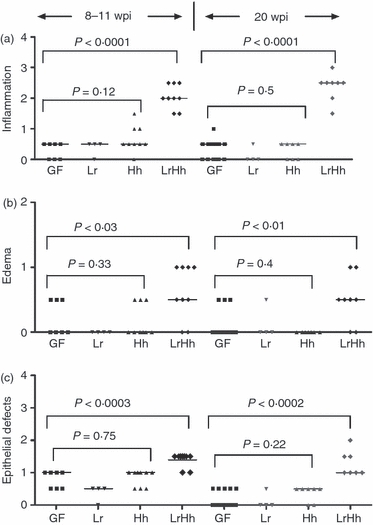
Germ-free B6.129P2-IL-10tm1Cgn (IL-10−/−) mice were colonized at 6–8 weeks of age with Lactobacillus reuteri (Lr) Helicobacter hepaticus (Hh) or L. reuteri followed in 1 week by H. hepaticus (LrHh) and killed and examined post-mortem at 8–11 or 20 weeks post-infection (p.i.). Median scores were significantly higher for (a) inflammation (P < 0.0001; P < 0.0001) (b) submucosal oedema (P < 0.03; P < 0.01) and (c) epithelial defects (P < 0.0003; P < 0.0002) at the caecal–colonic junction of IL-10−/− mice co-infected with L. reuteri and H. hepaticus (LrHh) for 8–11 or 20 weeks p.i. (respectively) compared with germ-free (GF) mice or mice mono-associated with L. reuteri (Lr) or H. hepaticus (Hh). There were no significant differences in inflammation, submucosal oedema or epithelial defects in mice that were germ-free or mono-associated with either L. reuteri or H. hepaticus (analysis of variance P-values as shown).
Figure 3.
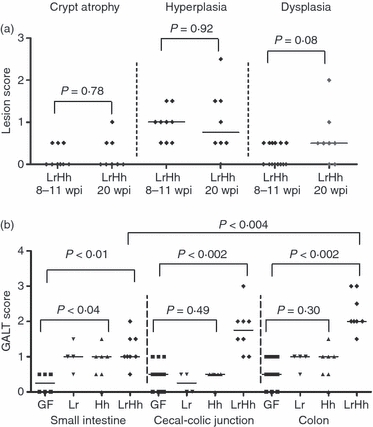
Germ-free B6.129P2-IL-10tm1Cgn (IL-10−/−) mice were colonized at 6–8 weeks of age with Lactobacillus reuteri (Lr) Helicobacter hepaticus (Hh) or L. reuteri followed in 1 week by H. hepaticus (LrHh), then killed and examined at post-mortem at 8–11 or 20 weeks post-infection (p.i.). (a) Crypt atrophy, epithelial hyperplasia and dysplasia developed only in co-infected IL-10−/− mice (LrHh); these features were absent in all other experimental groups so median scores were all significant for co-infected mice only (all P < 0.05). Differences between time-points were not significant. (b) Median gut-associated lymphoid tissue (GALT) scores for small intestine, caecal–colonic junction and the colon at 20 weeks p.i. were significantly greater in co-infected IL-10−/− mice (LrHh) compared with uninfected germ-free mice (GF) (P < 0.01, P < 0.002, P < 0.002, respectively). In co-infected mice the GALT scores were higher at the caecal–colonic junction and in representative sections of transverse and distal colon compared with small intestine (P < 0.004).
Colitis distal to the caecal–colonic junction developed as a result of co-infection with L. reuteri and H. hepaticus but was not observed in mice mono-infected with L. reuteri or H. hepaticus (colitis scores not shown). In co-infected mice, colitis was less severe in the mid to distal colon than at the more proximal caecal–colonic junction for inflammation (P < 0·0007), oedema (P < 0·012) and epithelial defects (P < 0·0015). Scores of the more distal colon for inflammation-driven crypt atrophy, epithelial hyperplasia and dysplasia were similar to scores observed for the caecal–colonic junction of co-infected mice.
In comparison to co-infected mice, significant inflammation did not develop at any level of the intestinal tract of mice mono-associated with L. reuteri or H. hepaticus. Compared with germ-free mice, GALT in the small intestine of mono-associated and co-infected mice had increased numbers of lymphoid mononuclear cells in Peyer's patches and in small, scattered foci within the lamina propria (Fig. 3b; P < 0·01). Consistent with minimal inflammation, GALT in the caecal–colonic junction and colon of mice mono-associated with L. reuteri or H. hepaticus were similar to those in germ-free mice with low lymphoid cell content (Figs 3b and 4). As an example, H. hepaticus-infected mice had a few small to medium-sized mucosal, and occasionally submucosal, foci of lymphoid aggregates and some distinct lymphoid follicles that contained mostly B cells (Fig. 4a), fewer T cells (Fig. 4c) and sparse macrophages (Fig. 4e). Proliferative (Ki67) activity in these lymphoid patches was minimal and was restricted to the basal aspect of the glandular acini and crypts (Fig. 4g). Mice co-infected with L. reuteri and H. hepaticus had similarly higher GALT scores at the caecal–colonic junction and in representative sections of transverse and distal colon (Fig. 3b; P < 0·002). Co-infection with L. reuteri and H. hepaticus stimulated multifocal mucosal and submucosal expansion of lymphoid follicles at the caecal–colonic junction that were predominantly B cells (Fig. 4b), indicative of B-cell expansion, with T cells surrounding the B-cell germinal centres (Fig. 4d). Small numbers of B cells and considerable numbers of T cells were distributed throughout the lamina propria. Macrophages were also increased in the lamina propria of the mucosa and submucosa (Fig. 4f). Intraepithelial T lymphocytes and macrophages were also increased compared with very low numbers observed in mice colonized with H. hepaticus (Fig. 4c) or L. reuteri alone (not shown). Consistent with epithelial hyperplasia induced by co-infection (Fig. 3a), Ki67 activity was prominently increased in the glands (Fig. 4h). Mild to moderate Ki67 activity was also present within the distinct lymphoid follicles in the mucosa and submucosa of co-infected mice and scattered Ki67+ cells were evident as part of the inflammatory infiltrates within the lamina propria and glands.
Figure 4.
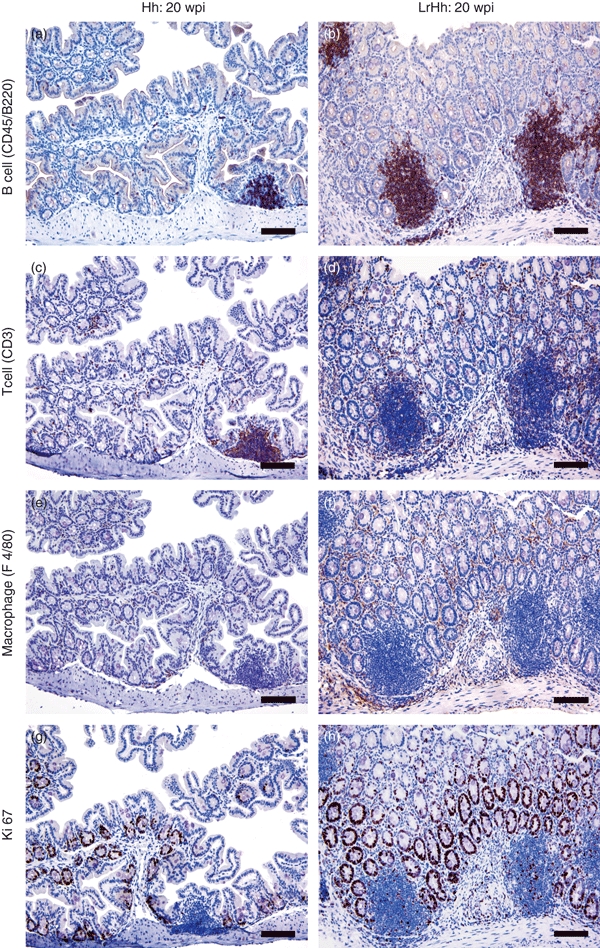
Immunohistochemistry to phenotype B cells (CD45/B220+), T cells (CD3+), macrophages (F4/80+) and proliferating cells (Ki67+) at the caecal–colonic junction of gnotobiotic IL-10−/− mice infected with Helicobacter hepaticus alone (Hh) or with H. hepaticus and Lactobacillus reuteri (LrHh) for 20 weeks post-infection (20 wpi). Germ-free controls and mice mono-associated with L. reuteri alone were similar to H. hepaticus alone and are not shown. Mice with H. hepaticus alone had a few foci of lymphoid aggregates and some distinct lymphoid follicles that contained mostly B cells (a), fewer T cells (c) and sparse macrophages (e). Ki67 activity was minimal and was restricted to the acini and crypts (g). The co-infected mice (LrHh) developed prominent lymphoid follicles that were predominantly B cells (b), with T cells surrounding the germinal centres (d). Small numbers of B cells, considerable numbers of T cells and macrophages (f) were also increased in the lamina propria. Intraepithelial T lymphocytes and macrophages were also increased. Ki67 activity was prominent in the glands (h), within lymphoid follicles and in the inflammatory infiltrates. Magnification × 200 (bar = 80 μm).
Lactobacillus reuteri colonization was suppressed in co-infected IL-10−/− mice but had no effect on H. hepaticus colonization dynamics
Colonization dynamics between L. reuteri and H. hepaticus were examined by assaying either CFU (L. reuteri) or the equivalent for H. hepaticus as estimated by qPCR and genome size. Lactobacillus reuteri was recovered by culture of caecal–colonic tissue from experimentally dosed mice at both 8–11 and 20 weeks p.i. The CFU of L. reuteri were determined in mice killed and examined post-mortem between 8 and 11 weeks p.i. and were significantly lower in mice co-colonized with L. reuteri and H. hepaticus compared with mice mono-associated with L. reuteri alone (P < 0·05, approximately 106 and 108 CFU per gram of tissue, respectively). We used qPCR to estimate H. hepaticus colonization levels because H. hepaticus grows in a mucoid film making colony counts impossible; there was no significant impact of L. reuteri on H. hepaticus colonization levels at the caecal–colonic junction at either 8–11 or 20 weeks p.i. (P = 0·22). Additionally, H. hepaticus colonization levels at 8–11 weeks p.i. persisted at similar levels until 20 weeks p.i. in mono-associated mice (mean ± SE of CFU equivalent of 1·15E+06 ± 2·59E+05 and 1·15E+06 ± 1·9E+05, respectively) and in co-infected IL-10−/− mice (1·82E+06 ± 8·68E+05 and 4·44E+06 ± 1·8E+06, respectively).
Pro-inflammatory IgG2c seroconversion to H. hepaticus was more robust in mice co-infected with L. reuteri
The Th1-associated IgG2c and Th2-associated IgG1 serological responses to H. hepaticus were measured as an index of the pro-inflammatory and anti-inflammatory host responses. Mice co-infected with L. reuteri and H. hepaticus developed higher Th1-associated IgG2c and Th2-associated IgG1 responses to H. hepaticus at 8–11 weeks p.i. compared with mice mono-associated with H. hepaticus (P < 0·004, P < 0·0005, respectively, Fig. 5a). At 20 weeks p.i., the Th1-associated IgG2c response of co-infected mice remained significantly higher than that of H. hepaticus mono-associated mice (P < 0·0005). The H. hepaticus mono-associated mice still had a robust anti-inflammatory associated IgG1 response, but a very weak pro-inflammatory IgG2c response, indicating that anti-inflammatory bias towards H. hepaticus was substantial. Notably the Th2-associated IgG1 to Th1-associated IgG2c ratio was higher and more variable in H. hepaticus mono-associated mice compared with co-infected mice, particularly at 20 weeks p.i. (mean ratio of 13 ± 15 (SD) versus 1·2 ± 0·6, respectively).
Figure 5.
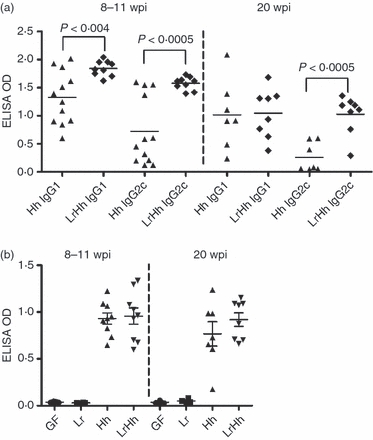
Germ-free B6.129P2-IL-10tm1Cgn (IL-10−/−) mice were colonized at 6–8 weeks of age with Lactobacillus reuteri (Lr), Helicobacter hepaticus (Hh) or L. reuteri followed in 1 week by H. hepaticus (LrHh) then killed and examined post-mortem at 8–11 or 20 weeks post-infection (p.i.). (a) The mean T helper type 2 (Th2)-associated IgG1 and Th1-associated IgG2c responses in co-infected mice were higher at 8–11 weeks p.i. compared with responses in H. hepaticus mono-associated mice (P < 0.004, <0.0005, respectively) and IgG2c was higher in co-infected mice at 20 weeks p.i. (P < 0.0005). The anti-inflammatory IgG1 response to H. hepaticus in mono-associated mice remained elevated through 20 weeks p.i. whereas the pro-inflammatory IgG2c response decreased over time. (b) Mucosal IgA responses to H. hepaticus were similarly robust in H. hepaticus mono-associated or co-infected mice (LrHh). Levels of IgA were similar at both time-points and as expected, germ-free and L. reuteri-infected mice had no detectable IgA to H. hepaticus.
Mucosal IgA responses were similar in mice mono-associated with H. hepaticus or co-infected with L. reuteri
Mucosal IgA responses to H. hepaticus were similarly robust in mice mono-associated with H. hepaticus or co-infected with L. reuteri and H. hepaticus (Fig. 5b). Levels of IgA were similar at both 8–11 and 20 weeks p.i. and, as anticipated, germ-free and L. reuteri-infected mice had no detectable IgA to H. hepaticus.
Typhlocolitis from L. reuteri and H. hepaticus co-infection of IL-10−/− mice was associated with elevated mRNA expression for innate and adaptive immune system genes
An expression array for innate and adaptive immune system gene expression was used to characterize the host inflammatory response to L. reuteri and H. hepaticus as independent and co-infections at 8–11 weeks p.i. With mRNA expression levels in germ-free control mice as a baseline for comparison, pro-inflammatory IL-1β, TNF-α, inducible nitric oxide synthase (iNOS) and anti-inflammatory transforming growth factor-β (TGF-β) were significantly elevated in caecal–colonic tissues obtained from IL-10−/− mice co-infected with L. reuteri and H. hepaticus compared with germ-free controls or mice mono-associated with L. reuteri or H. hepaticus (P < 0·05) (Fig. 6). The fold change increase was highest for TNF-α, followed by IL-1β, iNOS and TGF-β expression in response to the co-infection. Expression of mRNA for all four mediators was similarly low in L. reuteri or H. hepaticus mono-associated IL-10−/− mice.
Figure 6.
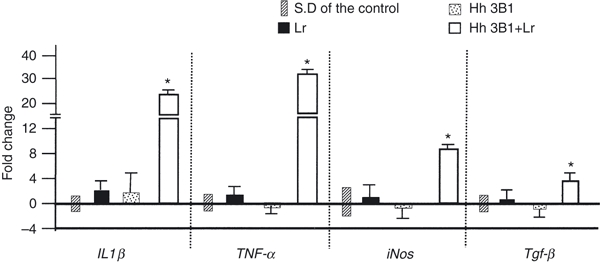
Germ-free B6.129P2-IL-10tm1Cgn (IL-10−/−) mice were colonized at 6–8 weeks of age with Lactobacillus reuteri (Lr) Helicobacter hepaticus (Hh) or L. reuteri followed in 1 week by H. hepaticus (LrHh) then killed and examined post-mortem at 8–11 or 20 weeks post-infection (p.i.). Mean fold-change of mRNA expression was measured in caecal–colonic tissue for interleukin-1β (IL-1β), tumour necrosis factor-α (TNF-α), inducible nitric oxide synthase (iNOS) and transforming growth factor-β (TGF-β) from mice infected with L. reuteri, H. hepaticus or co-infected with both for 8–11 weeks p.i. Co-infected mice had significant up-regulation in all four mediators compared with mono-associated mice (Lr or Hh) or germ-free controls (P < 0.05). SD = standard deviation which is shown for controls (baseline values set at zero) and for experimental groups as error bars. Significant comparisons are indicated by asterisk (*).
Compared with levels measured in germ-free mice, Toll-like receptor (TLR) gene expression was elevated less than twofold for TLR1, TLR2 or TLR3 in any of the L. reuteri and/or H. hepaticus infected IL-10−/− mice. There was a twofold or more increase for TLR4, TLR6, TLR8 and TLR9 in response to co-infection with L. reuteri and H. hepaticus (P < 0·05) (Fig. 7). The TLR4 gene expression also increased threefold in response to L. reuteri mono-association, which was similar to the fourfold increase noted in co-infected mice. Expression of TLR4 in response to H. hepaticus mono-infection was elevated less than twofold, so was low compared with L. reuteri mono-infection or L. reuteri and H. hepaticus co-infection (P < 0·02). The TLR9 mRNA expression was elevated to the greatest degree in L. reuteri and H. hepaticus co-infected mice, followed by TLR8 and TLR6. Notably there were no significant TLR responses other than the TLR4 response to L. reuteri in intestinal tissues of mice mono-infected with L. reuteri or H. hepaticus.
Figure 7.
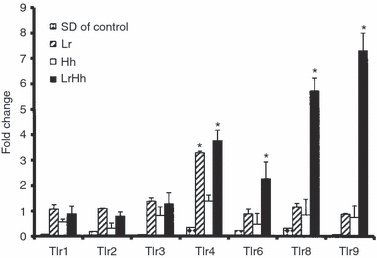
Germ-free B6.129P2-IL-10tm1Cgn (IL-10−/−) mice were colonized at 6–8 weeks of age with Lactobacillus reuteri (Lr), Helicobacter hepaticus (Hh) or L. reuteri followed in 1 week by H. hepaticus (LrHh) then killed and examined post-mortem at 8–11 weeks post-infection (p.i.). Mean fold-change of mRNA expression in caecal–colonic tissue for select toll-like receptors (TLR) from mice infected with L. reuteri, H. hepaticus or co-infected with both for 8–11 weeks p.i. TLR4, TLR6, TLR8 and TLR9 were up-regulated at least twofold by co-infection with LrHh compared with other infection groups (P < 0.05). Significant comparisons are indicated by asterisks (*). TLR4 gene expression increased threefold in response to L. reuteri mono-association which was similar to the fourfold increase noted in co-infected mice.
A broad variety of anti-microbially related genes of the immune system were activated twofold or more by L. reuteri and H. hepaticus co-infection (Table 1). Notably, genes that were up-regulated many-fold at the caecal–colonic junction of the co-infected mice included bactericidal virulence factors such as lactotransferrin, chemokine ligands for recruitment of inflammatory cells, transcription factors belonging to the IL-1 superfamily and lymphoid cell receptors that amplify the Th1 response such as Trem1 and IL-12rβ2.
Table 1.
Innate and adaptive immune system genes activated twofold or more by Lactobacillus reuteri and Helicobacter hepaticus co-infection
| Gene | L. reuteri | H. hepaticus | L. reuteri and H. hepaticus | Biological relevance |
|---|---|---|---|---|
| Casp4 (Caspase 4, apoptosis related cysteine peptidase) | 1·5 | 1·0 | 3·0 | Enzyme component of inflammasome-mediated apoptosis |
| Ccl2 [Chemokine (C-C motif) ligand 2 | 0·7 | 0·8 | 15·7 | Monocyte chemotaxis |
| Ccr3 Chemokine (C-C motif) receptor 3] | 1·1 | 0·9 | 19·0 | Mediates granulocyte activation and pro-inflammatory cytokine production |
| Cd14 (CD14 antigen) | 1·4 | 1·0 | 2·6 | Binds lipopolysaccharide and other PAMPs |
| Cfp (Complement factor properdin) | 1·1 | 0·9 | 4·2 | Alternative pathway for complement activation, stimulates phagocytosis |
| Clec7a (C-type lectin domain family 7, member α) | 1·1 | 0·7 | 3·8 | PAMP receptor |
| Cxcr4 (Chemokine receptor 4) | 0·8 | 0·5 | 4·8 | Receptor of CXCL12 mediating inflammatory cell recruitment |
| Cybb (Cytochrome β-245, β polypeptide) | 1·1 | 0·7 | 8·5 | Superoxide anion production by neutrophils |
| IL-12rβ2 (IL-12 receptor β 2) | 1·4 | 1·0 | 3·7 | Subunit of interleukin-12 binding receptor, up-regulated by interferon-γ |
| IL-1α (Interleukin 1α) | 1 | 0·8 | 3·6 | Pro-inflammatory cytokine secreted by macrophages |
| IL-1f6 (Family member 6) | 0·5 | 0·5 | 79·5 | Structural and functional homology with IL-1 superfamily |
| IL-1f8 (Family member 8) | 1·5 | 5·2 | 4·4 | Structural and functional homology with IL-1 superfamily |
| IL-1f9 (Family member 9) | 0·8 | 0·7 | 3·3 | Structural and functional homology with IL-1 superfamily |
| Il1r2 (Interleukin 1 receptor, type II) | 1·1 | 0·7 | 4·0 | Member of IL-1 receptor family |
| IL-1rl2 (Interleukin 1 receptor-like 2) | 1·1 | 0·7 | 2·3 | Member of IL-1 receptor family |
| IL-1rn (Receptor antagonist) | 1·0 | 1·0 | 4·7 | Blocks binding of IL-1α and IL-1β |
| Irf1 (Interferon regulatory factor 1) | 1·2 | 1·0 | 2·9 | Transcription factor regulating pro-inflammatory IL-18 |
| Ltf (Lactotransferrin) | 1·6 | 0·3 | 97·0 | Bactericidal, binds iron, inhibits bacterial adhesion |
| Lyz1 (Lysozyme 1) | 1·4 | 1·4 | 3·6 | Anti-bacterial muramidase released from neutrophils |
| Ncf4 (Neutrophil cytosolic factor 4) | 0·9 | 0·8 | 4·4 | Regulator of NADPH-oxidase production of superoxide by phagocytes |
| NfkBiα (Nf-κΒ inhibitor, α) | 1·1 | 0·9 | 2·1 | Binds and inhibits nuclear factor-κΒ |
| Pglyrp1 (Peptidoglycan recognition protein 1) | 1·8 | 0·9 | 3·7 | Neutrophil amidase targeting bacterial peptidoglycans |
| Pglyrp2 (Peptidoglycan recognition protein 2) | 1·7 | 0·9 | 4·4 | Neutrophil amidase targeting bacterial peptidoglycans |
| Prg2 (Proteoglycan 2, bone marrow) | 0·5 | 0·3 | 19·2 | Natural killer cell activation, constituent of eosinophil granules |
| Ptafr (Platelet activating factor receptor) | 0·9 | 1·2 | 4·9 | Binds PAF to signal recruitment of neutrophils |
| Serpina1a (Serine peptidase inhibitor, clade A, member 1a) | 0·8 | 0·6 | 3·4 | Serine protease inhibitor, protects host tissues from proteases |
| Stab 1 (Stablin 1) | 0·9 | 0·7 | 2·3 | Cell–cell and cell–matrix interactions in vascular function and inflammatory processes |
| Trem1 (Triggering receptor on myeloid cells 1) | 2·0 | 2·0 | 7·1 | Expressed on macrophages and amplifies T helper type 1 cytokine responses |
IL-1, interleukin-1; PAF, platelet-activating factor; PAMP.
Discussion
In this study, mono-association of gnotobiotic B6.129 IL-10−/− mice with H. hepaticus 3B1, the prototypic H. hepaticus strain containing CDT19 and a putative pathogenicity island,24 did not induce significant typhlocolitis, consistent with an earlier report that gnotobiotic, colitis-susceptible C57BL/6 × 129/Ola or 129/SvEv IL-10−/− mice developed minimal colitis when mono-associated with H. hepaticus 3B1 for 16 weeks.15 The lack of intestinal lesions from H. hepaticus mono-association may reflect the in vitro ability of H. hepaticus to suppress innate immune responsiveness of murine epithelial cells.31 Additionally, a type 6 secretion system in H. hepaticus32 has been shown to have a protective, anti-inflammatory activity that suppresses innate and adaptive immune responses during intestinal colonization. Nonetheless, with a complex microbiota in an SPF IL-10-deficient host, the same H. hepaticus 3B1 inoculum caused significant typhlocolitis within 11 weeks. Our study is similar to a report of gnotobiotic IL-10−/− mice that developed aggressive pancolitis when co-infected with non-pathogenic strains of Enterococcus faecalis and Escherichia coli that individually caused only mild inflammatory responses.5 Minimal inflammatory response to H. hepaticus in mono-associated IL-10−/− mice appears reproducible15 but nonetheless is striking given the deficiency of IL-10, an anti-inflammatory cytokine important for T regulatory cell function.33
This is a first report of mono-associated L. reuteri PTA-6475 persistently colonizing gnotobiotic IL-10−/− mice without producing intestinal lesions; however, L. reuteri co-colonized with H. hepaticus caused severe typhlocolitis. These findings contrast with the attenuation of intestinal lesions in H. hepaticus infected, SPF B6.129 IL-10−/− male mice when experimentally co-colonized with murine-derived L. paracasei 1602 and L. reuteri 6798.6 In the current study, typhlocolitis, epithelial defects, hyperplasia and dysplasia at the caecal–colonic junction lesions in co-infected mice were associated with elevated serum levels of Th1-associated IgG2c specific for H. hepaticus, possibly the result of an adjuvant effect of L. reuteri infection on the inflammatory response to H. hepaticus. Mucosal IgA responses to H. hepaticus were similar in mice mono-associated with H. hepaticus or co-infected with H. hepaticus and L. reuteri. Hence, H. hepaticus mono-associated mice developed a mucosal IgA response that was not accompanied by pro-inflammatory responses as demonstrated in co-infected mice that had elevated IL-1β, TNF-α, iNOS and TGF-β gene expression in addition to increased numbers of T cells and macrophages at the caecal–colonic junction, the main site of inflammation. These data are consistent with a report that IL-10−/− mice developed more robust gastritis and were colonized with higher levels of Helicobacter pylori when the gene for IgA was functionally inactivated,34 supporting the theory that IgA responses protected both the host and bacteria against inflammation.
Lactobacillus reuteri PTA-6475 was originally isolated from human milk, suggesting a role in GALT development in human babies.35 Until recently, only in vitro suppression of nuclear factor-κB signalling, reduced TNF-α activity and lymphoid apoptosis have been demonstrated for this L. reuteri strain.6,22,36 The L. reuteri may have suppressed nuclear factor-κB signalling and so suppressed innate and adaptive immune responses to H. hepaticus during early co-infection. In a neonatal rat model, L. reuteri PTA-6475 significantly reduced intestinal mucosal levels of KC/GRO (∼ IL-8) and IFN-γ and intestinal lesions in neonatal rats fed formula containing lipopolysaccharide.37 As H. hepaticus colonization became established, L. reuteri may have had an impact on the expression of virulence genes important for H. hepaticus colonization or virulence. Genome analysis 24 and infection studies with H. hepaticus strains containing sequence mutations in the cytolethal distending toxin gene cdt17,18 or deletions in a putative pathogenicity island (HHGI1)2,16 have demonstrated a genetic basis for the virulence variability of various H. hepaticus isolates. Hence, L. reuteri may have promoted the inflammatory response to H. hepaticus by enhancing expression of H. hepaticus CDT or genes within the putative HHGI1 pathogenicity island or possibly down-regulating the type 6 secretion system of H. hepaticus.
Paradoxically, select strains of probiotic lactobacilli have been associated with stimulating the innate immune response that can progress into a Th1-mediated inflammation38,39 in contrast to other lactobacilli that promote expansion of T regulatory cells through an adjuvant-like effect mediated by TLR9 signalling.40,41 Thus, in contrast to suppression of innate immunity, L. reuteri PTA-6475 may have alternatively primed an immune response to subsequent H. hepaticus infection, as noted in another L. reuteri strain (100–23) that was reported to provoke pro-inflammatory cytokine responses early in the course of colonizing mice.14,38,42 The order and timing of the serial infections of L. reuteri and H. hepaticus may have been important as early immune priming by L. reuteri may have led to further promotion of inflammatory responses to H. hepaticus in co-infected mice, particularly when superimposed on an IL-10-deficient tissue environment.
Typhlocolitis developed only in response to co-colonization of L. reuteri and H. hepaticus and both bacteria were found in significant numbers in the same ecological niche of the caecal–colonic junction. Although some L. reuteri strains release known and putative anti-microbial factors that may inhibit proliferation of important gastrointestinal pathogens,43L. reuteri PTA-6475 is a relatively low producer of reuterin44 and co-infected mice did not have H. hepaticus colonization levels different from those found in mono-associated mice. Colonization of L. reuteri in co-infected mice was reduced compared with L. reuteri mono-associated mice at 8–11 weeks p.i. and reduction of L. reuteri levels may have been secondary to typhlocolitis.
Chronic H. pylori gastritis and hepatitis in mice induced by a novel enterohepatic helicobacter species (MIT 96-1001) have been associated with de novo formation of organized tertiary lymphoid tissue.45,46 In this study, the H. hepaticus and L. reuteri induced typhlocolitis was accompanied by increased cellularity of GALT, observable as well-developed Peyer's patches in the small intestine accompanied by variably sized lymphoid aggregates, and was most evident at the caecal–colonic junction where lymphoid follicles were expanded by B and T cells. Intraepithelial T lymphocytes plus B cells, T cells and macrophages were also increased in the surrounding lamina propria. Consistent with higher scores for epithelial hyperplasia induced by co-infection, Ki67 activity was prominently increased in the glands as well as within lymphoid follicles in the mucosa and submucosa. Comparison of GALT sections between germ-free mice and mice mono-associated with H. hepaticus or L. reuteri showed that the mono-associated groups had variably increased but comparatively low numbers of B cells, T cells and macrophages in contrast to the higher numbers in co-infected mice.
Messenger RNA expression levels of genes reflecting activation of the innate and adaptive immune responses were elevated at least twofold in IL-10−/− mice co-infected with L. reuteri and H. hepaticus, reflecting a Th1-promoted inflammatory bowel disease as demonstrated for helicobacter-infected IL-10−/−mice with complex microbiota.2,3,6,47 In support of L. reuteri promoting the inflammatory response to H. hepaticus, IL-10−/− mice mono-associated with H. hepaticus exhibited no enhanced mRNA expression for inflammatory mediators and developed a higher anti-inflammatory-like IgG1–IgG2c ratio of isotype responses to the single infection compared with co-infected mice. Gene array data suggest that typhlocolitis was probably promoted principally by bacterial breakdown products, such as bacterial DNA CpG motifs activating TLR9 expression,48 which was the TLR gene most activated by co-infection. Although DNA from commensal microbiota, including probiotic species, promotes T regulatory cell development through TLR9 signalling, commensal DNA also has natural adjuvant effects on intestinal innate immune responses.40,49 A TLR9 polymorphism in humans linked to increased risk of H. pylori-induced pre-malignant gastric pathology has been associated with increased TLR9 transcription secondary to enhanced binding of nuclear factor-κB and subsequent increased host sensitivity to H. pylori CpG DNA.50 The increased TLR9 mRNA expression noted in our study may reflect enhanced intestinal permeability reported for IL-10−/− mice51 or enhanced sensitivity of the inflamed IL-10−/− tissue environment to CpG motifs released from both L. reuteri and H. hepaticus. In addition, the balance between the pro-inflammatory and anti-inflammatory effects of probiotics probably depends on host genetics and the type and breadth of bacterial species comprising the intestinal microbiota. For example, TLR9 transcriptional activity and increased nuclear factor-κΒ activation appeared higher in C57BL/6 mice compared with BALB/c.52Helicobacter felis-infected TLR9−/− mice had lower lymphoproliferative and IFN-γ responses and reduced neutrophilic gastritis, supporting our link between significantly up-regulated expression of TLR9 and other pro-inflammatory host genes in response to L. reuteri and H. hepaticus co-infection.
Gene expression levels of TLR4, TLR6, and TLR8 were also elevated in L. reuteri and H. hepaticus co-infected IL-10−/− mice. Although TLR4 mRNA expression was highest in co-infected mice, TLR4 mRNA was elevated in L. reuteri mono-associated mice even though TLR4 activation is most often associated with lipopolysaccharide activation. A role for TLR4 in T regulatory cell-mediated homeostasis in the H. hepaticus-infected IL-10−/−colitis model was suggested by earlier onset of colitis in H. hepaticus-infected TLR4−/− IL-10−/− mice that had aberrant T regulatory Foxp3+ cells in the lamina propria that secreted IFN-γ, IL-17, and RANTES (regulated on activation, normal, T-cell expressed and secreted).53 Little is known about the role of TLR6 in H. hepaticus-induced inflammation except that TLR6 complexed with TLR2 mediated IL-8 release from TLR2-transfected HEK293 cells incubated with H. hepaticus.54 Notably, typhlocolitis in the co-infected IL-10−/− mice may have been partially mediated through TLR8, as TLR8 activation has been shown in mice to reverse T regulatory cell suppression.55 Importantly, gene expression for TLRs was measured on whole thickness samples of the caecal–colonic junction and phenotypic evaluation of these receptors by cell type or signalling activity may yield more information on the significance of the mRNA elevation noted here in response to L. reuteri and H. hepaticus. Additionally, extension of these studies into evaluation of germ-free, TLR knockout mice should reveal whether these TLR-mediated signals are inflammatory or regulatory in the response to co-infection.
These data support that the development of typhlocolitis in H. hepaticus infected IL-10−/− mice required co-colonization with other microbiota and in this study, required only L. reuteri. Although the effects other microbiota may have on H. hepaticus virulence properties remain speculative, further investigations using this gnotobiotic model are now possible.
Acknowledgments
This work was supported in part by RO1-CA67529, R01DK052413, PO1CA26731, P30ESO2109 (J.G.F.) and R01DK065075 (J.G.F. and J.V.) from the National Institutes of Health. We thank Eamonn Connolly at Biogaia AB (Stockholm, Sweden) as the original source of human-derived Lactobacillus reuteri. We also thank Carlos Umana and Oscar Acevedo who contributed to colony maintenance and Melissa Mobley and Amanda Potter for technical assistance.
Disclosure
The authors have no conflicts of interest to declare.
References
- 1.Packey CD, Sartor RB. Commensal bacteria, traditional and opportunistic pathogens, dysbiosis and bacterial killing in inflammatory bowel diseases. Curr Opin Infect Dis. 2009;22:292–301. doi: 10.1097/QCO.0b013e32832a8a5d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ge Z, Sterzenbach T, Whary MT, et al. Helicobacter hepaticus HHGI1 is a pathogenicity island associated with typhlocolitis in B6.129-IL-10tm1Cgn mice. Microbes Infect. 2008;10:726–33. doi: 10.1016/j.micinf.2008.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kullberg MC, Rothfuchs AG, Jankovic D, Caspar P, Wynn TA, Gorelick PL, Cheever AW, Sher A. Helicobacter hepaticus-induced colitis in interleukin-10-deficient mice: cytokine requirements for the induction and maintenance of intestinal inflammation. Infect Immun. 2001;69:4232–41. doi: 10.1128/IAI.69.7.4232-4241.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sellon RK, Tonkonogy S, Schultz M, Dieleman LA, Grenther W, Balish E, Rennick DM, Sartor RB. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect Immun. 1998;66:5224–31. doi: 10.1128/iai.66.11.5224-5231.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim SC, Tonkonogy SL, Karrasch T, Jobin C, Sartor RB. Dual-association of gnotobiotic IL-10−/− mice with two non-pathogenic commensal bacteria induces aggressive pancolitis. Inflamm Bowel Dis. 2007;13:1457–66. doi: 10.1002/ibd.20246. [DOI] [PubMed] [Google Scholar]
- 6.Pena JA, Rogers AB, Ge Z, Ng V, Li SY, Fox JG, Versalovic J. Probiotic Lactobacillus spp. diminish Helicobacter hepaticus-induced inflammatory bowel disease in interleukin-10-deficient mice. Infect Immun. 2005;73:912–20. doi: 10.1128/IAI.73.2.912-920.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang L, Danon SJ, Grehan M, Chan V, Lee A, Mitchell H. Natural colonization with Helicobacter species and the development of inflammatory bowel disease in interleukin-10-deficient mice. Helicobacter. 2005;10:223–30. doi: 10.1111/j.1523-5378.2005.00314.x. [DOI] [PubMed] [Google Scholar]
- 8.Foltz CJ, Fox JG, Cahill R, Murphy JC, Yan L, Shames B, Schauer D. Spontaneous inflammatory bowel disease in multiple mutant mouse lines: association with colonization by Helicobacter hepaticus. Helicobacter. 1998;3:69–78. doi: 10.1046/j.1523-5378.1998.08006.x. [DOI] [PubMed] [Google Scholar]
- 9.Ward JM, Anver MR, Haines DC, Melhorn JM, Gorelick P, Yan L, Fox JG. Inflammatory large bowel disease in immunodeficient mice naturally infected with Helicobacter hepaticus. Lab Anim Sci. 1996;46:15–20. [PubMed] [Google Scholar]
- 10.Cahill RJ, Foltz CJ, Fox JG, Dangler CA, Powrie F, Schauer DB. Inflammatory bowel disease: an immunity-mediated condition triggered by bacterial infection with Helicobacter hepaticus. Infect Immun. 1997;65:3126–31. doi: 10.1128/iai.65.8.3126-3131.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Balfour SR. Bacteria in Crohn's disease: mechanisms of inflammation and therapeutic implications. J Clin Gastroenterol. 2007;41(Suppl. 1):S37–43. doi: 10.1097/MCG.0b013e31802db364. [DOI] [PubMed] [Google Scholar]
- 12.Izcue A, Coombes JL, Powrie F. Regulatory lymphocytes and intestinal inflammation. Annu Rev Immunol. 2009;27:313–38. doi: 10.1146/annurev.immunol.021908.132657. [DOI] [PubMed] [Google Scholar]
- 13.Kullberg MC, Ward JM, Gorelick PL, Caspar P, Hieny S, Cheever A, Jankovic D, Sher A. Helicobacter hepaticus triggers colitis in specific-pathogen-free interleukin-10 (IL-10)-deficient mice through an IL-12- and gamma interferon-dependent mechanism. Infect Immun. 1998;66:5157–66. doi: 10.1128/iai.66.11.5157-5166.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berg DJ, Davidson N, Kuhn R, et al. Enterocolitis and colon cancer in interleukin-10-deficient mice are associated with aberrant cytokine production and CD4+ TH1-like responses. J Clin Invest. 1996;98:1010–20. doi: 10.1172/JCI118861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dieleman LA, Arends A, Tonkonogy SL, et al. Helicobacter hepaticus does not induce or potentiate colitis in interleukin-10-deficient mice. Infect Immun. 2000;68:5107–13. doi: 10.1128/iai.68.9.5107-5113.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boutin SR, Shen Z, Rogers AB, et al. Different Helicobacter hepaticus strains with variable genomic content induce various degrees of hepatitis. Infect Immun. 2005;73:8449–52. doi: 10.1128/IAI.73.12.8449-8452.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Young VB, Knox KA, Pratt JS, Cortez JS, Mansfield LS, Rogers AB, Fox JG, Schauer DB. In vitro and in vivo characterization of Helicobacter hepaticus cytolethal distending toxin mutants. Infect Immun. 2004;72:2521–7. doi: 10.1128/IAI.72.5.2521-2527.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Young VB, Knox KA, Schauer DB. Cytolethal distending toxin sequence and activity in the enterohepatic pathogen Helicobacter hepaticus. Infect Immun. 2000;68:184–91. doi: 10.1128/iai.68.1.184-191.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pratt JS, Sachen KL, Wood HD, Eaton KA, Young VB. Modulation of host immune responses by the cytolethal distending toxin of Helicobacter hepaticus. Infect Immun. 2006;74:4496–504. doi: 10.1128/IAI.00503-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fox JG, Dewhirst FE, Tully JG, et al. Helicobacter hepaticus sp. nov., a microaerophilic bacterium isolated from livers and intestinal mucosal scrapings from mice. J Clin Microbiol. 1994;32:1238–45. doi: 10.1128/jcm.32.5.1238-1245.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fox JG, Li X, Yan L, Cahill RJ, Hurley R, Lewis R, Murphy JC. Chronic proliferative hepatitis in A/JCr mice associated with persistent Helicobacter hepaticus infection: a model of helicobacter-induced carcinogenesis. Infect Immun. 1996;64:1548–58. doi: 10.1128/iai.64.5.1548-1558.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin YP, Thibodeaux CH, Pena JA, Ferry GD, Versalovic J. Probiotic Lactobacillus reuteri suppress proinflammatory cytokines via c-Jun. Inflamm Bowel Dis. 2008;14:1068–83. doi: 10.1002/ibd.20448. [DOI] [PubMed] [Google Scholar]
- 23.Ward JM, Fox JG, Anver MR, et al. Chronic active hepatitis and associated liver tumors in mice caused by a persistent bacterial infection with a novel Helicobacter species. J Natl Cancer Inst. 1994;86:1222–7. doi: 10.1093/jnci/86.16.1222. [DOI] [PubMed] [Google Scholar]
- 24.Suerbaum S, Josenhans C, Sterzenbach T, et al. The complete genome sequence of the carcinogenic bacterium Helicobacter hepaticus. Proc Natl Acad Sci U S A. 2003;100:7901–6. doi: 10.1073/pnas.1332093100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Whary MT, Cline JH, King AE, Corcoran CA, Xu S, Fox JG. Containment of Helicobacter hepaticus by use of husbandry practices. Comp Med. 2000;50:78–81. [PubMed] [Google Scholar]
- 26.Ge Z, White DA, Whary MT, Fox JG. Fluorogenic PCR-based quantitative detection of a murine pathogen, Helicobacter hepaticus. J Clin Microbiol. 2001;39:2598–602. doi: 10.1128/JCM.39.7.2598-2602.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ullman-Cullere MH, Foltz CJ. Body condition scoring: a rapid and accurate method for assessing health status in mice. Lab Anim Sci. 1999;49:319–23. [PubMed] [Google Scholar]
- 28.Rogers AB, Cormier KS, Fox JG. Thiol-reactive compounds prevent non-specific antibody binding in immunohistochemistry. Lab Invest. 2006;86:526–33. doi: 10.1038/labinvest.3700407. [DOI] [PubMed] [Google Scholar]
- 29.Martin RM, Brady JL, Lew AM. The need for IgG2c specific antiserum when isotyping antibodies from C57BL/6 and NOD mice. J Immunol Methods. 1998;212:187–92. doi: 10.1016/s0022-1759(98)00015-5. [DOI] [PubMed] [Google Scholar]
- 30.Whary MT, Morgan TJ, Dangler CA, Gaudes KJ, Taylor NS, Fox JG. Chronic active hepatitis induced by Helicobacter hepaticus in the A/JCr mouse is associated with a Th1 cell-mediated immune response. Infect Immun. 1998;66:3142–8. doi: 10.1128/iai.66.7.3142-3148.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sterzenbach T, Lee SK, Brenneke B, von Goetz F, Schauer DB, Fox JG, Suerbaum S, Josenhans C. Inhibitory effect of enterohepatic Helicobacter hepaticus on innate immune responses of mouse intestinal epithelial cells. Infect Immun. 2007;75:2717–28. doi: 10.1128/IAI.01935-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chow J, Mazmanian SK. A pathobiont of the microbiota balances host colonization and intestinal inflammation. Cell Host Microbe. 2010;7:265–76. doi: 10.1016/j.chom.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee CW, Rao VP, Rogers AB, Ge Z, Erdman SE, Whary MT, Fox JG. Wild-type and interleukin-10-deficient regulatory T cells reduce effector T-cell-mediated gastroduodenitis in Rag2−/− mice, but only wild-type regulatory T cells suppress Helicobacter pylori gastritis. Infect Immun. 2007;75:2699–707. doi: 10.1128/IAI.01788-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Akhiani AA, Stensson A, Schon K, Lycke NY. IgA antibodies impair resistance against Helicobacter pylori infection: studies on immune evasion in IL-10-deficient mice. J Immunol. 2005;174:8144–53. doi: 10.4049/jimmunol.174.12.8144. [DOI] [PubMed] [Google Scholar]
- 35.Preidis GA, Versalovic J. Targeting the human microbiome with antibiotics, probiotics, and prebiotics: gastroenterology enters the metagenomics era. Gastroenterology. 2009;136:2015–31. doi: 10.1053/j.gastro.2009.01.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Iyer C, Kosters A, Sethi G, Kunnumakkara AB, Aggarwal BB, Versalovic J. Probiotic Lactobacillus reuteri promotes TNF-induced apoptosis in human myeloid leukemia-derived cells by modulation of NF-kappaB and MAPK signalling. Cell Microbiol. 2008;10:1442–52. doi: 10.1111/j.1462-5822.2008.01137.x. [DOI] [PubMed] [Google Scholar]
- 37.Liu Y, Fatheree NY, Mangalat N, Rhoads JM. Human derived probiotic Lactobacillus reuteri strains differentially reduce intestinal inflammation. Am J Physiol Gastrointest Liver Physiol. 2010;299:G1087–96. doi: 10.1152/ajpgi.00124.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mohamadzadeh M, Olson S, Kalina WV, Ruthel G, Demmin GL, Warfield KL, Bavari S, Klaenhammer TR. Lactobacilli activate human dendritic cells that skew T cells toward T helper 1 polarization. Proc Natl Acad Sci U S A. 2005;102:2880–5. doi: 10.1073/pnas.0500098102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dogi CA, Galdeano CM, Perdigon G. Gut immune stimulation by non-pathogenic Gram+ and Gram− bacteria. Comparison with a probiotic strain. Cytokine. 2008;41:223–31. doi: 10.1016/j.cyto.2007.11.014. [DOI] [PubMed] [Google Scholar]
- 40.Hall JA, Bouladoux N, Sun CM, et al. Commensal DNA limits regulatory T cell conversion and is a natural adjuvant of intestinal immune responses. Immunity. 2008;29:637–49. doi: 10.1016/j.immuni.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Di Giacinto C, Marinaro M, Sanchez M, Strober W, Boirivant M. Probiotics ameliorate recurrent Th1-mediated murine colitis by inducing IL-10 and IL-10-dependent TGF-beta-bearing regulatory cells. J Immunol. 2005;174:3237–46. doi: 10.4049/jimmunol.174.6.3237. [DOI] [PubMed] [Google Scholar]
- 42.Livingston M, Loach D, Wilson M, Tannock GW, Baird M. Gut commensal Lactobacillus reuteri 100-23 stimulates an immunoregulatory response. Immunol Cell Biol. 2010;88:99–102. doi: 10.1038/icb.2009.71. [DOI] [PubMed] [Google Scholar]
- 43.Spinler JK, Taweechotipatr M, Rognerud CL, Ou CN, Tumwasorn S, Versalovic J. Human-derived probiotic Lactobacillus reuteri demonstrate antimicrobial activities targeting diverse enteric bacterial pathogens. Anaerobe. 2008;14:166–71. doi: 10.1016/j.anaerobe.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jones SE, Versalovic J. Probiotic Lactobacillus reuteri biofilms produce antimicrobial and anti-inflammatory factors. BMC Microbiol. 2009;9:35. doi: 10.1186/1471-2180-9-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Winter S, Loddenkemper C, Aebischer A, Rabel K, Hoffmann K, Meyer TF, Lipp M, Hopken UE. The chemokine receptor CXCR5 is pivotal for ectopic mucosa-associated lymphoid tissue neogenesis in chronic Helicobacter pylori-induced inflammation. J Mol Med. 2010;88:1169–80. doi: 10.1007/s00109-010-0658-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shomer NH, Fox JG, Juedes AE, Ruddle NH. Helicobacter-induced chronic active lymphoid aggregates have characteristics of tertiary lymphoid tissue. Infect Immun. 2003;71:3572–7. doi: 10.1128/IAI.71.6.3572-3577.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kullberg MC, Jankovic D, Feng CG, et al. IL-23 plays a key role in Helicobacter hepaticus-induced T cell-dependent colitis. J Exp Med. 2006;203:2485–94. doi: 10.1084/jem.20061082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee J, Rachmilewitz D, Raz E. Homeostatic effects of TLR9 signaling in experimental colitis. Ann NY Acad Sci. 2006;1072:351–5. doi: 10.1196/annals.1326.022. [DOI] [PubMed] [Google Scholar]
- 49.Pagnini C, Saeed R, Bamias G, Arseneau KO, Pizarro TT, Cominelli F. Probiotics promote gut health through stimulation of epithelial innate immunity. Proc Natl Acad Sci U S A. 2009;107:454–59. doi: 10.1073/pnas.0910307107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ng MT, Van't Hof R, Crockett JC, et al. Increase in NF-kappaB binding affinity of the variant C allele of the toll-like receptor 9 -1237T/C polymorphism is associated with Helicobacter pylori-induced gastric disease. Infect Immun. 2010;78:1345–52. doi: 10.1128/IAI.01226-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arrieta MC, Madsen K, Doyle J, Meddings J. Reducing small intestinal permeability attenuates colitis in the IL-10 gene-deficient mouse. Gut. 2009;58:41–8. doi: 10.1136/gut.2008.150888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Anderson AE, Worku ML, Khamri W, Bamford KB, Walker MM, Thursz MR. TLR9 polymorphisms determine murine lymphocyte responses to Helicobacter: results from a genome-wide scan. Eur J Immunol. 2007;37:1548–61. doi: 10.1002/eji.200636562. [DOI] [PubMed] [Google Scholar]
- 53.Matharu KS, Mizoguchi E, Cotoner CA, et al. Toll-like receptor 4-mediated regulation of spontaneous Helicobacter-dependent colitis in IL-10 deficient mice. Gastroenterology. 2009;137:1380–90. doi: 10.1053/j.gastro.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mandell L, Moran AP, Cocchiarella A, Houghton J, Taylor N, Fox JG, Wang TC, Kurt-Jones EA. Intact gram-negative Helicobacter pylori, Helicobacter felis, and Helicobacter hepaticus bacteria activate innate immunity via toll-like receptor 2 but not toll-like receptor 4. Infect Immun. 2004;72:6446–54. doi: 10.1128/IAI.72.11.6446-6454.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Peng G, Guo Z, Kiniwa Y, et al. Toll-like receptor 8-mediated reversal of CD4+ regulatory T cell function. Science. 2005;309:1380–4. doi: 10.1126/science.1113401. [DOI] [PubMed] [Google Scholar]