

Abstract
The ability to alter the cytokine microenvironment has the potential to shape immune responses in many physiological settings, including the immunotherapy of tumours. We set out to develop a general approach in which cytokines could be functionally attenuated until activated. We report the development and initial characterization of fusion proteins in which human or mouse interleukin-2 (IL-2), a potent growth factor for immune cells, is joined to a specific IL-2 inhibitory binding component separated by a protease site. The rationale is that upon cleavage by a protease the cytokine is free to dissociate from the inhibitory component and becomes biologically more available. We describe the successful development of two attenuation strategies using specific binding: the first uses the mouse IL-2 receptor alpha chain as the inhibitory binding component whereas the second employs a human antibody fragment (scFv) reactive with human IL-2. We demonstrated that the fusion proteins containing a prostate-specific antigen or a matrix metalloproteinase (MMP) protease cleavage site are markedly attenuated in the intact fusion protein but had enhanced bioactivity of IL-2 in vitro when cleaved. Further, we showed that a fusion protein composed of the IL-2/IL-2 receptor alpha chain with an MMP cleavage site reduced tumour growth in vivo in a peritoneal mouse tumour model. This general strategy should be applicable to other proteases and immune modulators allowing site-specific activation of immunomodulators while reducing unwanted side-effects.
Keywords: cytokines, fusion proteins, interleukin-2, proteases, single chain antibodies, T cells, tumour microenvironment
Introduction
Considerable progress has been made in the treatment of cancer. However, a critical goal of cancer therapy remains the improved treatment of metastatic disease. Immunotherapy is conceptually attractive for the treatment of disseminated disease because cells of the immune system circulate throughout the organism and could in principle eliminate the widely distributed but relatively small metastases that originate from the primary tumour.1 T cells that recognize tumour-associated antigens have been clearly identified not only in experimental animals but also in human cancer patients and now many tumour-associated antigens have been molecularly characterized.2–5 However, despite the remarkable success at identifying tumour-associated antigens, the cellular immune response has generally not been successful at eliminating tumours. Generating clinically effective anti-tumour responses has long been a goal of tumour immunology and remains a challenge today.
One strategy for enhancing the immune response to tumours has been the use of cytokines. Investigators have not only focused on the use of cytokines to aid in the initiation of immune responses to tumours4,6 but also used them systemically as therapeutic agents.7 The cytokine interleukin-2 (IL-2) is currently approved to treat melanoma and renal cancer.7–9 However, cytokines can have serious side-effects when delivered systemically. Indeed, these undesired side effects have limited the use of IL-2 clinically, which has, in some cases, brought about remarkable and durable remissions.8 From a conceptual standpoint, these side-effects might be anticipated because many cytokines function physiologically in a paracrine fashion, over short distances between cells.6 An important challenge is to develop methods to deliver cytokines to tumour sites where they might enhance immune responses without producing undesirable systemic effects. Experimentally this has been achieved in a variety of ways including direct local injections of cytokines,10–12 injection of tumours with viruses encoding cytokine genes,13–15 or by transplanting genetically modified viable tumours into animals.16,17 These approaches have greatly contributed to our understanding of the effects of the local production of cytokines on the number and function of immune cells within the tumour microenvironment and also illustrated the considerable potential of cytokines to enhance anti-tumour immune responses. However, because metastatic lesions are often numerous and not easily accessible, translating these advances into a clinical setting remains a challenge. Hence, there remains a critical need to develop ways in which the cytokine milieu in the tumour microenvironment can be altered.
In our current work, we set out to develop a general strategy to construct cytokines that are biologically inactive but could be activated by proteases. Ultimately this approach could be used to deliver inactive cytokines systemically but have them activated locally by tumour-site-expressed proteases. In principle, this should reduce systemic side-effects but retain the enhancement of anti-tumour immune responses. The strategy we are developing uses a fusion protein approach that takes advantage of proteases that are secreted by tumours. As an initial test of this general strategy, we have used different proteases, prostate-specific antigen (PSA), matrix metalloproteinase 2 (MMP2) or MMP9. The expression of the protease PSA is highly restricted to prostate epithelial cells; PSA is produced by prostate tumours, and as such, is an excellent target protease for activating the cytokine fusion protein.18 The MMPs have been known to have critical and varied roles in tumour development and progression and are preferentially expressed in a variety of tumours.19 We have used IL-2 as the test cytokine in the fusion protein because it is a potent factor for T-cell and natural killer (NK) cell development20,21 and the local production of IL-2 within tumours has demonstrated anti-tumour immunological effects in animal models.16,17 Moreover, an IL-2-containing fusion protein might be able to be more easily translated to the treatment of human cancers because IL-2 is already Food and Drug Administration approved for the treatment of certain tumours.7–9 In this report, we examine several strategies of blocking the biological activity of IL-2, yet allowing it to be functionally activated by PSA or MMP proteases. We show that IL-2 biological activity can be markedly attenuated in the context of a fusion protein consisting of IL-2 and an inhibitory binding component separated by a protease cleavage site, but that the biological activity of the IL-2 increases after protease action. The significance and potential application of this approach for the treatment of tumours is also addressed.
Materials and methods
Construction of the mouse IL-2/PSAcs/IL-2Rα fusion protein
Interleukin-2 receptor alpha (IL-2Rα; generously provided by Dr Jim Miller, University of Rochester) in pcEVX-3 was PCR amplified using primers (Table 1) to add the KpnI and BamHI restriction sites, remove the hydrophobic transmembrane region and, for some constructs, addition of a 6 × Histidine tag (6 × His). This product was cloned into pBluescript (pBluescript IL-2Rα). The (GGGGS)x linker of various repeat lengths was either synthesized (GENEART Inc., Toronto, ON, Canada) or was made by annealing primers from complimentary oligonucleotides (Table 1) and then cloned into pBluescript using the EcoRI and KpnI restriction sites. The (GGGGS)x linker was excised and cloned into the pBluescript IL-2Rα plasmid. Τhe linker and IL-2Rα were excised using the EcoRI and BamHI sites and directionally cloned into the pBluescript IL-2/PSAcs plasmid containing murine IL-2 and the PSA cleavage sequence (HSSKLQ) resulting in the pBluescript IL-2/PSAcs/linker/IL-2Rα plasmid. This plasmid was then verified by sequencing and subsequently cloned into pcDNA3.1 (Invitrogen, Carlsbad, CA) using the XhoI and BamHI restriction sites to obtain flanking restriction enzyme sites so that it could be shuttled into pVL1392 for expression in the BD BaculoGold™ transfer vector system (BD Biosciences, San Jose, CA) using the XbaI and BamHI sites.
Table 1.
PCR primers for construction of fusion proteins
| Mouse IL-2 | Forward CATAGGTCGACATGTACAGCATGCAGCTCGCATCC |
| PSAcs reverse CATAGGGAATTCCTGCAGCTTGCTGCTGTGTTGAGGGCTTGTTGAGATGATGCT | |
| MMPcs reverse CGGGCGGAATTCAGCGGTGTAGTAAGCAGGGCTTTCACCGCTTTGAGGGCTTG TTGAGATGATGCT | |
| Mouse IL-2Rα | Forward GCGCGGGTACCGAACTGTGTCTGTATGACCCACCC |
| Reverse CGGCCGGATCCTCATTATGCTACCTTATACTCCATTGT | |
| Reverse 6 His CGGCCGGATCCTCATTAGTGGTGGTGGTGGTGGTGTGCTACCTTATACTCCATTGT | |
| Human IL-2 | Forward GATACGTCGACATGTACAGGATGCAACTCCTG |
| Reverse TCGGAGAATTCCTGCAGCTTGCTGCTGTGAGTCAGTGTTGAGATGATGCT | |
| Human scFv | External primers |
| Forward GCGCGGGTACCCAGTCTGTGCTGACTCAGCCA | |
| Reverse CCGGCGGATCCTGAGGAGACGGTGACCAGGGT | |
| Reverse 6 His CCGGCGGATCCGTGGTGGTGGTGGTGGTGTGAGGAGACCAGGGT | |
| Primers to insert stop codons and Not I site | |
| Forward GCGCCGCGGCCGCGTCGACATGTACAGGATGCAACTC | |
| Reverse GGCGCGGATCCTCATTATGAGGAGACGGTGACCAGGGTGCC | |
| Reverse 6 His CGCGCGGATCCTCATTAGTGGTGGTGGTGGTGGTGTGAGGAGACGGTGACCAGGGT | |
| Linker oligos | Forward GGCCGGAATTCGGTGGCGGTGGCTCTGGTGGCGGTGGCTCTGGTGGCGGTGGCTCT |
| ReverseGCGGGTACCAGAGCCACCGCCACCAGAGCCACCGCCACCAGAGCCACCGCCACCAGAGCC |
Construction of the mouse IL-2/MMPcs/IL-2Rα fusion protein
To change the cleavage sequence (cs) from HSSKLQ (PSAcs) to SGESPAYYTA (MMPcs) the pBluescript plasmid containing the mouse IL-2 and the PSAcs portion of the fusion protein was linearized using NotI and PCR was performed using the IL-2 forward primer and the MMPcs reverse primer (Table 1). This PCR product was then digested with SalI and EcoRI restriction endonucleases and cloned into pBluescript to create the pBluescript IL-2/MMPcs plasmid. The pVL1392 vector containing the mouse IL-2/PSAcs/(GGGGS)4/IL-2Rα + 6 × His fusion protein was digested with EcoRI and BamHI and the fragment containing the (GGGGS)4 linker and IL-2Rα was isolated and cloned into the pBluescript IL-2/MMPcs plasmid using the EcoRI and BamHI sites. The fragment encoding the entire fusion protein was then shuttled into pcDNA3.1 using the XhoI and BamHI sites and subsequently shuttled into pVL1392 using XbaI and BamHI for expression.
Use of human phage display library to identify and characterize IL-2 reactive scFv
A human phage display library constructed from peripheral blood lymphocytes was used to screen for phage expressing single-chain fragments of antibodies capable of binding to human IL-2 on their surface (phscFvs). The library was generated in the pAP-III6 vector,22,23 a monovalent display vector, by PCR amplification of VL and VH immunoglobulin domains from peripheral blood lymphocyte cDNA prepared from approximately 100 donors. The variable regions were PCR amplified with primers that encode a 14-amino-acid linker between the VL and VH domains, and then cloned into pAP-III6. The library consists of approximately 2 × 109 independent transformants and was screened using a modified ELISA as described previously22 using recombinant human IL-2 (Peprotech, Rocky Hill, NJ) adsorbed to plates as the target antigen. After several rounds of phage panning purification, a small panel of phage expressing scFv (phscFv) was tested for the ability to bind human IL-2 in the presence of a neutralizing anti-human IL-2 monoclonal antibody (eBioscience, San Diego, CA). A recombinant form of a Plasmodium falciparum protein (accession number XM_001347271) and the phscFv from SGPP (structural genomics of parasitic protozoa) that reacts with it,24 was used as a control to check for specificity of inhibition with the anti-human IL-2 neutralizing antibody. In brief, 0·5 μg/ml human IL-2 or SGPP in PBS was used to coat the ELISA plate, the wells were washed and 2 μg/ml anti-human IL-2 neutralizing antibody (MQ1-17H12; eBioscience), or blocking buffer was added. Supernatants containing individual phscFv clones were then added and phage binding was detected using an anti-M13 phage horseradish peroxidase (HRP) -conjugated antibody (GE Healthcare, Buckinghamshire, UK). The ELISA plate was developed by adding 50 μl o-phenylenediamine (Sigma-Aldrich, St Louis, MO) in 0·1 m citrate buffer pH 4·5 and 0·04% H2O2, stopped by adding 50 μl/well 2 m H2SO4 and the absorbance was read at 490 nm. The DNA from phscFv-2 was isolated and used as the starting material for the construction of the scFv human IL-2 fusion construct.
Construction of the human IL-2/PSAcs/human scFv fusion protein
The human IL-2 cDNA in pBR322 (ATCC, Manassas, VA) was PCR amplified using primers (Table 1) which added an N-terminal SalI site, the PSAcs (HSSKLQ) and a C-terminal EcoRI restriction site. This insert was then directionally cloned into pBluescript (Stratagene, La Jolla, CA) using the SalI and EcoRI restriction sites. The (GGGGS)x linker of various repeat lengths was cloned into pBluescript using the EcoRI and KpnI restriction sites. The human IL-2 scFv was PCR amplified (Table 1) from the M13 phage DNA from the phage clone scFv-2 and the 6 × His tag and the KpnI and BamHI restriction sites were added. This insert was then cloned into the pBluescript human IL-2/PSAcs/linker plasmid and shuttled into pcDNA 3.1 and subsequently cloned into the pVL1392 expression plasmid as described above.
Baculovirus production of fusion proteins
The generation of recombinant baculoviruses for the expression of proteins in insect cells has been described previously.25,26 Recombinant viruses were created using the pVL1392 transfer vector and the BD BaculoGold™ transfer vector system (BD Biosciences) as described by the manufacturer. Initial virus production was performed in Spodoptera frugiperda (Sf-9) cells cultured in Sf-900 II SFM media (Gibco®; Invitrogen) and after several passages a high-titre stock was obtained. For final production of fusion proteins, Trichoplusia ni cells (Invitrogen) cultured in Express Five® SFM media (Gibco®; Invitrogen) plus 2 mm l-Glutamine High Five™ were propagated in 300-ml shaking cultures in 1-l flasks (125 rpm, 27°) and were infected with the high-titre stock and incubated with shaking for 72 hr at 27°. The supernatant was used directly after clarification in some experiments, or in some cases, the fusion proteins were purified via the 6 × Histidine tag using Nickel-NTA agarose beads (Qiagen, Valencia, CA) and Poly-Prep® Chromatography columns (BioRad, Hercules, CA) using the manufacturer's recommendations.
Detection of mouse IL-2 and IL-2Rα in fusion proteins by ELISA
Interleukin-2 or the IL-2Rα chain was detected using either the anti-IL-2 monoclonal antibody (JES6-1A12; BD Pharmingen) or the anti-mouse IL-2Rα monoclonal antibody (PC61; BD Pharmingen), respectively. Wells of a 96-well plate were coated with either antibody (2·5 μg/ml) in PBS. Wells were blocked with 5% non-fat milk in PBS with 0·2% Tween (PBS-M-Tw) and fusion proteins were added for 1–2 hr at 37°. After washing, an anti-mouse IL-2 biotin-labelled antibody (JES5H4; BD Pharmingen) was added and binding was detected using Strepavidin HRP (Southern Biotechnology Associates, Birmingham, AL). The ELISA plate was developed by adding 50 μl o-phenylenediamine (Sigma-Aldrich) in 0·1 m citrate buffer pH 4·5 and 0·04% H2O2, stopped by adding 50 μl/well 2 M H2SO4 and the absorbance was read at 490 nm.
Mouse IL-2, IL-2Rα, 6 × Histidine and MMP Immunoblot analyses
Immunoblot analyses were performed as reported previously with minor modifications.27 The following monoclonal antibodies were used: rat anti-mouse IL-2 antibody (JES6-1A12; BD Pharmingen), rat anti-mouse IL-2Rα (PC61; BD Pharmingen), and mouse anti-6 × His monoclonal antibody (MM5-156P; Covance, Princeton, NJ). Detection was performed using a goat anti-rat HRP-conjugated antibody (Jackson Immuno Research, West Grove, PA) and developed using the Amersham ECL Plus Western blotting detection reagent (GE Healthcare) following the manufacturer's recommendations. A determination of fusion protein concentration was established using immunoblot analyses and quantitative densitometry and compared with recombinant IL-2. For MMP immunoblot analyses, extracts or supernatants were probed with goat anti-mouse MMP2 or MMP9 antibodies (R&D Systems, Minneapolis, MN).
In vitro digestion conditions for fusion proteins
Fusion proteins were digested with PSA (Cortex Biochem, San Leandro, CA) or prostate extracts in 50 mm Tris–HCl, 100 mm NaCl pH 7·8 at 37°. For digestion of the fusion protein containing the MMP cleavage sequence, MMP9 or MMP2 (R&D Systems) was activated with p-aminophenylmercuric acetate and this activated protease or equivalent amount of activating solution without the protease was used to digest the fusion protein for 1 hr at 37° for MMP9 and 10 min for MMP2. Aliquots of digests were loaded on 15% Laemmli gels for Western blotting. The rat anti-mouse IL-2 primary antibody (JES6-1A12; BD Pharmingen) and goat anti-rat HRP-conjugated secondary antibody (Jackson Immuno Research) were used and blots were developed as described above. Aliquots of digests were also used in the IL-2 functional assay described below.
IL-2 functional assay
Functional IL-2 was measured using CTLL-2 cells (ATCC) as described elsewhere28 with minor modifications. In brief, digested samples were serially diluted 1 : 2, then 50 μl of test supernatant was added to 3·5 × 104 to 5·0 × 104 CTLL-2 cells per well in 100 μl medium in a 96-well plate and incubated at 37° in 5% CO2 for 18–22 hr. At the end of this period, 75 μg/well Thiazolyl Blue Tetrazolium Bromide (MTT) (Sigma-Aldrich) was added and the plate was incubated for 8 hr at 37° in 5% CO2. Cells were lysed with 100 μl/well 10% SDS (Gibco®; Invitrogen) acidified with HCl, incubated at 37° in 5% CO2 overnight, and absorbance 570 nm was read.29 Recombinant human IL-2 standard (Peprotech) was serially diluted with 0·5 ng delivered to CTLL-2 cells in the first well.
Mice
All animal experiments were performed in accordance with guidelines established by the National Institutes of Health and the University Committee on Animal Resources at the University of Rochester. C57BL/6J mice were purchased from The Jackson Laboratory (Bar Harbor, ME). Human PSA transgenic mice were backcrossed onto the C57BL/6J background and were used as a source of PSA-expressing prostate tissue.30
Prostate explant cultures and preparation of prostate extracts
Ventral prostates from wild-type C57BL/6J (Jackson) (non-transgenic; NTG) and PSA transgenic C57BL/6J (TG) mice were surgically removed and placed in 600 μl Dulbecco's modified Eagle's medium (Gibco®; Invitrogen) supplemented with 0·005 mg/ml bovine insulin (Sigma-Aldrich), 10 nmtrans-dehydroandrosterone (Sigma-Aldrich), 5% fetal calf serum (Hyclone, Logan, UT), 5% Nu-serum IV (BD Biosciences), and 0·05% penicillin/streptomycin (Sigma-Aldrich). Fusion protein was added to explant culture and incubated at 37° in 5% CO2 and 100 μl aliquots were removed at 1, 12, 24 and 48 hr and stored at −20° until use. Prostate extracts were made using ventral prostates homogenized in a Dounce homogenizer in 100 μl of 50 mm Tris–HCl, 100 mm NaCl pH 7·8. Extracts were centrifuged to remove debris and the supernatants were stored at −20°. Total protein concentration was determined using the Bio Rad Protein Assay (Bio Rad) according to the manufacturer's recommendation and equal amounts of protein extracts were used for fusion protein digestions described earlier. The PSA levels in culture supernatants or in the prostate extracts were measured using a capture ELISA as described previously31 with minor modifications.
Detection of human IL-2 by Immunoblot analyses
Human IL-2 was detected by standard Western blot technique using a rabbit anti-human IL-2 antibody (Leinco, St Louis, MO; 1·0 μg/ml) in TBS-M-Tw followed by a goat anti-rabbit HRP-conjugated antibody (Leinco; 0·2 μg/ml) in TBS-M-Tw. The blot was developed using the Amersham ECL Plus Western blotting detection system (GE Healthcare) according to the manufacturer's recommendations.
Tumour growth experiments
For the tumour growth experiments, 5 × 105 Colon 38 cells were injected intraperitoneally into syngeneic mice and allowed to attach for 24 hr. Groups of mice were treated daily for 6 days with fusion protein, treated with vehicle, or untreated as indicated in the legend of Fig. 6. On day 7, the animals were killed; the omenta were removed and treated with collagenase, then stained for flow cytometry as described previously with minor modifications.32 Preliminary experiments were performed using normal omental cells, tumour cells and a reconstructed mixture of tumour cells and omental cells to establish the gates shown. Colony-forming assays were performed as described previously.33 Statistical analyses testing for significance were performed as indicated in the figure legends.
Figure 6.
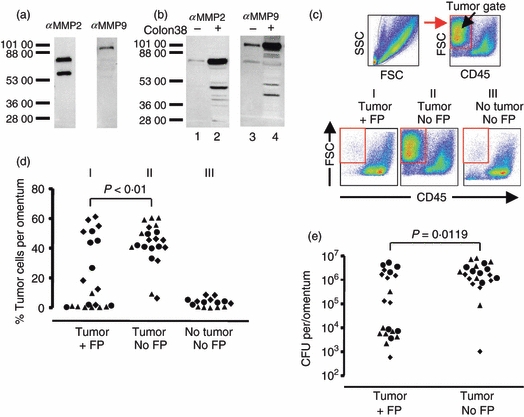
Fusion protein treatment reduces Colon 38 tumour growth in vivo. (a) In vitro expression of matrix metalloproteinase 2 (MMP2) and MMP9 using the Colon 38 tumour cell line. Immunoblot analyses of in vitro grown Colon 38 supernatants using the indicated antibodies. (b) In vivo expression of MMP2 and MMP9 from omental lysates with and without Colon 38 tumour present. Lanes 1 and 2 were probed with the anti-MMP2 antibody. Lane 1 contains omental lysate from an untreated mouse with no tumour. Lane 2 contains lysate from an omentum that had Colon 38 grown in vivo. Lanes 3 and 4 are replicates of lanes 1 and 2 except they were probed with an anti-MMP9 antibody. Dashes and numbers indicate apparent molecular weights. Note lower molecular weight bands probably represent cleavage products. (c) Gating scheme and representative flow analyses used to identify tumour cells (high forward scatter, CD45 negative) growing in vivo on the omentum. Reconstitution experiments in which tumour cells were added to omental cells were used to establish the tumour gate, which is indicated by the box. Bottom panels are examples of flow analyses of omenta from mice which received tumour and MMP fusion protein treatment (panel I: tumour +FP), mice which had received tumour but no treatment with MMP fusion protein (panel II: tumour No FP), or mice which received neither tumour nor fusion protein (panel III: no tumour, No FP). (d) Compiled analyses of tumour cells detected on the omentum by flow cytometry. Each symbol represents an individual mouse. Different symbols indicate mice from three experiments. The P-value between the indicated groups was calculated using the Kruskal–Wallis test. (e) Viable tumour cells were determined by colony-forming assay. Symbols indicate individual mice. P-value was calculated using the Mann–Whitney U-test.
Results
Construction and characterization of IL-2/PSAcs/IL-2Rα fusion proteins
We set out to create a cytokine fusion protein that could be cleaved by a tumour cell expressed protease so that it becomes more active after cleavage. We initially tested a strategy based on steric hindrance by constructing a fusion protein consisting of IL-2 and Mip1-a separated by a PSA cleavage sequence.34 We hypothesized that both immunomodulatory proteins would be largely inactive in the fusion protein because of their close proximity, but would become more active if the fusion protein could be successfully cleaved, thereby separating the two proteins. Although the fusion protein could be expressed and cleaved by PSA, IL-2 did not appear to be attenuated in the intact fusion protein and the biological activity of the IL-2 did not increase after cleavage (data not shown). Hence, simply joining two molecules, even very closely, does not necessarily interfere with their functional activity. However, we reasoned that if we constructed a molecule in which the putative inhibitory portion of the fusion protein bound the cytokine specifically, this would be more likely to inhibit its activity. As we describe below, we used two distinct strategies to inhibit the biological activity of IL-2. The first strategy employed a cytokine receptor whereas the second used an antibody fragment (scFv). The first strategy using specific inhibition employed IL-2 and a portion of the IL-2 receptor is illustrated schematically in Fig. 1(a). We used mouse IL-2 cDNA and took advantage of the alpha chain of the IL-2 receptor (IL-2Rα) which can bind IL-2 in the absence of the other subunits (β and γ) of the high-affinity IL-2 receptor.35 In this construct, we eliminated the transmembrane and cytoplasmic region of the IL-2Rα chain, creating a soluble form of the receptor. To increase flexibility and allow the IL-2Rα portion of the molecule to fold back and inhibit IL-2, we also introduced a repeating Gly–Ser linker consisting of (GGGGS)2 (designated 2 ×), or (GGGGS)436 (designated 4 ×), and in some cases also added a 6 × His tag. These plasmids were used to construct recombinant baculoviruses to mediate expression in insect cells as described in the Materials and methods. As shown in Fig. 1(b), we examined the fusion proteins with a capture ELISA using antibodies reactive with IL-2Rα and IL-2. Also, the immunoblot analysis in Fig. 1(c) showed that the fusion protein is at the predicted apparent molecular weight (MW) of approximately 50 000 and is reactive with anti-IL-2, IL-2Rα and 6 × His antibodies. These data show that the fusion proteins are produced, secreted and contain both IL-2 and IL-2Rα on the same molecule.
Figure 1.
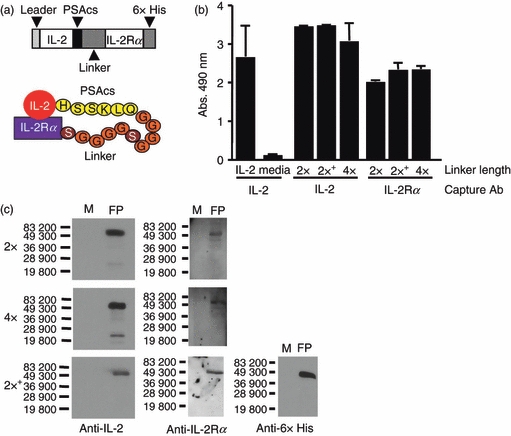
Characterization of mouse IL-2/PSAcs/IL-2Rα fusion proteins. (a) Schematic diagram of mouse IL-2/PSAcs/linker/IL-2Rα fusion proteins. The prostate-specific antigen cleavage sequence (PSAcs) indicates the amino acid sequence HSSKLQ that can be cleaved by the PSA protease followed by the 2 × (GGGGS)2 (shown) or 4 × (GGGGS)4 linker lengths. (b) ELISA analysis of fusion proteins using capture antibodies to interleukin-2 (IL-2; JES6-1A12) or IL-2 receptor alpha chain (IL-2Rα; PC61) and a different biotin-labelled anti-IL-2-detecting antibody (JES5H4) showing that both IL-2 and the IL-2Rα moieties are present on the same molecule. 2 × indicates (GGGGS)2 linker, 4 × indicates (GGGGS)4 linker, and (+) indicates that the construct contains the 6 × His tag. (c) Immunoblot analyses of the fusion proteins using antibodies reactive with IL-2 (JES6-1A12), IL-2Rα (PC61) and 6 × His epitope tag (MM5-156P). Molecular weight standards are indicated. The full length fusion proteins (FP) have an approximate molecular weight of 50 000 MW and medium was run as a negative control (M).
PSA cleavage of the IL-2/PSAcs/IL-2Rα fusion proteins results in increased accessibility to antibodies and biologically active IL-2
We characterized the IL-2/PSAcs/IL-2Rα fusion proteins biochemically before and after cleavage with the protease PSA. Immunoblot analyses revealed that the fusion proteins could be cleaved by PSA and that there was an increase in intensity of the predicted low-molecular-weight cleavage product of approximately 20 000 MW reactive with an anti-IL-2 antibody (Fig. 2a). The degree of cleavage was dependent upon the amount of PSA as well as the time of incubation (Fig. 2b,c). Interestingly, when we analysed the fusion protein before and after PSA treatment by ELISA, we found that the apparent amount of IL-2 was increased after PSA cleavage (Fig. 2d). In this experiment, there was an approximately twofold or fourfold increase in the amount of IL-2 detected using this sandwich ELISA depending on the construct, suggesting that the detection antibody binding was partially hindered in the intact fusion protein. We also analysed aliquots of the same samples shown in Fig. 2(a) after PSA treatment for functional IL-2 using the CTLL-2 cell line. As seen in Fig. 2(e,f) there is an increase in the amount of biologically active IL-2 after PSA cleavage. After protease treatment, the apparent amount of biologically available IL-2 increased approximately 3·5-fold for the fusion protein with the 2 × linker and ninefold for the fusion protein with the 4 × linker. Hence, the above data show that after PSA cleavage there is an increase in the predicted low-molecular-weight cleavage fragment of approximately 20 000 MW that is reactive with an anti-IL-2 antibody, an increase in antibody accessibility, and most importantly, an increase in the amount of biologically active IL-2. Because the 4 × linker fusion protein had a larger fold increase in biologically active IL-2, this fusion protein was used in subsequent experiments.
Figure 2.
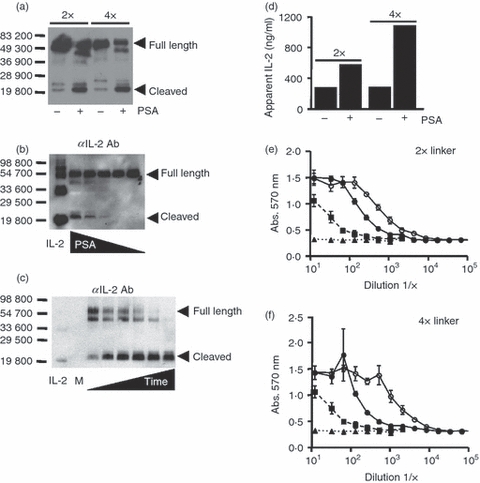
Prostate-specific antigen (PSA) treatment enhances antibody accessibility and interleukin-2 (IL-2) functional activity in mouse IL-2/PSAcs/IL-2Rα fusion proteins. Fusion proteins (2 × and 4 × linker lengths) were treated with PSA or buffer controls for 1 hr at 37° and aliquots were analysed by immunoblot, ELISA, or by functional analysis. (a) Anti-IL-2 immunoblot analysis of fusion proteins before and after treatment with PSA. Bars indicate molecular weight markers, (+) indicates treatment with PSA, (−) indicates treatment with control buffer, and full length and predicted cleavage product containing IL-2 have been denoted (arrowheads). (b) Titration of PSA using mouse IL-2/PSAcs/2 × linker/IL-2Rα fusion protein at 37° for 1 hr and immunoblot analysis using an anti-mouse IL-2 antibody (JES6-1A12). Bars and numbers indicate molecular weight markers. Full length and cleaved fusion proteins have been denoted. The amount of PSA used in the reaction is as follows: 11·25, 5·6, 2·8, 1·4 and 0 μg. (c) PSA time–course using mouse IL-2/PSAcs/4 × linker/IL-2Rα fusion protein digested with 5 μg for 0, 1, 3, 6, 12 and 24 hr at 37° analysed by immunoblot with an anti-mouse IL-2 antibody (JES6-1A12). Medium was run as negative control (M). (d) An ELISA used to measure the amount of IL-2 before and after treatment with PSA. An apparent increase in IL-2 can be seen after PSA incubation for both fusion proteins tested. (e, f) Functional analyses of IL-2 before and after cleavage. Biologically active IL-2 from the fusion proteins was measured using the CTLL-2 functional assay. Fusion protein treated with PSA (○), or with buffer control (•), IL-2 standard (▪), and medium control (▴). The same amount of fusion protein was present in the PSA-treated and untreated samples used in the CTLL-2 assay. Points represent the average of three replicates and error bars indicate standard deviation. Representative of three independent experiments.
Prostate explants or extracts expressing human PSA can cleave the IL-2/PSAcs/4 × linker/IL-2Rα fusion protein and increase the biological activity of IL-2
To examine the cleavage of the fusion protein in the context of prostate tissue that expresses a complex mixture of proteases, we took advantage of TG mice that express human PSA30 in prostate explants. Because conventional mice do not express PSA or any close homologue of human PSA, NTG mouse prostates served as a control for the expression of a variety of other proteases produced in the prostates that might cleave the fusion protein. The prostates were removed from TG mice and their NTG counterparts and placed into culture medium containing the IL-2/PSAcs/IL-2Rα fusion protein. At various times, samples were removed and analysed biochemically for cleavage and functionally for IL-2 activity. Supernatants from the explant cultures were analysed for PSA expression using a PSA-specific ELISA, which shows that the media from explants from TG mice contained PSA whereas those from NTG mice did not (Fig. 3a). These same explant culture supernatants were also analysed by immunoblot using the anti-IL-2 monoclonal antibody JES6-1A12 and by functional analysis using the IL-2-dependent cell line CTLL-2. In Fig. 3(b), a lower apparent molecular weight band of approximately 20 000 MW reactive with anti-IL-2 (cleaved) increased with time of culture in the TG explant cultures, but not in the NTG cultures. These data suggest that other proteases that might be expressed by prostate cells did not cleave the IL-2/PSAcs/IL-2Rα fusion protein effectively but that human PSA derived from the prostate cells in the TG mouse could cleave the fusion protein. These same supernatants were also analysed for functional IL-2 activity (Fig. 3c). The amount of biologically active IL-2 was approximately eightfold higher in the TG explant cultures compared with the NTG cultures. This experiment has been repeated three times with the degree of enhancement of IL-2 activity ranging from fivefold to tenfold. As an additional, and perhaps more stringent, test of specific cleavage of the fusion protein by PSA but not by other proteases found in the prostate, we made extracts of prostates from PSA TG and NTG mice, and examined the ability of these extracts to cleave the fusion protein in the absence of any protease inhibitors that might be found in fetal calf serum. As shown in Fig. 3(d), the TG prostate extracts contain large amounts of PSA in comparison to the NTG extracts. As can be seen in the immunoblot analysis in Fig. 3(e), the extracts from the PSA TG mice effectively cleaved the fusion protein, whereas the NTG extracts did not. Importantly, there was an increase in the functional activity of the IL-2 assessed by the CTLL-2 assay after incubation with the PSA-containing TG extracts compared with the NTG extracts (Fig. 3f).
Figure 3.
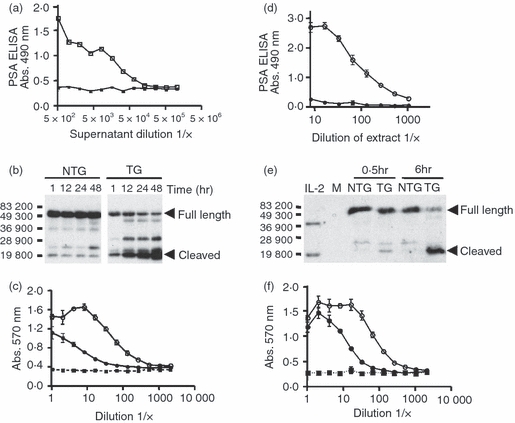
Interleukin-2 (IL-2) bioactivity increases when IL-2/PSAcs/4 × linker/IL-2Rα fusion protein is cultured with explanted prostates or homogenized prostate extracts. Prostates were removed from non-transgenic (NTG) or prostate-specific antigen (PSA) transgenic (TG) C57BL/6J mice, cultured at 37° with fusion protein and aliquots of media containing fusion protein were removed at 1, 12, 24, 48 hr for analysis. (a) Detection of PSA in TG (□) or NTG (▪) supernatant aliquots at 48 hr by ELISA. (b) Immunoblot analysis using an anti-IL-2 antibody of the culture supernatants at the indicated time-points. Bars and numbers indicate molecular weight markers. The full length and the predicted cleavage products containing IL-2 are indicated by arrowheads. (c) IL-2 functional assay at 48 hr. Supernatant from cultures containing TG prostate explants (○), NTG explants (•), media control (▪). The same amount of culture supernatant was used for the CTLL-2 assay. (d) Analysis of PSA by ELISA in homogenized prostate extracts from TG (○) and NTG (•) mice. The first well represents approximately 40 ng of NTG or TG extract. (b) Immunoblot analysis using an anti-IL-2 antibody of the samples containing prostate extracts and the fusion protein at 0·5 or 6 hr at 37°. Bacterially derived non-glycosylated recombinant mouse IL-2 (10 ng) was used as a standard and has an approximate molecular weight of 18 500 MW. The band at approximately 37 000 MW in the control lane probably represents a dimer of IL-2. Medium-only control (M). Full-length fusion protein and the predicted cleavage product containing IL-2 have been denoted by arrowheads. (f) IL-2 functional assay of fusion proteins after incubation for 6 hr at 37° with prostate extracts. TG prostate extracts (○), NTG extracts (•) and media control (▪). Equivalent amounts of fusion protein were present in the PSA-treated and untreated samples used in the CTLL-2 assay.
Construction and analysis of human IL-2/PSAcs/human scFv fusion proteins
The previous approach used a receptor as the inhibitory component in the fusion protein. We also investigated the ability of a single-chain Fv antibody fragment (scFv) to bind and inhibit IL-2. This strategy examines the importance of specific binding in the protease-activated cytokine approach by using a totally different binding component. The use of an scFv also has some potential theoretical advantages as we delineate in the discussion. The scFv constructs we developed are outlined schematically in Fig. 4(a). Here we were able to take advantage of an scFv phage display library previously constructed using human VH and VL gene segments.22,23 As this phage display library expressed human scFv, we used it to identify phages (phscFv) that bound human IL-2 (M. Sullivan, unpublished data) so that the components of the fusion protein constructed would all be derived from one species. From the small panel of phscFv that bound human IL-2 in a modified ELISA, we chose a phage (scFv-2) whose binding to IL-2 could be inhibited by a neutralizing anti-IL-2 antibody (Fig. 4b); the rationale for this choice was that the scFv-2 might also recognize a neutralizing epitope. The anti-IL-2 antibody blocked the binding of the scFv-2 phage by approximately 70%. As a control, we used a non-IL-2-reactive scFv-expressing phage. We found that this same anti-IL-2 neutralizing monoclonal antibody did not block the binding of this non-IL-2-reactive phscFv to its cognate antigen (designated SGPP), thereby illustrating that the antibody blocking we observed was indeed specific for human IL-2 (Fig. 4b). The antibody variable regions of scFv-2 were sub-cloned and used to create the fusion proteins outlined in Fig. 4(a), which were then expressed in insect cells via recombinant baculoviruses as described in the Materials and methods. Analogous to the IL-2Rα chain constructs, we made the scFv-2 fusion proteins with 2 × and 4 × linker lengths. As preliminary experiments suggested the fusion protein with the 2 × and 4 × linker length were similar in terms of their expression and their ability to be cleaved (data not shown), for subsequent experiments we focused on the fusion protein containing the scFv-2 with the 2 × linker length. As can be seen in Fig. 4c using the human IL-2/PSAcs/human scFv-2 with the 2 × linker fusion protein, a lower-molecular-weight fragment of approximately 20 000 MW reactive with an anti-IL-2 antibody resulted after cleavage with purified PSA. We also used the IL-2-dependent cell line CTLL-2 and the MTT assay to assess the biological effect of PSA cleavage on the same samples. Samples were incubated with or without purified PSA and assessed for functional activity. The cleavage of the scFv-2 fusion protein with PSA resulted in an increase in biologically active IL-2 (Fig. 4d).
Figure 4.
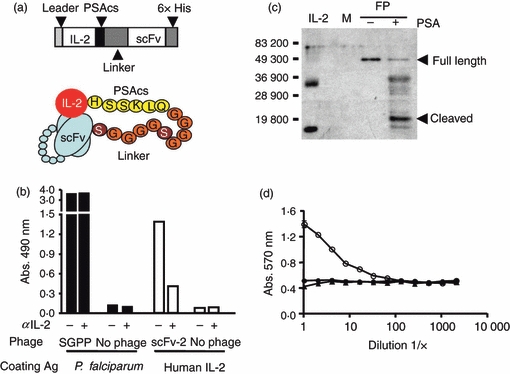
Characterization and analyses of human IL-2/PSAcs/scFv fusion proteins. (a) Schematic diagram of human IL-2/PSAcs/scFv fusion proteins containing human interleukin-2 (IL-2) fused to the prostate-specific antigen cleavage sequence (PSAcs), a (GGGGS)2 or (GGGGS)4 linker unit followed by VL and VH fragments of an antibody tethered together by a linker (scFv) and a 6 × His carboxyl tag. (b) A modified ELISA using scFv phage was performed and a phage clone expressing scFv (phscFv) that binds human IL-2 (scFv-2) was screened for the ability to be inhibited by the anti-human IL-2 neutralizing antibody (MQ1-17H12). Black columns indicate recombinant Plasmodium falciparum protein (SGPP) coating antigen, white columns indicate human IL-2 as the coating antigen. A phage which bound SGPP served as a control and this binding was not inhibited by the anti-human IL-2 neutralizing antibody whereas the phage clone scFv-2 could be partially blocked by the antibody. (c) Anti-human IL-2 immunoblot analysis of fusion protein treated with purified PSA or with control PSA buffer treatment. Bars and numbers indicate molecular weight markers. Full-length fusion protein (FP) and the predicted cleavage product containing IL-2 have been denoted with arrowheads. Media negative control (M). Bacterially derived non-glycosylated recombinant human IL-2 (50 ng) was used as a standard and has an approximate molecular weight of 15 500. The fusion protein IL-2 was derived from insect cells and may be post-translationally modified accounting for its slightly higher molecular weight. (d) IL-2 functional assay on fusion protein. Treatment with PSA (○), control buffer (•) or media control (▴). Equal amounts of fusion protein were used in the CTLL-2 assay.
Alteration of the protease cleavage site: use of an MMP cleavage sequence
To extend the potential utility of the fusion protein approach, we have also investigated whether the concept of activating cytokines by proteases might be applied to other proteases. For this purpose we have substituted an MMP cleavage site that can be cleaved by MMP2 and MMP9 (37 and our unpublished data) in place of the PSA cleavage site used in the IL-2/PSAcs/IL-2Rα fusion protein. This construct encoding the MMP cleavage sequence was expressed using the baculovirus system in insect cells and the resulting fusion protein was tested for its ability to be cleaved using MMP9 and MMP2 and analysed by immunoblot analyses. As can be seen in Fig. 5(a,c), the fusion protein can be cleaved by MMP2 or by MMP9. After incubation with the proteases, a product with low apparent molecular weight of approximately 20 000 MW reactive with an anti-IL-2 antibody resulted, consistent with the release of IL-2 from the fusion protein. Figure 5(b,d) compares the functional activity of the fusion protein before and after cleavage with MMP2 or MMP9 and illustrates that the functional level of IL-2 assessed by CTLL-2 is increased after cleavage. Taken together, these data support the concept that the specific inhibitory moiety can markedly inhibit the functional activity of the cytokine in the intact fusion protein, but the cytokine activity increases upon protease cleavage.
Figure 5.
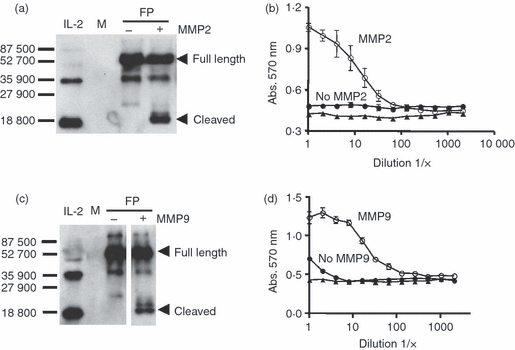
Evaluation of mouse IL-2/MMPcs/4 × linker/IL-2Rα + 6 × His fusion proteins digested with matrix metalloproteinase 2 (MMP2) or MMP9. The fusion protein containing the MMP cleavage sequence was incubated with either MMP9 or MMP2 or buffer treated and the resulting material was tested by Western blotting analysis and for activity using the CTLL-2 assay. (a) Immunoblot analyses of the fusion protein digested with MMP2 using an anti-interleukin-2 (IL-2) antibody. Bars and numbers indicate molecular weight markers. The full length and the predicted cleavage product containing IL-2 are indicated by arrowheads. Media control (M). (b) Fusion protein digests were run in the CTLL-2 assay using equal amounts of fusion protein for the MMP2 treated or untreated fusion protein. Fusion protein plus MMP2 (○), fusion protein no treatment (•), media control (▴). (c) Immunoblot analyses of the fusion protein digested with MMP9 using anti-IL-2 antibody. Bars and numbers indicate molecular weight markers. The full length and the predicted cleavage product containing IL-2 are indicated by arrowheads. Note that the + and – MMP9 digests were run in the same gel and exposed for the same amount of time but some intervening lanes were removed. (d) Fusion protein digests were run in the CTLL-2 assay using equal amounts of fusion protein for the MMP9 treated or untreated fusion protein. Fusion protein plus MMP9 (○), fusion protein no teatment (•), media control (▴). Note: in some cases error bars are not visible in CTLL-2 assay because of the size of symbols.
In vivo delivery of a protease activated fusion protein results in decreased tumour growth
We next examined whether a fusion protein could have biological effects in vivo. For these experiments, we used a system developed previously, in which tumour cells injected intraperitoneally rapidly and preferentially attach and grow initially on the milky spots, a series of organized immune aggregates found on the omentum.38 This system offers a convenient way to examine the effects of fusion protein treatment on tumour growth because fusion protein can be delivered intraperitoneally multiple times and tumour growth can be analysed by examining the dissociated omental cells. For these experiments we used the Colon 38 cell line, a rapidly growing tumour cell line that expresses both MMP2 and MMP9 in vitro (Fig. 6a). The omental tissue normally expresses a relatively small amount of MMP2 and MMP9 but when Colon 38 tumour is present on the omentum, MMP levels increase (Fig. 6b). Using this tumour model, we examined the ability of the IL-2/MMPcs/IL-2Rα fusion protein to affect tumour growth. Colon 38 cells were injected intraperitoneally, allowed to attach and grow for 1 day, and then treated daily with fusion protein intraperitoneally. At day 7 the animals were killed and the omenta were examined for tumour growth using flow cytometry and by a colony-forming assay (Fig. 6c–e). Figure 6(c) illustrates the gating scheme employed to analyse the tumour population present on the omentum by flow cytometry and panels I, II and III represent plots of single mice from each of the three test groups studied. Figure 6(d) illustrates the compiled flow cytometry data obtained from the individual mice.
We found that treatment with the fusion protein can reduce tumour growth in vivo. In the mice that received tumour and fusion protein treatment (group I), there was a significant decrease (P < 0·01) in the percentage of tumour cells detected on the omenta compared with the mice, which were inoculated with tumour but not treated with fusion protein (group II Fig. 6d). As expected, there was a substantial fraction of cells in the tumour gate in mice that received tumour but were not treated with fusion protein (Fig. 6c panel II) and a very low fraction of cells in the tumour gate of mice that did not receive tumour (Fig. 6c panel III). Similar results were obtained when the presence of tumour cells was assessed using a colony-forming assay33 in which cells isolated from the omentum were tested for their ability to form colonies in vitro. These compiled data are shown in Fig. 6(e). Again, a significant difference was observed (P = 0·0119) between the fusion-protein-treated mice and the vehicle-treated mice in the number of viable tumour cells present on the omenta. Hence, in both the flow cytometry and the colony-forming assays there was a clear decrease in the tumour burden with fusion protein treatment although it should be noted that the decrease was not evident in all the treated animals. Taken together these data illustrate that the fusion protein can affect tumour growth in vivo.
Discussion
The current work illustrates the feasibility of using proteases to activate cytokines in the context of novel fusion proteins. We demonstrated the protease-activated cytokine approach with mouse and human IL-2 and two specific binding components, the IL-2Rα and an inhibitory scFv. The specific binding component appears important in this strategy as both of the fusion proteins with the specific binding moieties (IL-2 Rα or the scFv) showed enhancement of IL-2 activity comparing the cleaved with the uncleaved fusion proteins (Figs 2 and 4). In contrast, an approach that relied solely on steric hindrance using IL-2 and Mip1α resulted in a slight decrease in IL-2 activity after protease cleavage, supporting the importance of specific binding (data not shown). Moreover, we could also show that the biological activity of IL-2 is attenuated > 50-fold in the intact fusion protein (IL-2/MMPcs/IL-2Rα fusion protein) when comparing the cleaved and uncleaved fusion proteins. We further show that the protease-activated cytokine can function with different protease cleavage sites in a cassette fashion. We successfully used cleavage sites tailored for different proteases, including PSA, MMP9 and MMP2, in the context of an IL-2/IL-2Rα fusion protein. These proteases are relevant to tumour immunotherapy as the MMP family of proteases plays an important role in the development of a variety of tumours19,39,40 and because tumour cells, as well as host cells such as activated macrophages, can contribute to over-expression of MMPs at the tumour site.41–43 The prostate-expressed protease PSA is also potentially useful for the protease-activated cytokine approach. It is produced almost exclusively by prostate epithelial cells, and the cancers that arise from them. Whereas PSA can be found in serum, its expression is typically low even in cancer patients (ng/ml range) and it can complex with serum protease inhibitors.44 The prostate is typically removed or ablated as part of the treatment for prostate cancer,45 but metatstatic prostate cancer cells often continue to express PSA and so could be targets for a PSA-activated fusion protein.
Our finding that cleavage of the fusion protein results in increased biological activity might initially be surprising because the IL-2 could remain bound to the alpha chain or the scFv after cleavage. Moreover, even if dissociated, the inhibitory component could potentially rebind free IL-2. Indeed, others have speculated that IL-2 receptor alpha chain shed by cells such as activated T cells may have a regulatory role in dampening the immune response.46 However, there is probably competition for the free IL-2 derived from the fusion protein by cellular IL-2 receptors. In this light, it is useful to consider that the alpha chain used in the fusion protein has a Kd of approximately 10 nm.47 In contrast, the βγ IL-2 receptor on resting NK cells and memory phenotype T cells has a Kd of approximately 1 nm whereas activated T cells express the αβγ receptor, which has a Kd of approximately 10 pm (approximately 1000-fold lower than the isolated alpha chain). These differences should favour the binding of the IL-2 to cellular receptors. Consistent with this idea, we found that the CTLL-2 cell line, an IL-2-dependent T-cell line which expresses high levels of the alpha chain characteristic of the high-affinity receptor (αβγ) on activated T cells, can compete for the IL-2 released after cleavage of the fusion protein as seen in Figs 2–5.
Given the attenuated bioactivity of the intact fusion protein in vitro, an important issue is whether the fusion proteins would have any biological activity in vivo. We examined the activity of a fusion protein on tumour growth on the omentum,32,38 a common site of intraperitoneal tumour growth and metastases. This model system has a number of features that make it attractive for the initial testing of the protease-activated cytokine strategy. The peritoneal cavity, particularly in the context of growing tumours, contains a number of immunosuppressive cells and factors often found at other tumour sites. However, there are also a variety of leucocytes in the peritoneal fluid as well as a number of immune aggregates or milky spots on the omentum, which function in many respects like lymph nodes. The milky spots are particularly intriguing because they contain organized collagen structures that appear to aid in tumour cell attachment and they are also highly vascular and pro-angiogenic, which promotes tumour cell growth.32,38 However, they also contain many immune effectors including macrophages, B cells, T cells and NK cells that in principle could be activated in an anti-tumour response (48 reviewed in ref. 32). Despite these immune cells, tumours typically grow rapidly on the milky spots.38,49,50 Tumours growing on the omentum express high levels of MMPs as a result of their intrinsic production as well as contributions by host cells including macrophages. Hence, this experimental model of tumour metastases has a number of technical and conceptual features that make it amenable for testing the protease-activated cytokine strategy. We showed that the fusion protein significantly reduced tumour growth on the omentum (Fig. 6) illustrating that it can have biological activity in vivo. Future studies are needed to determine the immune cells involved in the anti-tumour response as well as a variety of pharmacokinetic parameters including the maximum tolerated dose, optimal dosing regimen and potential immunogenicity. However, because the fusion protein is composed of IL-2, it is likely that it will function in many, although perhaps not all, respects like free IL-2, and activate NK and T cells. It remains to be determined how the fusion protein compares with free IL-2 in terms of efficacy. However, it is likely that a major improvement would be its lower off-site toxicity given its attenuation. Interestingly, we were able to show that a fusion protein can decrease the tumour burden in some, although not all mice. These data are consistent with previous studies in clinical treatment of tumours found in the peritoneum showing the benefit of the IL-2 but also heterogeneity in the effects of treatment.51 The reason for this heterogeneity is not known, although it might reflect differences in the relative balance of effector cells and regulatory T cells.52
There is a great deal of interest in manipulating the immune response at specific sites exploiting the biological activity of cytokines. One innovative approach takes advantage of monoclonal antibodies to tumour-associated antigens (e.g. anti-HER-2/neu or anti-ganglioside GD2) that may have anti-tumour activities themselves, and genetically fuses them to cytokines (e.g. IL-2 or IL-12), which are then expressed and infused in vivo.53–55 Although the antibody fused to the cytokine diffuses throughout the recipient, it eventually accumulates at the tumour site as a result of antibody binding and retention so the cytokine concentration increases at tumour sites. This approach differs fundamentally from the one presented in the current work. In the current study the antibody does not bind the tumour but rather serves to inhibit the cytokine. The cytokine in the anti-tumour-associated antigen–antibody fusion is constitutively active and so may have unwanted effects. In contrast, in the approach demonstrated here, the cytokine activity is attenuated because of the specific binding component and increases only when activated by proteases. Another interesting strategy employs the latency-associated protein (LAP) of transforming growth factor-β (TGF-β) that is genetically fused to interferon-β (IFN-β) via a cleavable linker recognized by an MMP such that the IFN-β becomes more active when the linker is cleaved. In this method, unlike the specific inhibition presented here, the LAP protein sterically shields the IFN-β from its receptor. This approach has been used to down-regulate inflammatory responses in a mouse model of arthritis.56
A variety of cytokines have been tested for their ability to act as adjuvants in the context of anti-tumour responses. Interestingly, while some studies found that immunization with irradiated or mitomycin-treated transfected tumour cells expressing IL-2 can aid in initiating anti-tumour responses,57,58 other studies showed more modest effects.59 In contrast, viable tumour cells expressing even relatively low amounts of IL-2 within the tumour microenvironment can have dramatic immune effects and even result in tumour rejection.17,58,60,61 It is therefore likely that IL-2 produced by transfected growing tumours at the tumour site is largely acting locally, probably by enhancing T-cell and NK cell responses at the tumour site.60–62 The protease-activated cytokine fusion is similarly designed so that cytokines become biologically available at the site of tumours, which in turn should also enhance the T cells or NK cells present. These effectors could arise naturally as the tumours develop, such as the T cells seen in many melanoma patients,2,63,64 or from intentional immunization with tumour-associated antigens,2–4 or could even be T cells that have been expanded and even genetically modified in vitro and adoptively transferred.65,66 Hence, although we have shown effects of the fusion protein as a single agent, probably enhancing innate responses and the endogenous T-cell response, we hypothesize that the fusion protein would be even more effective in conjunction with immunization schemes. In this context there are a wide variety of innovative approaches for initiating anti-tumour cellular immune responses that show substantial promise (reviewed in refs 1 and 67) as well as recent clinical successes in patients with prostate cancer.68,69
The data presented here represent the first ‘proof of principle’ of the protease-activated cytokine approach using specific inhibition. Importantly, the tethered cytokine strategy using specific inhibition is a platform technology that could be employed with different immunomodulatory agents to either promote (e.g. IL-12) or inhibit (IFN-β or IL-10) cellular immune responses. This would be particularly useful for cytokines that have potent anti-tumour effects like IL-12 but systemic side-effects limit their usefulness when given systemically.11,70 The scFv format is particularly flexible in this regard. An scFv could be developed against almost any target molecule given the extremely large antibody repertoire in the scFv library and could be made against immunomodulators such as chemokines where the receptor approach is not easily implemented. It is also important to consider that the cytokine environment in the tumour would probably be affected in a cascade fashion as the infiltrating cells change. As a result, it may be possible to alter the balance of cytokines from the generally suppressive environment of the tumour, rich in a variety of immunosuppressive factors, enzymes and cells,1,71–74 to one that is conducive to an ongoing immune response leading to the eradication of tumours.
Acknowledgments
The authors would like to thank Drs Edward Messing and Baek Kim for encouragement and helpful suggestions, Dr Robert Rose and Christopher Lane for helpful advice on insect cell expression of proteins, and Drs Barth, Leddy, Courtney, Simon, Valentino and Cohen for comments on the manuscript. This work was made possible by generous gifts from Steven and Alison Krausz and F.C. Blodgett. John Puskas, Denise Skrombolas and Abigail Sedlacek were supported by 5T32AI00728 from the National Institutes of Health.
Glossary
Abbreviations
- 6His
6 × Histidine
- APMA
p-aminophenylmercuric acetate
- i.p.
intraperitoneal
- IL-2
interleukin-2
- IL-2Rα
interleukin-2 receptor alpha subunit
- LAP
latency-associated protein
- MMP9
matrix metalloproteinase 9
- MMPcs
matrix metalloproteinase cleavage sequence
- MTT
Thiazolyl Blue Tetrazolium Bromide
- NTG
non-transgenic mouse
- PBS-M-Tw
phosphate buffered saline plus 5% w/v milk and 0.2% Tween-20
- PCR
polymerase chain reaction
- phscFvs
phage displaying scFv on the cell surface
- PSA
prostate-specific antigen
- PSAcs
prostate-specific antigen cleavage sequence
- scFv
single-chain variable region of antibodies
- SGPP
structural genomics of parasitic protozoa
- TBS
Tris-buffered saline
- TG
transgenic mouse
Disclosures
None of the authors involved with this work has any financial interests or any other conflict of interest to disclose.
References
- 1.Finn OJ. Cancer immunology. N Engl J Med. 2008;358:2704–15. doi: 10.1056/NEJMra072739. [DOI] [PubMed] [Google Scholar]
- 2.Boon T, Coulie PG, Van den Eynde BJ, van der Bruggen P. Human T cell responses against melanoma. Annu Rev Immunol. 2006;24:175–208. doi: 10.1146/annurev.immunol.24.021605.090733. [DOI] [PubMed] [Google Scholar]
- 3.Parmiani G, De Filippo A, Novellino L, Castelli C. Unique human tumor antigens: immunobiology and use in clinical trials. J Immunol. 2007;178:1975–9. doi: 10.4049/jimmunol.178.4.1975. [DOI] [PubMed] [Google Scholar]
- 4.Berzofsky JA, Terabe M, Oh S, Belyakov IM, Ahlers JD, Janik JE, Morris JC. Progress on new vaccine strategies for the immunotherapy and prevention of cancer. J Clin Invest. 2004;113:1515–25. doi: 10.1172/JCI21926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Slingluff CL, Jr, Chianese-Bullock KA, Bullock TN, et al. Immunity to melanoma antigens: from self-tolerance to immunotherapy. Adv Immunol. 2006;90:243–95. doi: 10.1016/S0065-2776(06)90007-8. [DOI] [PubMed] [Google Scholar]
- 6.Pardoll DM. Paracrine cytokine adjuvants in cancer immunotherapy. Annu Rev Immunol. 1995;13:399–415. doi: 10.1146/annurev.iy.13.040195.002151. [DOI] [PubMed] [Google Scholar]
- 7.Rosenberg SA. Progress in human tumour immunology and immunotherapy. Nature. 2001;411:380–4. doi: 10.1038/35077246. [DOI] [PubMed] [Google Scholar]
- 8.Rosenberg SA, Yang JC, Topalian SL, et al. Treatment of 283 consecutive patients with metastatic melanoma or renal cell cancer using high-dose bolus interleukin 2. JAMA. 1994;271:907–13. [PubMed] [Google Scholar]
- 9.Den Otter W, Jacobs JJ, Battermann JJ, et al. Local therapy of cancer with free IL-2. Cancer Immunol Immunother. 2008;57:931–50. doi: 10.1007/s00262-008-0455-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hanes J, Sills A, Zhao Z, et al. Controlled local delivery of interleukin-2 by biodegradable polymers protects animals from experimental brain tumors and liver tumors. Pharm Res. 2001;18:899–906. doi: 10.1023/a:1010963307097. [DOI] [PubMed] [Google Scholar]
- 11.Egilmez NK, Jong YS, Sabel MS, Jacob JS, Mathiowitz E, Bankert RB. In situ tumor vaccination with interleukin-12-encapsulated biodegradable microspheres: induction of tumor regression and potent antitumor immunity. Cancer Res. 2000;60:3832–7. [PubMed] [Google Scholar]
- 12.Sabel MS, Arora A, Su G, Mathiowitz E, Reineke JJ, Chang AE. Synergistic effect of intratumoral IL-12 and TNF-alpha microspheres: systemic anti-tumor immunity is mediated by both CD8+ CTL and NK cells. Surgery. 2007;142:749–60. doi: 10.1016/j.surg.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 13.Sangro B, Mazzolini G, Ruiz J, et al. Phase I trial of intratumoral injection of an adenovirus encoding interleukin-12 for advanced digestive tumors. J Clin Oncol. 2004;22:1389–97. doi: 10.1200/JCO.2004.04.059. [DOI] [PubMed] [Google Scholar]
- 14.Triozzi PL, Allen KO, Carlisle RR, Craig M, LoBuglio AF, Conry RM. Phase I study of the intratumoral administration of recombinant canarypox viruses expressing B7.1 and interleukin 12 in patients with metastatic melanoma. Clin Cancer Res. 2005;11:4168–75. doi: 10.1158/1078-0432.CCR-04-2283. [DOI] [PubMed] [Google Scholar]
- 15.Addison CL, Braciak T, Ralston R, Muller WJ, Gauldie J, Graham FL. Intratumoral injection of an adenovirus expressing interleukin 2 induces regression and immunity in a murine breast cancer model. Proc Natl Acad Sci U S A. 1995;92:8522–6. doi: 10.1073/pnas.92.18.8522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gansbacher B, Zier K, Daniels B, Cronin K, Bannerji R, Gilboa E. Interleukin 2 gene transfer into tumor cells abrogates tumorigenicity and induces protective immunity. J Exp Med. 1990;172:1217–24. doi: 10.1084/jem.172.4.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McAdam AJ, Pulaski BA, Harkins SS, Hutter EK, Lord EM, Frelinger JG. Synergistic effects of co-expression of the TH1 cytokines IL-2 and IFN-gamma on generation of murine tumor-reactive cytotoxic cells. Int J Cancer. 1995;61:628–34. doi: 10.1002/ijc.2910610508. [DOI] [PubMed] [Google Scholar]
- 18.Balk SP, Ko YJ, Bubley GJ. Biology of prostate-specific antigen. J Clin Oncol. 2003;21:383–91. doi: 10.1200/JCO.2003.02.083. [DOI] [PubMed] [Google Scholar]
- 19.Chambers AF, Matrisian LM. Changing views of the role of matrix metalloproteinases in metastasis. J Natl Cancer Inst. 1997;89:1260–70. doi: 10.1093/jnci/89.17.1260. [DOI] [PubMed] [Google Scholar]
- 20.Malek TR. The biology of interleukin-2. Annu Rev Immunol. 2008;26:453–79. doi: 10.1146/annurev.immunol.26.021607.090357. [DOI] [PubMed] [Google Scholar]
- 21.Bachmann MF, Oxenius A. Interleukin 2: from immunostimulation to immunoregulation and back again. EMBO Rep. 2007;8:1142–8. doi: 10.1038/sj.embor.7401099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haidaris CG, Malone J, Sherrill LA, Bliss JM, Gaspari AA, Insel RA, Sullivan MA. Recombinant human antibody single chain variable fragments reactive with Candida albicans surface antigens. J Immunol Methods. 2001;257:185–202. doi: 10.1016/s0022-1759(01)00463-x. [DOI] [PubMed] [Google Scholar]
- 23.Malone J, Sullivan MA. Analysis of antibody selection by phage display utilizing anti-phenobarbital antibodies. J Mol Recognit. 1996;9:738–45. doi: 10.1002/(sici)1099-1352(199634/12)9:5/6<738::aid-jmr333>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 24.Mehlin C, Boni E, Buckner FS, et al. Heterologous expression of proteins from Plasmodium falciparum: results from 1000 genes. Mol Biochem Parasitol. 2006;148:144–60. doi: 10.1016/j.molbiopara.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 25.Rose RC, Bonnez W, Strike DG, Reichman RC. Expression of the full-length products of the human papillomavirus type 6b (HPV-6b) and HPV-11 L2 open reading frames by recombinant baculovirus, and antigenic comparisons with HPV-11 whole virus particles. J Gen Virol. 1990;71(Pt 11):2725–9. doi: 10.1099/0022-1317-71-11-2725. [DOI] [PubMed] [Google Scholar]
- 26.Rose RC, Bonnez W, Da Rin C, McCance DJ, Reichman RC. Serological differentiation of human papillomavirus types 11, 16 and 18 using recombinant virus-like particles. J Gen Virol. 1994;75(Pt 9):2445–9. doi: 10.1099/0022-1317-75-9-2445. [DOI] [PubMed] [Google Scholar]
- 27.Turner MJ, Abdul-Alim CS, Willis RA, Fisher TL, Lord EM, Frelinger JG. T-cell antigen discovery (T-CAD) assay: a novel technique for identifying T cell epitopes. J Immunol Methods. 2001;256:107–19. doi: 10.1016/s0022-1759(01)00436-7. [DOI] [PubMed] [Google Scholar]
- 28.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 29.Young FM, Phungtamdet W, Sanderson BJ. Modification of MTT assay conditions to examine the cytotoxic effects of amitraz on the human lymphoblastoid cell line, WIL2NS. Toxicol In Vitro. 2005;19:1051–9. doi: 10.1016/j.tiv.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 30.Wei C, Willis RA, Tilton BR, Looney RJ, Lord EM, Barth RK, Frelinger JG. Tissue-specific expression of the human prostate-specific antigen gene in transgenic mice: implications for tolerance and immunotherapy. Proc Natl Acad Sci U S A. 1997;94:6369–74. doi: 10.1073/pnas.94.12.6369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fisher TL, Nocera M, Willis RA, et al. Generation of monoclonal antibodies specific for human kallikrein 2 (hK2) using hK2-expressing tumors. Prostate. 2002;51:153–65. doi: 10.1002/pros.10071. [DOI] [PubMed] [Google Scholar]
- 32.Sorensen EW, Gerber SA, Sedlacek AL, Rybalko VY, Chan WM, Lord EM. Omental immune aggregates and tumor metastasis within the peritoneal cavity. Immunol Res. 2009;45:185–194. doi: 10.1007/s12026-009-8100-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lord EM, Burkhardt G. Assessment of in situ host immunity to syngeneic tumors utilizing the multicellular spheroid model. Cell Immunol. 1984;85:340–50. doi: 10.1016/0008-8749(84)90248-x. [DOI] [PubMed] [Google Scholar]
- 34.Denmeade SR, Lou W, Lovgren J, Malm J, Lilja H, Isaacs JT. Specific and efficient peptide substrates for assaying the proteolytic activity of prostate-specific antigen. Cancer Res. 1997;57:4924–30. [PMC free article] [PubMed] [Google Scholar]
- 35.Minami Y, Kono T, Miyazaki T, Taniguchi T. The IL-2 receptor complex: its structure, function, and target genes. Annu Rev Immunol. 1993;11:245–68. doi: 10.1146/annurev.iy.11.040193.001333. [DOI] [PubMed] [Google Scholar]
- 36.Trinh R, Gurbaxani B, Morrison SL, Seyfzadeh M. Optimization of codon pair use within the (GGGGS)3 linker sequence results in enhanced protein expression. Mol Immunol. 2004;40:717–22. doi: 10.1016/j.molimm.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 37.Chen EI, Kridel SJ, Howard EW, Li W, Godzik A, Smith JW. A unique substrate recognition profile for matrix metalloproteinase-2. J Biol Chem. 2002;277:4485–91. doi: 10.1074/jbc.M109469200. [DOI] [PubMed] [Google Scholar]
- 38.Gerber SA, Rybalko VY, Bigelow CE, Lugade AA, Foster TH, Frelinger JG, Lord EM. Preferential attachment of peritoneal tumor metastases to omental immune aggregates and possible role of a unique vascular microenvironment in metastatic survival and growth. Am J Pathol. 2006;169:1739–52. doi: 10.2353/ajpath.2006.051222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hofmann UB, Westphal JR, Van Muijen GN, Ruiter DJ. Matrix metalloproteinases in human melanoma. J Invest Dermatol. 2000;115:337–44. doi: 10.1046/j.1523-1747.2000.00068.x. [DOI] [PubMed] [Google Scholar]
- 40.Bergers G, Brekken R, McMahon G, et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol. 2000;2:737–44. doi: 10.1038/35036374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nikkola J, Vihinen P, Vuoristo MS, Kellokumpu-Lehtinen P, Kahari VM, Pyrhonen S. High serum levels of matrix metalloproteinase-9 and matrix metalloproteinase-1 are associated with rapid progression in patients with metastatic melanoma. Clin Cancer Res. 2005;11:5158–66. doi: 10.1158/1078-0432.CCR-04-2478. [DOI] [PubMed] [Google Scholar]
- 42.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161–74. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 43.Bremer C, Tung CH, Weissleder R. In vivo molecular target assessment of matrix metalloproteinase inhibition. Nat Med. 2001;7:743–8. doi: 10.1038/89126. [DOI] [PubMed] [Google Scholar]
- 44.Lilja H. Biology of prostate-specific antigen. Urology. 2003;5(Suppl. 1):27–33. doi: 10.1016/s0090-4295(03)00775-1. [DOI] [PubMed] [Google Scholar]
- 45.Schroder FH, Roach M, III, Scardino P. Clinical decisions. Management of prostate cancer. N Engl J Med. 2008;359:2605–9. doi: 10.1056/NEJMclde0805491. [DOI] [PubMed] [Google Scholar]
- 46.Heaney ML, Golde DW. Soluble cytokine receptors. Blood. 1996;87:847–57. [PubMed] [Google Scholar]
- 47.Smith KA. The structure of IL2 bound to the three chains of the IL2 receptor and how signaling occurs. Med Immunol. 2006;5:3. doi: 10.1186/1476-9433-5-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shimotsuma M, Shields JW, Simpson-Morgan MW, Sakuyama A, Shirasu M, Hagiwara A, Takahashi T. Morpho-physiological function and role of omental milky spots as omentum-associated lymphoid tissue (OALT) in the peritoneal cavity. Lymphology. 1993;26:90–101. [PubMed] [Google Scholar]
- 49.Oosterling SJ, van der Bij GJ, Bogels M, van der Sijp JR, Beelen RH, Meijer S, van Egmond M. Insufficient ability of omental milky spots to prevent peritoneal tumor outgrowth supports omentectomy in minimal residual disease. Cancer Immunol Immunother. 2006;55:1043–51. doi: 10.1007/s00262-005-0101-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Krist LF, Kerremans M, Broekhuis-Fluitsma DM, Eestermans IL, Meyer S, Beelen RH. Milky spots in the greater omentum are predominant sites of local tumour cell proliferation and accumulation in the peritoneal cavity. Cancer Immunol Immunother. 1998;47:205–12. doi: 10.1007/s002620050522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vlad AM, Budiu RA, Lenzner DE, et al. A phase II trial of intraperitoneal interleukin-2 in patients with platinum-resistant or platinum-refractory ovarian cancer. Cancer Immunol Immunother. 2009;59:293–301. doi: 10.1007/s00262-009-0750-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Curiel TJ. Regulatory T cells and treatment of cancer. Curr Opin Immunol. 2008;20:241–6. doi: 10.1016/j.coi.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Becker JC, Varki N, Gillies SD, Furukawa K, Reisfeld RA. Long-lived and transferable tumor immunity in mice after targeted interleukin-2 therapy. J Clin Invest. 1996;98:2801–4. doi: 10.1172/JCI119107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dela Cruz JS, Huang TH, Penichet ML, Morrison SL. Antibody–cytokine fusion proteins: innovative weapons in the war against cancer. Clin Exp Med. 2004;4:57–64. doi: 10.1007/s10238-004-0039-y. [DOI] [PubMed] [Google Scholar]
- 55.Schrama D, Reisfeld RA, Becker JC. Antibody targeted drugs as cancer therapeutics. Nat Rev Drug Discov. 2006;5:147–59. doi: 10.1038/nrd1957. [DOI] [PubMed] [Google Scholar]
- 56.Adams G, Vessillier S, Dreja H, Chernajovsky Y. Targeting cytokines to inflammation sites. Nat Biotechnol. 2003;21:1314–20. doi: 10.1038/nbt888. [DOI] [PubMed] [Google Scholar]
- 57.Allione A, Consalvo M, Nanni P, et al. Immunizing and curative potential of replicating and nonreplicating murine mammary adenocarcinoma cells engineered with interleukin (IL)-2, IL-4, IL-6, IL-7, IL-10, tumor necrosis factor alpha, granulocyte–macrophage colony-stimulating factor, and gamma-interferon gene or admixed with conventional adjuvants. Cancer Res. 1994;54:6022–6. [PubMed] [Google Scholar]
- 58.Porgador A, Gansbacher B, Bannerji R, Tzehoval E, Gilboa E, Feldman M, Eisenbach L. Anti-metastatic vaccination of tumor-bearing mice with IL-2-gene-inserted tumor cells. Int J Cancer. 1993;53:471–7. doi: 10.1002/ijc.2910530320. [DOI] [PubMed] [Google Scholar]
- 59.Dranoff G, Jaffee E, Lazenby A, et al. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte–macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc Natl Acad Sci U S A. 1993;90:3539–43. doi: 10.1073/pnas.90.8.3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Roth C, Mir LM, Cressent M, Quintin-Colonna F, Ley V, Fradelizi D, Kourilsky P. Inhibition of tumor growth by histoincompatible cells expressing interleukin-2. Int Immunol. 1992;4:1429–36. doi: 10.1093/intimm/4.12.1429. [DOI] [PubMed] [Google Scholar]
- 61.McAdam AJ, Pulaski BA, Storozynsky E, Yeh KY, Sickel JZ, Frelinger JG, Lord EM. Analysis of the effect of cytokines (interleukins 2, 3, 4, and 6, granulocyte–monocyte colony-stimulating factor, and interferon-gamma) on generation of primary cytotoxic T lymphocytes against a weakly immunogenic tumor. Cell Immunol. 1995;165:183–92. doi: 10.1006/cimm.1995.1204. [DOI] [PubMed] [Google Scholar]
- 62.Woods ML, McAdam AJ, Frelinger JG, Lord EM. Isolation and expansion of tumor-infiltrating lymphocytes. BioTechniques. 1993;15:970–2. [PubMed] [Google Scholar]
- 63.Lee PP, Yee C, Savage PA, et al. Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nat Med. 1999;5:677–85. doi: 10.1038/9525. [DOI] [PubMed] [Google Scholar]
- 64.Yee C, Savage PA, Lee PP, Davis MM, Greenberg PD. Isolation of high avidity melanoma-reactive CTL from heterogeneous populations using peptide–MHC tetramers. J Immunol. 1999;162:2227–34. [PubMed] [Google Scholar]
- 65.Morgan RA, Dudley ME, Wunderlich JR, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–9. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008;8:299–308. doi: 10.1038/nrc2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McKarney I. Sipuleucel-T (Provenge): active cellular immunotherapy for advanced prostate cancer. Issues Emerg Health Technol. 2007;101:1–4. [PubMed] [Google Scholar]
- 68.Kantoff PW, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411–22. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 69.Longo DL. New therapies for castration-resistant prostate cancer. N Engl J Med. 363:479–81. doi: 10.1056/NEJMe1006300. ???? [DOI] [PubMed] [Google Scholar]
- 70.Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3:133–46. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- 71.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–74. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol. 2009;182:4499–506. doi: 10.4049/jimmunol.0802740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007;25:267–96. doi: 10.1146/annurev.immunol.25.022106.141609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer. 2005;5:263–74. doi: 10.1038/nrc1586. [DOI] [PubMed] [Google Scholar]