

Abstract
R-type calcium channels in postsynaptic spines signal through functional calcium microdomains to regulate a calcium/calmodulin-sensitive potassium channel that in turn regulates postsynaptic hippocampal long-term potentiation (LTP). Here, we ask whether R-type calcium channels in presynaptic terminals also signal through calcium microdomains to control presynaptic LTP. We focus on presynaptic LTP at parallel fiber to Purkinje cell synapses in the cerebellum (PF-LTP), which is mediated by calcium/calmodulin-stimulated adenylyl cyclases. Although most presynaptic calcium influx is through N-type and P/Q-type calcium channels, blocking these channels does not disrupt PF-LTP, but blocking R-type calcium channels does. Moreover, global calcium signaling cannot account for the calcium dependence of PF-LTP because R-type channels contribute modestly to overall calcium entry. These findings indicate that, within presynaptic terminals, R-type calcium channels produce calcium microdomains that evoke presynaptic LTP at moderate frequencies that do not greatly increase global calcium levels.
Introduction
Calcium can activate proteins within neurons either by widespread but modest increases in global calcium levels (Caglobal) or by larger localized calcium increases in microdomains near calcium-permeable channels (Calocal) (Fogelson and Zucker, 1985; Simon and Llinas, 1985; Augustine and Neher, 1992; Stern, 1992; Zucker and Regehr, 2002; Berridge, 2006; Rizzuto and Pozzan, 2006; Higley and Sabatini, 2008). Calocal signaling allows specific types of calcium channels to selectively activate calcium-activated potassium channels (BK and SK) (Fakler and Adelman, 2008). Calcium microdomains can also allow calcium sources to initiate calcium-sensitive pathways within cells (Berridge, 2006; Rizzuto and Pozzan, 2006). Although it is straightforward to measure Caglobal signals with standard fluorescence techniques, measuring calcium signals associated with calcium microdomains is exceedingly difficult. Consequently, there is much left to learn about the regulation of cellular processes by calcium microdomains.
Calcium microdomains have been implicated in the induction of long-term potentiation (LTP). In the spines of hippocampal pyramidal cells, Calocal near R-type calcium channels (CaV2.3) activate SK channels (Bloodgood and Sabatini, 2007). SK channels also control the magnitude of calcium signals within spines and thereby regulate LTP at the CA3 to CA1 synapse (Stackman et al., 2002; Bond et al., 2004; Ngo-Anh et al., 2005; Hammond et al., 2006). This suggests that functional calcium microdomains near R-type calcium channels can regulate the induction of postsynaptic hippocampal LTP. Such Calocal signals can allow a type of calcium channel to strongly influence a neuron, even if it accounts for a small fraction of Caglobal. Although it is known that R-type calcium channels can regulate presynaptic LTP at some synapses (Breustedt et al., 2003; Dietrich et al., 2003), it is not known whether this is a result of spatially localized signaling through calcium microdomains.
Presynaptic LTP mediated by calcium/calmodulin-stimulated adenylyl cyclases (Ca-ACs, type 1 and 8, AC1 and AC8) is well suited to testing this possibility. This form of plasticity has been studied at mossy fiber (MF) synapses onto CA3 pyramidal cells (MF-LTP) (Zalutsky and Nicoll, 1990; Nicoll and Schmitz, 2005), parallel fiber (PF) synapses onto Purkinje cells (PCs) (PF-LTP) (Salin et al., 1996; van Beugen et al., 2006), and other synapses (Maffei et al., 2002; Liu et al., 2004; Fourcaudot et al., 2008; Bender et al., 2009; Wang et al., 2009). Mice that lack AC1 and AC8 do not express either MF-LTP (Wong et al., 1999; Wang et al., 2003) or PF-LTP (Storm et al., 1998) and exhibit deficits in hippocampal and cerebellar behaviors (Wu et al., 1995; Shan et al., 2008; Zhang et al., 2008). MF and PF boutons contain multiple types of voltage-gated calcium channels (P/Q, N, and R) (Castillo et al., 1994; Mintz et al., 1995), but only R-type calcium channels play a crucial role in MF-LTP (Breustedt et al., 2003; Dietrich et al., 2003). It is not known whether R-type calcium channels play a similar role in Ca-AC-dependent LTP at other synapses. With regard to Caglobal versus Calocal signaling, MF-LTP is thought to be triggered by Caglobal (Dietrich et al., 2003), but the nature of the calcium signal that triggers PF-LTP is not known. This is of particular interest because PF-LTP is readily induced by a moderate-frequency train (8 Hz, 15 s) that may not elevate Caglobal sufficiently to activate Ca-AC (Olwin et al., 1984; Cimler et al., 1985; Masada et al., 2009).
Here, we ask whether R-type calcium channels are important for PF-LTP and whether they signal through Caglobal or Calocal. We find that R-type calcium channels play a crucial role in the induction of PF-LTP. Moreover, we find that Caglobal cannot account for the calcium dependence of PF-LTP. Instead, R-type calcium channels induce PF-LTP by elevating Calocal to activate Ca-AC. These findings indicate that by signaling through functional calcium microdomains, R-type calcium channels can control presynaptic forms of LTP.
Materials and Methods
Tissue preparation.
Animals were handled in accordance with federal guidelines and protocols approved by Harvard University. Sprague Dawley rats (postnatal day 17–19) were deeply anesthetized with isoflurane and decapitated. Brains were removed and placed in an ice-cold dissecting solution containing the following (in mm): 82.7 NaCl, 65 sucrose, 23.8 NaHCO3, 23.7 glucose, 6.8 MgCl2, 2.4 KCl, 1.4 NaH2PO4, and 0.5 CaCl2. Parasagittal (for electrophysiology) or transverse (for presynaptic calcium imaging) slices (250 μm thick) were made from the cerebellum in ice-cold dissecting solution. Slices were then incubated at 32°C for 30 min in a saline solution containing the following (in mm): 125 NaCl, 26 NaHCO3, 25 glucose, 2.5 KCl, 2 CaCl2, 1.25 NaH2PO4, and 1 MgCl2, adjusted to 315 mOsm. Slices were then allowed to equilibrate to room temperature for an additional 30 min. Slices were constantly bubbled with 95% O2/5% CO2. Most experiments were performed at 25 ± 1°C and flow rates were 2 ml/min. A few experiments were performed at 34 ± 1°C and flow rates were 4 ml/min.
Electrophysiology.
Whole-cell recordings of PCs were obtained with borosilicate glass electrodes of 1–1.5 MΩ resistances. The internal solution contained the following (in mm): 110 Cs2SO4, 15 HEPES, 10 EGTA, 5.5 MgSO4, 4 CaCl2, 2 Na2-ATP, 1 MgCl2, and 0.2 D600, adjusted to 315 mOsm and pH 7.3. PCs were visualized with a BX51 upright microscope equipped with a 60× 0.9 numerical aperture (NA) water-immersion lens (Olympus). Recordings were made with a Multiclamp 700B amplifier (Molecular Devices). For LTP experiments, two borosilicate glass electrodes (2–3 MΩ resistances) filled with saline were placed in the molecular layer (>75 μm apart) to activate independent PF pathways. PFs were stimulated with brief (0.2 ms) current pulses (10–25 μA). This protocol was repeated every 10 s to evoke EPSCs in PCs. Stimulation to each pathway was staggered by 5 s. To induce LTP, an 8 Hz, 15 s train was delivered to only one pathway. The other pathway was not stimulated during this time. For LTP experiments in the presence of ω-agatoxin IVA, the second electrode was elevated slightly above the slice and connected to ground to reduce the stimulus artifact.
Presynaptic calcium imaging.
Calcium transients in PFs were measured as described previously (Regehr and Tank, 1991; Mintz et al., 1995; Regehr and Atluri, 1995). Briefly, Magnesium Green AM (Invitrogen) was pressure loaded into PFs for 5–10 min and then allowed to equilibrate throughout PFs for 1 h. Fluorescence signals were visualized on a BX50 microscope equipped with a 60× 0.9 NA water-immersion lens (Olympus). Magnesium Green AM was excited by a 470 nm LED (Thorlabs) and fluorescence was detected by a custom-built photodiode. A 50-μm-diameter imaging site was selected >200 μm from the loading site. Because the molecular layer is mostly comprised of PFs, this imaging site effectively measures Caglobal signals in thousands of PFs (Sabatini and Regehr, 1998; Yuste and Konnerth, 2005). A single borosilicate glass electrode filled with saline (∼500 kΩ resistance) was placed in the molecular layer >150 μm from the imaging site. For low-frequency stimulation, single 0.2 ms (50–100 μA) pulses were repeated at 0.1 Hz. Each calcium transient was normalized to the baseline fluorescence and expressed as ΔF/F.
There were several differences in the way calcium transients during 8 Hz, 15 s trains were analyzed. First, the entire fluorescence signal was normalized to the amplitude of the first peak in the train. This allowed us to quantify the buildup of calcium during the train. Second, light exposure was limited to a brief (20 ms) window around the stimulus to reduce photobleaching. The blanked times between stimuli in the train were extrapolated using a straight line for integral measurements. Finally, 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide (NBQX, 10 μm) was included in these experiments.
Pharmacology.
All experiments were performed in the presence of picrotoxin (20 μm) to block GABAA-mediated currents. Experiments investigating evoked EPSCs were also performed in the presence of 8-cyclopentyl-1,3-dipropylxanthine (DPCPX, 5 μm) to block adenosine A1 receptors, (2S)-3-[(15)-1-(3,4-dichlorophenyl)ethyl]amino-2-hydroxypropyl)(phenylmethyl) phosphinic acid (CGP 55845, 2 μm) to block GABAB receptors, and N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (AM 251, 2 μm) to block cannabinoid CB1 receptors (CB1Rs). This prevented regulation of presynaptic G-protein-coupled receptor signaling that could regulate Ca-AC (van Beugen et al., 2006). Experiments investigating spontaneous miniature EPSCs (mEPSCs) were performed in the presence of octahydro-12-(hydroxymethyl)-2-imino-5,9:7,10a-dimethano-10aH-[1,3]dioxocino[6,5-d]pyrimidine-4,7,10,11,12-pentol (TTX, 1 μm) to block voltage-gated sodium channels. For experiments using peptide toxins ω-conotoxin GVIA, ω-agatoxin IVA, or SNX-482, recirculation was used to reduce the amount of toxin used. Drugs were dissolved in dimethylsulfoxide at 5000–50,000 times the final concentration except for NBQX, TTX, and peptide toxins, which were dissolved in water at 1000 times the final concentration. All drugs were purchased from Tocris Bioscience, except for ω-conotoxin GVIA and ω-agatoxin IVA, which were from Peptides International, and SNX-482, which was from Alomone Labs. All other chemicals were purchased from Sigma.
Experiments at near-physiological temperatures.
Experiments performed at near-physiological temperatures differed from those performed at 25°C in the following ways. LTP experiments were performed at 34 ± 1°C. In some experiments, LTP was induced with a 30 s tetanus and DPCPX, CGP 55845, and AM 251 were omitted. Presynaptic calcium imaging experiments were performed at 32 ± 1°C and DPCPX, CGP 55845, and AM 251 were omitted.
Data acquisition and analysis.
Data were acquired on a personal computer using an ITC-18 interface (Instrutech) at 10 and 1 kHz for electrophysiology and fluorescence data, respectively. Data were then digitally filtered using an eight-pole Bessel filter at 5 kHz for electrophysiology data and 500 Hz for fluorescence data. All data were analyzed using Igor Pro (Wavemetrics). Averages are presented as mean ± SEM. One-way ANOVA or paired Student's t test was performed to determine statistical significance (p < 0.05) where indicated.
Results
LTP at parallel fiber to Purkinje cell synapses is sensitive to changes in external calcium concentration
We studied PF-LTP by activating two independent PF pathways (0.1 Hz) (Fig. 1A). Resulting EPSCs were monitored with a whole-cell recording electrode in a PC. A tetanus was delivered to one pathway (8 Hz, 15 s) and the other pathway served as a control. After the tetanus, 0.1 Hz stimulation of both pathways was resumed. When the external calcium concentration (Cae) was 2 mm, EPSC amplitudes in the tetanized pathway were potentiated by 22 ± 8% (Fig. 1E, filled circles) and the control pathway was unchanged (0 ± 7%, n = 10, p < 0.05, ANOVA) (Fig. 1E, open circles).
Figure 1.
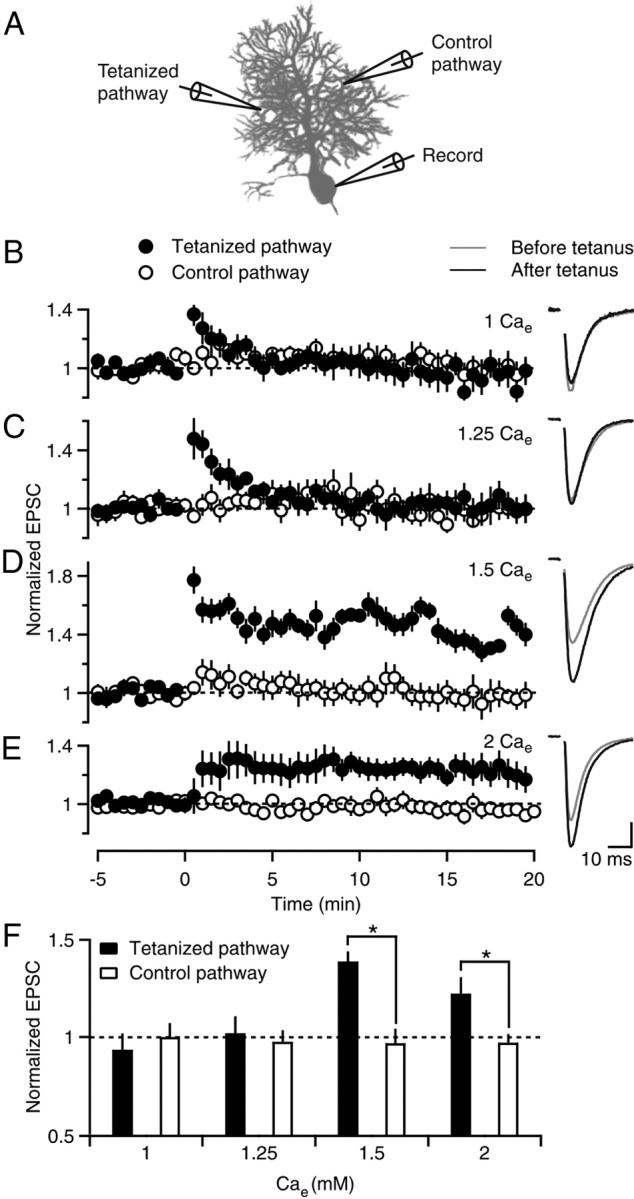
Sensitivity of PF-LTP to external calcium concentration. A, A schematic of the recording configuration shows two extracellular electrodes that were used to stimulate independent PF pathways (0.1 Hz) and a third electrode that was used to measure the amplitude of EPSCs from a PC soma. After a baseline response was determined, a tetanus (8 Hz, 15 s, t = 0) was delivered to one pathway, and the other pathway served as a control for recording stability. B–E, Left, Experiments were performed in the indicated external calcium concentration (Cae), and EPSCs were normalized to the amplitude before tetanic stimulation. Six to 10 experiments were averaged for each experimental condition. Filled and open circles indicate tetanized and control pathways, respectively. B–E, Right, EPSCs before (−5–0 min, gray line) and after (15–20 min, black line) tetanic stimulation are shown for representative experiments in the indicated Cae. Vertical scale bar (in pA): B, 38; C, 56; D, 71; E, 172. F, Experiments in B–E are summarized, where normalized EPSC amplitudes are plotted for the tetanized pathway (filled bars) and control pathway (open bars). *p < 0.05 (tetanized pathway vs control, one-way ANOVA).
We determined the calcium sensitivity of PF-LTP by measuring the magnitude of potentiation in different Cae (Fig. 1B–D). Reducing Cae decreases influx through calcium channels without discriminating between calcium channel types (Mintz et al., 1995). In 1.5 mm Cae the tetanized pathway was potentiated (39 ± 5%) compared with the control pathway (−3 ± 8%, n = 8, p < 0.05, ANOVA) (Fig. 1D). Although the magnitude of this potentiation was not significantly different from in 2 mm Cae (p = 0.07), it appeared to be somewhat larger, perhaps because it is easier to induce LTP when the initial probability of release is slightly decreased. In contrast, in 1.25 and 1 mm Cae, the tetanus only produced a short-lived enhancement that was likely due to post-tetanic potentiation (Zucker and Regehr, 2002; Beierlein et al., 2007) and LTP was not observed (1.25 mm Cae, 2 ± 9%, n = 6; 1 mm Cae, −7 ± 8%, n = 10) (Fig. 1B,C). Thus, PF-LTP is steeply dependent on Cae (Fig. 1F), which suggests that the extent of enhancement is exquisitely sensitive to the magnitude of calcium influx.
Calcium influx through R-type calcium channels is required for PF-LTP
Multiple types of calcium channels are present on PF boutons: P/Q-type [blocked by ω-agatoxin IVA (AgTx)], N-type [blocked by ω-conotoxin GVIA (CgTx)], and a component that is insensitive to these toxins (Mintz et al., 1995). T-type calcium channels are not present in cerebellar granule cells (Randall and Tsien, 1995) and L-type calcium channels are not present in PF boutons (Mintz et al., 1995), indicating that this AgTx/CgTx-insensitive component is mediated by R-type calcium channels, which are also known as α1E or CaV2.3 calcium channels. R-type calcium channels are blocked by Ni2+ and selectively but incompletely blocked by the peptide toxin SNX-482 (SNX) (Newcomb et al., 1998; Breustedt et al., 2003; Dietrich et al., 2003).
We determined which calcium channel types are important for PF-LTP by examining whether selective antagonists influence LTP induction in 2 mm Cae (Fig. 2). Robust LTP was induced when N-type calcium channels were blocked with CgTx (36 ± 7%, n = 7, p < 0.05, ANOVA) (Fig. 2A) and when P/Q-type calcium channels were blocked with AgTx (23 ± 4%, n = 18, p < 0.05, ANOVA) (Fig. 2B). This indicates that calcium entry through either N-type or P/Q-type calcium channels is not required for PF-LTP. We examined the component mediated by R-type calcium channels. Ni2+ prevented the induction of LTP (−10 ± 8%, n = 7) (Fig. 2C). Although Ni2+ can also block T-type calcium channels (Lee et al., 1999; Chow et al., 2003; Obejero-Paz et al., 2008), because they are not present in cerebellar granule cells (Randall and Tsien, 1995), the Ni2+-sensitivity of PF-LTP is consistent with the involvement of R-type calcium channels. SNX also prevented the induction of LTP (−19 ± 5%, n = 7) (Fig. 2D), further indicating that calcium influx through R-type calcium channels, but not N-type or P/Q-type calcium channels, is required to induce PF-LTP (Fig. 2E).
Figure 2.

Sensitivity of PF-LTP to calcium channel antagonists. A–D, Time courses of normalized EPSCs (left) and example EPSC traces (right) are shown for experiments that were conducted as in Figure 1, except that Cae was 2 mm and 500 nm ω-conotoxin GVIA (A), 200 nm ω-agatoxin IVA (B), 100 μm NiCl2 (C), or 500 nm SNX-482 (D) was present. Vertical scale bar (in pA): A, 138; B, 19; C, 77; D, 191. E, Experiments in A–D are summarized. *p < 0.05 (tetanized pathway vs control, one-way ANOVA). For ω-agatoxin IVA experiments, the tetanized pathway was compared with an idealized value of 1.
cAMP-dependent plasticity is not sensitive to calcium influx
Previous studies have shown that the induction of PF-LTP is triggered by elevations of presynaptic cAMP that stimulate PKA (Salin et al., 1996), which in turn phosphorylates the active zone protein RIM1α to increase the probability of release (Lonart et al., 2003; but see Kaeser et al., 2008). It is possible to bypass the initial calcium trigger by using forskolin to activate adenylyl cyclase, which elevates presynaptic cAMP levels to enhance both evoked release and spontaneous release (Salin et al., 1996; Chen and Regehr, 1997). We examined forskolin-stimulated mEPSCs to determine whether manipulations that prevent the induction of LTP act on the initial calcium trigger, or act downstream of cAMP production (Fig. 3). In 2 mm Cae, forskolin elevated mEPSC frequency (236 ± 39% of control, n = 5) (Fig. 3A,B) to a much larger extent than mEPSC amplitude (126 ± 10% of control) (Fig. 3C, gray bars). Forskolin elevated mEPSC frequency to a similar extent in 1 mm Cae (240 ± 27% of control, n = 5) in the presence of Ni2+ (235 ± 30% of control, n = 5) and in the presence of SNX (236 ± 39% of control, n = 5) (Fig. 3B). These findings indicate that forskolin-stimulated enhancement is unaffected by manipulations that prevent the induction of LTP (Fig. 3C), suggesting that these manipulations suppress LTP by reducing the calcium signal that stimulates Ca-AC.
Figure 3.

Calcium-sensitivity of cAMP-stimulated mEPSCs. A, Example traces of mEPSCs recorded from a PC in control conditions (top) and after application of forskolin (bottom). B, Time courses of normalized mEPSC frequency in (from top to bottom) 2 mm Cae, 1 mm Cae, and in 2 mm Cae with either 500 nm SNX-482 or 100 μm NiCl2. C, Experiments in B are summarized. mEPSC frequency (black bars) and amplitude (gray bars) 15–20 min after forskolin application were normalized to control conditions (−5 to 0 min).
R-type calcium channels contribute to synaptic transmission at PF to PC synapses
Our findings thus far indicate that R-type calcium channels play a crucial role in regulating PF-LTP, just as they do for MF-LTP. In MFs, low-frequency stimulation evokes narrow action potentials that are largely ineffective at opening R-type calcium channels (Alle et al., 2001; Li et al., 2007). As a result, blockade of R-type calcium channels in MFs with either Ni2+ or SNX only modestly reduces presynaptic calcium entry and does not reduce synaptic strength evoked by low-frequency stimulation (Dietrich et al., 2003). During high-frequency stimulation (100 Hz) used to induce MF-LTP, presynaptic action potentials broaden and become more effective at opening R-type calcium channels. As a result, there is significantly more influx through R-type calcium channels during the train compared with low-frequency stimulation. It is thought that facilitation of calcium influx through R-type calcium channels during the induction protocol is important for LTP induction (Dietrich et al., 2003). The role of R-type calcium channels in synaptic transmission has not been determined at PF to PC synapses. We therefore investigated presynaptic calcium signaling by R-type calcium channels in PFs to gain insight into the manner in which R-type calcium channels regulate PF-LTP.
We determined the calcium dependence of synaptic transmission evoked by low-frequency stimulation (0.1 Hz) (Fig. 4) by measuring the relationship between synaptic strength (EPSC amplitude) (Fig. 4A) and presynaptic calcium influx (Cainflux) (Fig. 4B) under the manipulations we used in our LTP experiments. We measured presynaptic calcium levels using the low-affinity calcium indicator Magnesium Green as described previously (Mintz et al., 1995; Regehr and Atluri, 1995). These signals provide a measure of the global presynaptic calcium levels in a population of presynaptic boutons and are referred to as Caglobal (expressed as ΔF/F). They do not provide a measure of local signals (Calocal) near open calcium channels. Previous studies have established that the change in Caglobal evoked by a single action potential is proportional to the total Cainflux per action potential (Sabatini and Regehr, 1995). In general, a reduction in calcium entry was correlated with a larger reduction in EPSC amplitude (note the different y-axis scales between Fig. 4, A and B). A double-logarithmic plot of EPSC amplitude versus Cainflux (normalized to values obtained in 2 mm Cae) illustrates that the relationship between Cainflux and synaptic strength can be well approximated by EPSC = k(Cainflux)n, where k is a constant, and the peak of the Caglobal transient following a single action potential provides a measure of Cainflux (Fig. 4C). When Cainflux was altered by changing Cae (Fig. 4A,B), the relationship between synaptic strength (EPSC amplitude) and Cainflux was approximated by the above equation with n ∼ 4 (Fig. 4C, open circles). We next investigated the contributions of different types of calcium channels. P/Q-type calcium channels (AgTx-sensitive), which accounted for ∼50% of Cainflux, were effective at triggering vesicular release (n ∼ 4), whereas N-type calcium channels (CgTx-sensitive) contributed a smaller percentage of Cainflux (∼25%) and were less effective at triggering release (n ∼ 2). In contrast to MF synapses, R-type calcium channels at PF synapses contribute to low-frequency synaptic transmission. Ni2+ and SNX reduced EPSC amplitude by 38 ± 5% (n = 8) and 20 ± 2% (n = 6), respectively, and Cainflux by 24 ± 2% (n = 6) and 9 ± 2% (n = 6), respectively, such that 2 < n < 3 (Fig. 4C). The smaller effect of SNX compared with Ni2+ likely reflects a less complete block of R-type calcium channels by SNX (Newcomb et al., 1998). However, the finding that R-type calcium channels influence Cainflux and synaptic strength suggests that R-type calcium channels may function differently in MFs and PFs.
Figure 4.
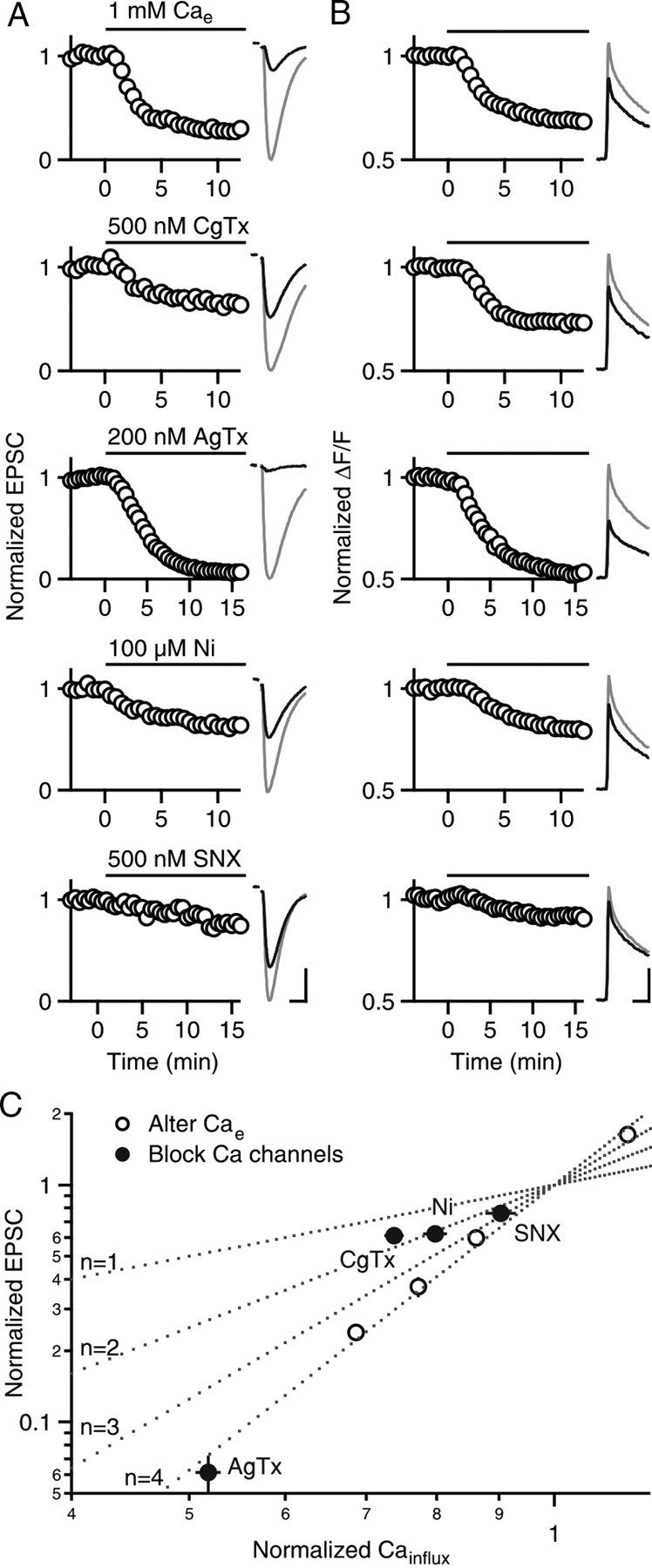
Relationship between calcium influx and synaptic strength. A, B, Normalized EPSCs (A) and presynaptic calcium influx (B) are shown for (from top to bottom) reducing Cae from 2 to 1 mm or in 2 mm Cae and application of one of the following: 500 nm ω-conotoxin GVIA, 200 nm ω-agatoxin IVA, 100 μm NiCl2, or 500 nm SNX-482. Average time courses (left) and example traces (right) before (gray lines) and after (black lines) are shown for each manipulation (5–7 experiments per manipulation, error bars are occluded by the circles). Horizontal scale bars: A, 10 ms; B, 20 ms. Vertical scale bars: A (in pA, from top to bottom), 240, 164, 230, 196, and 151; B (in ΔF/F, from top to bottom), 2.1, 2.1, 1.5, 1.1, and 1.8. C, Normalized EPSCs in A are plotted against normalized calcium influx (Cainflux) in B on log10 scales. Open circles indicate alterations of Cae and filled circles indicate manipulations in 2 mm Cae. Dotted lines indicate EPSC = k(Cainflux)n, where n = 1, 2, 3, and 4.
R-type calcium channels are not potentiated during the induction protocol
We went on to measure Caglobal evoked by the induction protocol (Fig. 5). The stimulus train evoked a series of Caglobal transients that rapidly decayed to an elevated plateau level between stimuli. After the train, Caglobal gradually returned to prestimulus levels (Fig. 5A). In control conditions, the peak Caglobal levels were 56 ± 8% larger than the peak Caglobal signal evoked by a single stimulus (n = 8).
Figure 5.

Presynaptic calcium signals during the induction protocol. PFs were stimulated (8 Hz, 15 s) and global presynaptic calcium signals (Caglobal) were measured. A, An example Caglobal transient during the induction protocol is shown. Breaks in the trace indicate times where the excitation was turned off to reduce photobleaching. B, Example quantification of Caglobal during the train in 2 mm Cae (left) and after washing in 1 mm Cae (right) is shown (n = 8, error bars are occluded by the symbols). Circles, Peaks during the train; triangles, buildup of plateau calcium during the train. C, Quantification of Caglobal signals before (left) and after (right) application of 500 nm ω-conotoxin GVIA and 200 nm ω-agatoxin IVA. The Cainflux per spike during the train was determined by subtracting peak values by plateau values for each spike (normalized to the value of the first spike) and is plotted as a function of time for control conditions (lower left) and in the presence of ω-conotoxin GVIA and ω-agatoxin IVA (lower right, n = 5). Each symbol represents an average of five stimuli.
We measured the peak of each transient in the train as well as the plateau and normalized them to the value of the first peak (Fig. 5B, left). Then we performed the manipulations of Cainflux, as described in Figure 4, and reanalyzed the train, normalizing values to the first peak in control conditions (2 mm Cae). An example of reducing Cae to 1 mm is shown in Figure 5B. The peak Caglobal at the end of the train and integral of Caglobal during the train were reduced by ∼25% in 1 mm Cae, which is slightly less than the reduction in Cainflux evoked by a single stimulus (∼30%) (Fig. 6A).
Figure 6.

The calcium dependence of PF-LTP. A, Effects of manipulations of calcium entry are summarized for Cainflux during single stimuli (white bars, from Fig. 4), for the peak Caglobal reached in the train (gray bars), and the integral of Caglobal during the train (black bars). Values are normalized to measurements made in 2 mm Cae and represent the averages of four to seven experiments. B–D, Normalized EPSCs from LTP experiments (Figs. 1, 2) are plotted against Cainflux for single stimuli (B), peak Caglobal during the induction protocol (C), and the integral of Caglobal during the induction protocol (D). Open circles connected by lines, Manipulations of Cae; filled circles, blockade of specific calcium channels.
We determined whether our induction protocol preferentially increased calcium entry through R-type calcium channels, as is the case at MF synapses (Dietrich et al., 2003). In these experiments, we evoked trains in control conditions and measured the resulting Caglobal transients, applied a combination of CgTx and AgTx and again measured the Caglobal transients evoked by the train. Based on the insensitivity to CgTx and AgTx and the sensitivity to Ni2+ and SNX, the remaining component is dominated by calcium entry through R-type calcium channels. CgTx and AgTx substantially reduced the peak and plateau Caglobal signals during the train. We subtracted plateau values from peak values to obtain the magnitude of Cainflux for each stimulus in the train (Fig. 5C, bottom). In control conditions and in the presence of the toxins, there was a small enhancement of the Cainflux by the end of the train. The signal evoked by the last 5 stimuli in the train divided by the signal evoked by the first stimulus was 1.06 ± 0.04 and 1.17 ± 0.15 in control conditions and in the presence of CgTx and AgTx, respectively (n = 5). These findings suggest that there is a very small (11%) increase in the calcium entry through R-type calcium channels during the PF-LTP induction protocol.
Global calcium signaling does not control PF-LTP
The magnitudes of global presynaptic calcium signals (Caglobal) and LTP recorded under different conditions allowed us to assess the role of Caglobal in the induction of LTP. The effects of altering Cae and blocking specific types of calcium channels on Cainflux evoked by single action potentials (Fig. 4), and on the peak Caglobal and integral of Caglobal during the induction protocol (Fig. 5), were determined for a variety of experimental conditions (Fig. 6A). In some instances, there were differences in the effects of the manipulations on calcium signaling between single stimuli and the induction protocol. For example, CgTx had a larger effect on Cainflux evoked by single stimuli than on the peak Caglobal signal evoked by the induction protocol.
We tested whether the calcium dependence of PF-LTP could be explained by changes in Caglobal. Our fluorescence measurements of Caglobal reflect global calcium rather than Calocal (which is exceedingly difficult to measure). We plotted the normalized EPSCs for LTP experiments from Figures 1 and 2 as a function of Cainflux of single action potentials from Figure 4 (Fig. 6B), as a function of the peak Caglobal levels reached during the train (Fig. 6C), and as a function of the integral of Caglobal evoked by a train (Fig. 6D). If the extent of LTP is controlled by Caglobal, then the magnitude of LTP will depend only upon Caglobal and there will be a single function describing the relationship between synaptic enhancement and Caglobal. This was not the case (Fig. 6B–D). When calcium entry was reduced by lowering Cae there was a predicted relationship between plasticity and the global calcium signal (Fig. 6B–D, open circles and lines). When calcium entry through specific types of calcium channels was targeted, the extent of LTP did not match the predicted relationship. When P/Q-type calcium channels were blocked with AgTx, there was an extremely large reduction in Caglobal, but plasticity remained. When R-type calcium channels were targeted with SNX or Ni2+, there was a rather small effect on Caglobal, even though LTP was completely blocked (Fig. 6B–D). In the presence of Ni2+, increasing calcium influx through N-type and P/Q-type calcium channels by elevating Cae from 2 mm to 2.5 mm did not rescue LTP. Thus, the calcium dependence of LTP cannot be accounted for by global presynaptic calcium signaling. The lack of a role for N-type and P/Q-type calcium channels, combined with the crucial role for R-type calcium channels, suggests that calcium entry through R-type calcium channels must act locally to stimulate Ca-AC and ultimately induce PF-LTP.
The calcium dependence of LTP at near-physiological temperatures
To this point, as in previous studies of presynaptic PF-LTP (Salin et al., 1996; van Beugen et al., 2006), experiments were performed at 25°C. We went on to test the properties at presynaptic LTP at near-physiological temperatures (34 ± 1°C). At these temperatures, tetanic stimulation for 15 s at 8 Hz produced a large transient enhancement that lasted for several minutes, but 15–20 min following the tetanus the extent of the enhancement was small (7 ± 6% compared with −5 ± 6% for the control pathway, n = 9, p = 0.09, paired t test) (Fig. 7A). Prolonged tetanic stimulation (8 Hz, 30 s), as used to examine LTP at PF to stellate cell synapses (Bender et al., 2009), resulted in more pronounced enhancement (18 ± 10% compared with −5 ± 5% for the control pathway, n = 13, p < 0.05, paired t test) (Fig. 7B, top).
Figure 7.
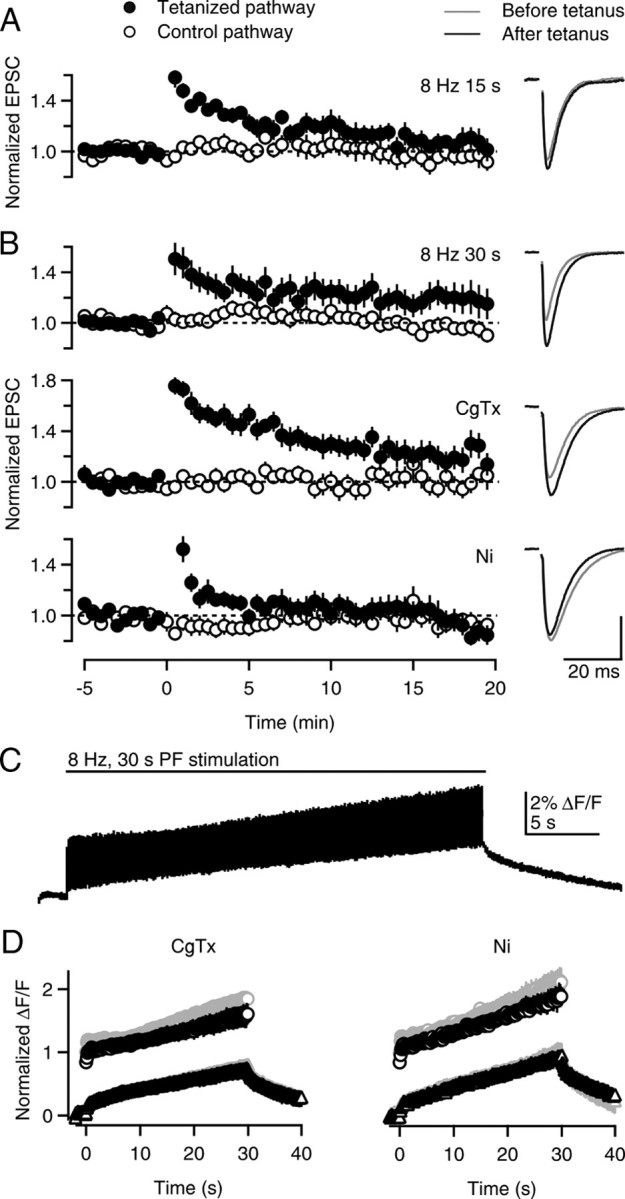
PF-LTP and presynaptic calcium signals at near-physiological temperatures. A, B, Time courses of normalized EPSCs (left) and example EPSC traces (right) are shown for LTP experiments conducted at 34 ± 1°C. A, LTP was induced with an 8 Hz, 15 s tetanus at t = 0. B, LTP was induced with an 8 Hz, 30 s tetanus at t = 0 in control conditions (top), in the presence of 500 nm ω-conotoxin GVIA (middle), or 100 μm NiCl2 (bottom). Vertical scale bar: A, 95 pA; B (from top to bottom), 293, 186, and 176 pA. C, An example Caglobal transient during an 8 Hz, 30 s tetanus is shown. D, Quantification of Caglobal peaks during the train (circles) and buildup of plateau calcium during the train (triangles) before (gray symbols) and after (black symbols) application of 500 nm ω-conotoxin GVIA (left) or 100 μm NiCl2 (right).
We next examined the roles of different types of calcium channels in LTP at near-physiological temperatures. We found that at elevated temperatures, LTP was intact in the presence of the N-type calcium channel blocker CgTx (22 ± 9% compared with 1 ± 9% for the control pathway, n = 12, p < 0.01, paired t test) (Fig. 7B, middle), but was absent in the presence of the R-type calcium channel antagonist Ni2+ (−5 ± 7% compared with −5 ± 6% for the control pathway, n = 13, p = 0.97, paired t test) (Fig. 7B, bottom). These results are consistent with R-type calcium channels but not N-type calcium channels playing a crucial role in LTP at 34 ± 1°C, as was observed at 25 ± 1°C. It was impractical to examine the role of P/Q-type calcium channels because the synaptic currents remaining in the presence of P/Q-type blockers were too small, and the stability of synaptic currents was inadequate at elevated temperatures.
We went on to examine the contributions of N-type and R-type calcium channels on Caglobal during the induction protocol to gain insight into the calcium dependence of LTP at elevated temperatures. An example trace shows a typical presynaptic Caglobal transient evoked by an 8 Hz, 30 s train in control conditions at near-physiological temperatures (Fig. 7C). Stimulus trains were applied first in control conditions and then the presence of a calcium channel antagonist (Fig. 7D). We found that the peak Caglobal levels reached in the presence of CgTx and Ni2+ relative to control conditions were 0.86 ± 0.08 (n = 5) and 0.89 ± 0.07 (n = 5), respectively. These results are summarized in Figure 7D, where the average peak Caglobal signals evoked during the train in the presence of calcium channel blockers are expressed relative to the amplitude of the Caglobal signal evoked by low-frequency stimulation in control conditions. In contrast, the magnitude of calcium signals evoked by the second of two trains in control conditions was unchanged (peak calcium levels at the end of the second train, 1.03 ± 0.06 of the amplitude at the end of the first train; n = 8; data not shown).
Together, these results indicate that the crucial role of R-type but not N-type calcium channels in the induction of LTP cannot be explained by Caglobal, because CgTx and Ni2+ have approximately the same effect on global presynaptic calcium signals. Thus, the basic finding that local calcium signaling through R-type calcium triggers PF-LTP holds at near-physiological temperatures.
Discussion
We found that, although most presynaptic calcium enters PFs through N-type and P/Q-type calcium channels, neither channel regulates the induction of PF-LTP under our experimental conditions. Instead, calcium entry through R-type calcium channels induces PF-LTP, even though these channels contribute a small fraction of overall calcium entry during the induction protocol. Modulation of global calcium signaling cannot account for the observed calcium dependence of PF-LTP. We conclude that R-type calcium channels act within presynaptic boutons to activate Ca-AC through functional calcium microdomains, thereby inducing PF-LTP.
The calcium sensitivity of PF-LTP
Experiments in which presynaptic calcium influx was systematically altered by changing Cae revealed that PF-LTP is steeply calcium-dependent. Reducing Cae from 1.5 to 1.25 mm decreased Cainflux through all calcium channels by just 10%, but prevented the induction of PF-LTP (Fig. 1B,C). This steep calcium dependence is compatible with the high cooperativity of the activation of Ca-AC by calmodulin, which has four calcium binding sites (Olwin et al., 1984; Iida and Potter, 1986).
Using an approach that has been used previously to distinguish between Calocal and Caglobal signaling (Yasuda et al., 2003; Berridge, 2006; Bloodgood and Sabatini, 2007), we established that calcium microdomains near R-type calcium channels play a crucial role in presynaptic PF-LTP. The relationship between the magnitude of synaptic enhancement and the magnitude of Caglobal indicated that regulation of global presynaptic calcium signaling does not explain the calcium dependence of PF-LTP. Blocking P/Q-type calcium channels with AgTx, which reduced Caglobal by almost 50%, did not block LTP. However, blocking R-type calcium channels with SNX, which only reduced Caglobal by 9%, completely blocked LTP. These findings indicate that LTP is evoked by calcium influx specifically through R-type calcium channels. Moreover, they establish that calcium-stimulation of AC1 and AC8 must reflect local calcium microdomains and suggest that R-type calcium channels and Ca-AC are closely associated within PF boutons.
Local calcium signaling and Ca-AC activation
A direct association between Ca-AC and R-type calcium channels has not been reported, but there is evidence that AC1 and AC8 can be functionally localized to sources of calcium influx that stimulate them (Crossthwaite et al., 2005; Hall et al., 2007; Piggott et al., 2008). In heterologous systems, AC1 and AC8 are stimulated by capacitative calcium entry, but not by calcium released from intracellular stores (Masada et al., 2009; Willoughby et al., 2010). AC1 and AC8 are also sensitive to calcium influx through voltage-gated calcium channels (Fagan et al., 2000), but the subtype specificity of this mechanism has not been investigated.
The manner in which a calcium microdomain produced by R-type calcium channels stimulates Ca-AC under physiological conditions is complex. Both AC1 and AC8 can be activated by modest, sustained increases in calcium (EC50 of 150–190 nm and 320–560 nm, respectively) (Cimler et al., 1985; Wayman et al., 1994). These values are substantially lower than what likely occurs at the pore of the channel (10–100 μm) (Fogelson and Zucker, 1985; Simon and Llinas, 1985; Augustine and Neher, 1992; Stern, 1992; Zucker and Regehr, 2002). However, it is unlikely that, in our studies, the transient elevations of Caglobal produced by the induction protocol are long enough to reach steady-state activation of Ca-AC. This is consistent with our observation that global calcium signals produced by such trains are not sufficient to explain the calcium dependence of PF-LTP (Fig. 6B–D). Instead, it appears that Calocal near R-type calcium channels is large enough to activate Ca-AC sufficiently to induce PF-LTP. The modest calcium requirement for activation of Ca-AC also suggests that R-type calcium channels and Ca-AC might not need to be particularly closely associated, and that a local calcium signal at ∼100 nm from an R-type calcium channel might be sufficient to activate Ca-AC (Fogelson and Zucker, 1985; Simon and Llinas, 1985; Augustine and Neher, 1992; Zucker and Regehr, 2002). The properties of R-type calcium channel microdomain activation that we report here for Ca-AC in presynaptic terminals is similar in many ways to the activation of SK channels in postsynaptic spines (Bloodgood and Sabatini, 2007). In both cases, the calcium sensor is calmodulin, activation is steeply calcium-dependent, and half-maximal steady-state activation occurs for calcium increases of several hundred nanomolar (Wu et al., 1993; Xia et al., 1998; Li and Aldrich, 2009).
R-type calcium channels and cAMP-dependent presynaptic LTP
Our finding that R-type calcium channels are involved in PF-LTP, along with previous studies establishing the involvement of R-type calcium channels in MF-LTP (Breustedt et al., 2003; Dietrich et al., 2003), suggests a general role for R-type calcium channels in cAMP-dependent presynaptic LTP. There are, however, important differences in the contributions of R-type calcium channels to synaptic transmission and presynaptic calcium signaling between MF and PF synapses. First, R-type calcium channels in MFs are not effectively opened by low-frequency stimulation and do not contribute to basal synaptic transmission (Dietrich et al., 2003; but see Gasparini et al., 2001), whereas R-type calcium channels in PFs contribute to low-frequency transmission with 2 < n < 3 (Fig. 4). Second, during the tetanic stimulation used to induce MF-LTP, presynaptic spike broadening occurs, which more effectively opens R-type calcium channels (Li et al., 2007). This allows R-type calcium channels in MFs to make a more substantial contribution to calcium entry by the end of the train compared with low-frequency stimulation. In contrast, Cainflux through R-type calcium channels in PFs is increased by just 11% by the end of the induction protocol.
Finally, MF-LTP is thought to be controlled by a Caglobal (Dietrich et al., 2003), whereas we find that PF-LTP is regulated by Calocal. The apparent differences in the roles of Caglobal and Calocal between MF and PF synapses may be a consequence of the stimulus frequency used to induce LTP (100 Hz at MF synapses vs 8 Hz at PF synapses). During high-frequency trains, Caglobal may be sufficient to activate Ca-AC and Calocal signals near open R-type calcium channels may not be essential. It may be that only moderate frequency induction protocols require local calcium increases to activate Ca-AC and induce LTP. This raises the possibility that Calocal might also be crucial to induce MF-LTP with moderate frequency induction trains.
Implications for the regulation of PF-LTP
G-protein-coupled receptors could regulate PF-LTP by inhibiting cAMP production either through direct actions on Ca-AC, or indirectly by reducing the calcium signal available to activate Ca-AC (van Beugen et al., 2006). Our findings, combined with our previous finding that CB1R activation reduces calcium entry through R-type calcium channels (Brown et al., 2004), suggest that the modulation of R-type calcium channels may contribute to the disruption of PF-LTP by activation of presynaptic CB1Rs. Moreover, it seems likely that any neuromodulator that inhibits presynaptic R-type calcium channels (Dittman and Regehr, 1996) will also regulate the induction of PF-LTP.
Regulation of presynaptic LTP by calcium microdomains
Our major finding is that local calcium signaling through microdomains near R-type calcium channels is important for presynaptic LTP. The fact that R-type calcium channels can control LTP postsynaptically (Stackman et al., 2002; Bond et al., 2004; Ngo-Anh et al., 2005; Hammond et al., 2006) and presynaptically as a consequence of local calcium interactions with calmodulin suggest a widespread role for R-type calcium channels in regulating calmodulin-sensitive cellular processes.
Footnotes
This work was supported by National Institutes of Health Grants DA024090 and R37NS032405 to W.G.R. and DA025450 to M.H.M. We thank M. Antal, A. Best, Y. Chu, J. Crowley, D. Fioravante, C. Hull, S. Jackman, T. Pressler, and M. Thanawala for valuable comments on this manuscript.
References
- Alle H, Jonas P, Geiger JR. PTP and LTP at a hippocampal mossy fiber-interneuron synapse. Proc Natl Acad Sci U S A. 2001;98:14708–14713. doi: 10.1073/pnas.251610898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustine GJ, Neher E. Neuronal Ca2+ signalling takes the local route. Curr Opin Neurobiol. 1992;2:302–307. doi: 10.1016/0959-4388(92)90119-6. [DOI] [PubMed] [Google Scholar]
- Beierlein M, Fioravante D, Regehr WG. Differential expression of posttetanic potentiation and retrograde signaling mediate target-dependent short-term synaptic plasticity. Neuron. 2007;54:949–959. doi: 10.1016/j.neuron.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender VA, Pugh JR, Jahr CE. Presynaptically expressed long-term potentiation increases multivesicular release at parallel fiber synapses. J Neurosci. 2009;29:10974–10978. doi: 10.1523/JNEUROSCI.2123-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ. Calcium microdomains: organization and function. Cell Calcium. 2006;40:405–412. doi: 10.1016/j.ceca.2006.09.002. [DOI] [PubMed] [Google Scholar]
- Bloodgood BL, Sabatini BL. Nonlinear regulation of unitary synaptic signals by CaV(2.3) voltage-sensitive calcium channels located in dendritic spines. Neuron. 2007;53:249–260. doi: 10.1016/j.neuron.2006.12.017. [DOI] [PubMed] [Google Scholar]
- Bond CT, Herson PS, Strassmaier T, Hammond R, Stackman R, Maylie J, Adelman JP. Small conductance Ca2+-activated K+ channel knock-out mice reveal the identity of calcium-dependent afterhyperpolarization currents. J Neurosci. 2004;24:5301–5306. doi: 10.1523/JNEUROSCI.0182-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breustedt J, Vogt KE, Miller RJ, Nicoll RA, Schmitz D. Alpha1E-containing Ca2+ channels are involved in synaptic plasticity. Proc Natl Acad Sci U S A. 2003;100:12450–12455. doi: 10.1073/pnas.2035117100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown SP, Safo PK, Regehr WG. Endocannabinoids inhibit transmission at granule cell to Purkinje cell synapses by modulating three types of presynaptic calcium channels. J Neurosci. 2004;24:5623–5631. doi: 10.1523/JNEUROSCI.0918-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo PE, Weisskopf MG, Nicoll RA. The role of Ca2+ channels in hippocampal mossy fiber synaptic transmission and long-term potentiation. Neuron. 1994;12:261–269. doi: 10.1016/0896-6273(94)90269-0. [DOI] [PubMed] [Google Scholar]
- Chen C, Regehr WG. The mechanism of cAMP-mediated enhancement at a cerebellar synapse. J Neurosci. 1997;17:8687–8694. doi: 10.1523/JNEUROSCI.17-22-08687.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow KY, Wu C, Sui GP, Fry CH. Role of the T-type Ca2+ current on the contractile performance of guinea pig detrusor smooth muscle. Neurourol Urodyn. 2003;22:77–82. doi: 10.1002/nau.10081. [DOI] [PubMed] [Google Scholar]
- Cimler BM, Andreasen TJ, Andreasen KI, Storm DR. P-57 is a neural specific calmodulin-binding protein. J Biol Chem. 1985;260:10784–10788. [PubMed] [Google Scholar]
- Crossthwaite AJ, Seebacher T, Masada N, Ciruela A, Dufraux K, Schultz JE, Cooper DM. The cytosolic domains of Ca2+-sensitive adenylyl cyclases dictate their targeting to plasma membrane lipid rafts. J Biol Chem. 2005;280:6380–6391. doi: 10.1074/jbc.M411987200. [DOI] [PubMed] [Google Scholar]
- Dietrich D, Kirschstein T, Kukley M, Pereverzev A, von der Brelie C, Schneider T, Beck H. Functional specialization of presynaptic Cav2.3 Ca2+ channels. Neuron. 2003;39:483–496. doi: 10.1016/s0896-6273(03)00430-6. [DOI] [PubMed] [Google Scholar]
- Dittman JS, Regehr WG. Contributions of calcium-dependent and calcium-independent mechanisms to presynaptic inhibition at a cerebellar synapse. J Neurosci. 1996;16:1623–1633. doi: 10.1523/JNEUROSCI.16-05-01623.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan KA, Graf RA, Tolman S, Schaack J, Cooper DM. Regulation of a Ca2+-sensitive adenylyl cyclase in an excitable cell: role of voltage-gated versus capacitative Ca2+ entry. J Biol Chem. 2000;275:40187–40194. doi: 10.1074/jbc.M006606200. [DOI] [PubMed] [Google Scholar]
- Fakler B, Adelman JP. Control of K(Ca) channels by calcium nano/microdomains. Neuron. 2008;59:873–881. doi: 10.1016/j.neuron.2008.09.001. [DOI] [PubMed] [Google Scholar]
- Fogelson AL, Zucker RS. Presynaptic calcium diffusion from various arrays of single channels: implications for transmitter release and synaptic facilitation. Biophys J. 1985;48:1003–1017. doi: 10.1016/S0006-3495(85)83863-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fourcaudot E, Gambino F, Humeau Y, Casassus G, Shaban H, Poulain B, Lüthi A. cAMP/PKA signaling and RIM1alpha mediate presynaptic LTP in the lateral amygdala. Proc Natl Acad Sci U S A. 2008;105:15130–15135. doi: 10.1073/pnas.0806938105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasparini S, Kasyanov AM, Pietrobon D, Voronin LL, Cherubini E. Presynaptic R-type calcium channels contribute to fast excitatory synaptic transmission in the rat hippocampus. J Neurosci. 2001;21:8715–8721. doi: 10.1523/JNEUROSCI.21-22-08715.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall DD, Davare MA, Shi M, Allen ML, Weisenhaus M, McKnight GS, Hell JW. Critical role of cAMP-dependent protein kinase anchoring to the L-type calcium channel Cav1.2 via A-kinase anchor protein 150 in neurons. Biochemistry. 2007;46:1635–1646. doi: 10.1021/bi062217x. [DOI] [PubMed] [Google Scholar]
- Hammond RS, Bond CT, Strassmaier T, Ngo-Anh TJ, Adelman JP, Maylie J, Stackman RW. Small-conductance Ca2+-activated K+ channel type 2 (SK2) modulates hippocampal learning, memory, and synaptic plasticity. J Neurosci. 2006;26:1844–1853. doi: 10.1523/JNEUROSCI.4106-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higley MJ, Sabatini BL. Calcium signaling in dendrites and spines: practical and functional considerations. Neuron. 2008;59:902–913. doi: 10.1016/j.neuron.2008.08.020. [DOI] [PubMed] [Google Scholar]
- Iida S, Potter JD. Calcium binding to calmodulin: cooperativity of the calcium-binding sites. J Biochem. 1986;99:1765–1772. doi: 10.1093/oxfordjournals.jbchem.a135654. [DOI] [PubMed] [Google Scholar]
- Kaeser PS, Kwon HB, Blundell J, Chevaleyre V, Morishita W, Malenka RC, Powell CM, Castillo PE, Südhof TC. RIM1alpha phosphorylation at serine-413 by protein kinase A is not required for presynaptic long-term plasticity or learning. Proc Natl Acad Sci U S A. 2008;105:14680–14685. doi: 10.1073/pnas.0806679105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Gomora JC, Cribbs LL, Perez-Reyes E. Nickel block of three cloned T-type calcium channels: low concentrations selectively block alpha1H. Biophys J. 1999;77:3034–3042. doi: 10.1016/S0006-3495(99)77134-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Bischofberger J, Jonas P. Differential gating and recruitment of P/Q-, N-, and R-type Ca2+ channels in hippocampal mossy fiber boutons. J Neurosci. 2007;27:13420–13429. doi: 10.1523/JNEUROSCI.1709-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Aldrich RW. Activation of the SK potassium channel-calmodulin complex by nanomolar concentrations of terbium. Proc Natl Acad Sci U S A. 2009;106:1075–1080. doi: 10.1073/pnas.0812008106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HN, Kurotani T, Ren M, Yamada K, Yoshimura Y, Komatsu Y. Presynaptic activity and Ca2+ entry are required for the maintenance of NMDA receptor-independent LTP at visual cortical excitatory synapses. J Neurophysiol. 2004;92:1077–1087. doi: 10.1152/jn.00602.2003. [DOI] [PubMed] [Google Scholar]
- Lonart G, Schoch S, Kaeser PS, Larkin CJ, Südhof TC, Linden DJ. Phosphorylation of RIM1alpha by PKA triggers presynaptic long-term potentiation at cerebellar parallel fiber synapses. Cell. 2003;115:49–60. doi: 10.1016/s0092-8674(03)00727-x. [DOI] [PubMed] [Google Scholar]
- Maffei A, Prestori F, Rossi P, Taglietti V, D'Angelo E. Presynaptic current changes at the mossy fiber-granule cell synapse of cerebellum during LTP. J Neurophysiol. 2002;88:627–638. doi: 10.1152/jn.2002.88.2.627. [DOI] [PubMed] [Google Scholar]
- Masada N, Ciruela A, Macdougall DA, Cooper DM. Distinct mechanisms of regulation by Ca2+/calmodulin of type 1 and 8 adenylyl cyclases support their different physiological roles. J Biol Chem. 2009;284:4451–4463. doi: 10.1074/jbc.M807359200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mintz IM, Sabatini BL, Regehr WG. Calcium control of transmitter release at a cerebellar synapse. Neuron. 1995;15:675–688. doi: 10.1016/0896-6273(95)90155-8. [DOI] [PubMed] [Google Scholar]
- Newcomb R, Szoke B, Palma A, Wang G, Chen X, Hopkins W, Cong R, Miller J, Urge L, Tarczy-Hornoch K, Loo JA, Dooley DJ, Nadasdi L, Tsien RW, Lemos J, Miljanich G. Selective peptide antagonist of the class E calcium channel from the venom of the tarantula Hysterocrates gigas. Biochemistry. 1998;37:15353–15362. doi: 10.1021/bi981255g. [DOI] [PubMed] [Google Scholar]
- Ngo-Anh TJ, Bloodgood BL, Lin M, Sabatini BL, Maylie J, Adelman JP. SK channels and NMDA receptors form a Ca2+-mediated feedback loop in dendritic spines. Nat Neurosci. 2005;8:642–649. doi: 10.1038/nn1449. [DOI] [PubMed] [Google Scholar]
- Nicoll RA, Schmitz D. Synaptic plasticity at hippocampal mossy fibre synapses. Nat Rev Neurosci. 2005;6:863–876. doi: 10.1038/nrn1786. [DOI] [PubMed] [Google Scholar]
- Obejero-Paz CA, Gray IP, Jones SW. Ni2+ block of CaV3.1 (alpha1G) T-type calcium channels. J Gen Physiol. 2008;132:239–250. doi: 10.1085/jgp.200809988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olwin BB, Edelman AM, Krebs EG, Storm DR. Quantitation of energy coupling between Ca2+, calmodulin, skeletal muscle myosin light chain kinase, and kinase substrates. J Biol Chem. 1984;259:10949–10955. [PubMed] [Google Scholar]
- Piggott LA, Bauman AL, Scott JD, Dessauer CW. The A-kinase anchoring protein Yotiao binds and regulates adenylyl cyclase in brain. Proc Natl Acad Sci U S A. 2008;105:13835–13840. doi: 10.1073/pnas.0712100105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall A, Tsien RW. Pharmacological dissection of multiple types of Ca2+ channel currents in rat cerebellar granule neurons. J Neurosci. 1995;15:2995–3012. doi: 10.1523/JNEUROSCI.15-04-02995.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regehr WG, Atluri PP. Calcium transients in cerebellar granule cell presynaptic terminals. Biophys J. 1995;68:2156–2170. doi: 10.1016/S0006-3495(95)80398-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regehr WG, Tank DW. Selective fura-2 loading of presynaptic terminals and nerve cell processes by local perfusion in mammalian brain slice. J Neurosci Methods. 1991;37:111–119. doi: 10.1016/0165-0270(91)90121-f. [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Pozzan T. Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol Rev. 2006;86:369–408. doi: 10.1152/physrev.00004.2005. [DOI] [PubMed] [Google Scholar]
- Sabatini BL, Regehr WG. Detecting changes in calcium influx which contribute to synaptic modulation in mammalian brain slice. Neuropharmacology. 1995;34:1453–1467. doi: 10.1016/0028-3908(95)00129-t. [DOI] [PubMed] [Google Scholar]
- Sabatini BL, Regehr WG. Optical measurement of presynaptic calcium currents. Biophys J. 1998;74:1549–1563. doi: 10.1016/S0006-3495(98)77867-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salin PA, Malenka RC, Nicoll RA. Cyclic AMP mediates a presynaptic form of LTP at cerebellar parallel fiber synapses. Neuron. 1996;16:797–803. doi: 10.1016/s0896-6273(00)80099-9. [DOI] [PubMed] [Google Scholar]
- Shan Q, Chan GC, Storm DR. Type 1 adenylyl cyclase is essential for maintenance of remote contextual fear memory. J Neurosci. 2008;28:12864–12867. doi: 10.1523/JNEUROSCI.2413-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon SM, Llinás RR. Compartmentalization of the submembrane calcium activity during calcium influx and its significance in transmitter release. Biophys J. 1985;48:485–498. doi: 10.1016/S0006-3495(85)83804-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stackman RW, Hammond RS, Linardatos E, Gerlach A, Maylie J, Adelman JP, Tzounopoulos T. Small conductance Ca2+-activated K+ channels modulate synaptic plasticity and memory encoding. J Neurosci. 2002;22:10163–10171. doi: 10.1523/JNEUROSCI.22-23-10163.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern MD. Buffering of calcium in the vicinity of a channel pore. Cell Calcium. 1992;13:183–192. doi: 10.1016/0143-4160(92)90046-u. [DOI] [PubMed] [Google Scholar]
- Storm DR, Hansel C, Hacker B, Parent A, Linden DJ. Impaired cerebellar long-term potentiation in type I adenylyl cyclase mutant mice. Neuron. 1998;20:1199–1210. doi: 10.1016/s0896-6273(00)80500-0. [DOI] [PubMed] [Google Scholar]
- van Beugen BJ, Nagaraja RY, Hansel C. Climbing fiber-evoked endocannabinoid signaling heterosynaptically suppresses presynaptic cerebellar long-term potentiation. J Neurosci. 2006;26:8289–8294. doi: 10.1523/JNEUROSCI.0805-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Pineda VV, Chan GC, Wong ST, Muglia LJ, Storm DR. Type 8 adenylyl cyclase is targeted to excitatory synapses and required for mossy fiber long-term potentiation. J Neurosci. 2003;23:9710–9718. doi: 10.1523/JNEUROSCI.23-30-09710.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Chen G, Gao W, Ebner T. Long-term potentiation of the responses to parallel fiber stimulation in mouse cerebellar cortex in vivo. Neuroscience. 2009;162:713–722. doi: 10.1016/j.neuroscience.2009.01.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayman GA, Impey S, Wu Z, Kindsvogel W, Prichard L, Storm DR. Synergistic activation of the type I adenylyl cyclase by Ca2+ and Gs-coupled receptors in vivo. J Biol Chem. 1994;269:25400–25405. [PubMed] [Google Scholar]
- Willoughby D, Wachten S, Masada N, Cooper DM. Direct demonstration of discrete Ca2+ microdomains associated with different isoforms of adenylyl cyclase. J Cell Sci. 2010;123:107–117. doi: 10.1242/jcs.062067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong ST, Athos J, Figueroa XA, Pineda VV, Schaefer ML, Chavkin CC, Muglia LJ, Storm DR. Calcium-stimulated adenylyl cyclase activity is critical for hippocampus-dependent long-term memory and late phase LTP. Neuron. 1999;23:787–798. doi: 10.1016/s0896-6273(01)80036-2. [DOI] [PubMed] [Google Scholar]
- Wu Z, Wong ST, Storms DR. Modification of the calcium and calmodulin sensitivity of the type I adenylyl cyclase by mutagenesis of its calmodulin binding domain. J Biol Chem. 1993;268:23766–23768. [PubMed] [Google Scholar]
- Wu ZL, Thomas SA, Villacres EC, Xia Z, Simmons ML, Chavkin C, Palmiter RD, Storm DR. Altered behavior and long-term potentiation in type I adenylyl cyclase mutant mice. Proc Natl Acad Sci U S A. 1995;92:220–224. doi: 10.1073/pnas.92.1.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia XM, Fakler B, Rivard A, Wayman G, Johnson-Pais T, Keen JE, Ishii T, Hirschberg B, Bond CT, Lutsenko S, Maylie J, Adelman JP. Mechanism of calcium gating in small-conductance calcium-activated potassium channels. Nature. 1998;395:503–507. doi: 10.1038/26758. [DOI] [PubMed] [Google Scholar]
- Yasuda R, Sabatini BL, Svoboda K. Plasticity of calcium channels in dendritic spines. Nat Neurosci. 2003;6:948–955. doi: 10.1038/nn1112. [DOI] [PubMed] [Google Scholar]
- Yuste R, Konnerth A. Imaging in neuroscience and development: a laboratory manual. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory; 2005. pp. 307–314. [Google Scholar]
- Zalutsky RA, Nicoll RA. Comparison of two forms of long-term potentiation in single hippocampal neurons. Science. 1990;248:1619–1624. doi: 10.1126/science.2114039. [DOI] [PubMed] [Google Scholar]
- Zhang M, Moon C, Chan GC, Yang L, Zheng F, Conti AC, Muglia L, Muglia LJ, Storm DR, Wang H. Ca-stimulated type 8 adenylyl cyclase is required for rapid acquisition of novel spatial information and for working/episodic-like memory. J Neurosci. 2008;28:4736–4744. doi: 10.1523/JNEUROSCI.1177-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]