

Abstract
We report comprehensive structure activity relationship studies on a novel series of c-Jun N-terminal kinase (JNK) inhibitors. Intriguingly, the compounds have a dual inhibitory activity by functioning as both ATP and JIP mimetics, possibly by binding to both the ATP binding site and to the docking site of the kinase. Several of such novel compounds display potent JNK inhibitory profiles both in vitro and in cell.
The c-Jun N-terminal kinases (JNKs) have been originally identified in the early 1990s as a family of serine/threonine protein kinases that are activated by a range of stimuli. The activation of each of the JNK genes results in the phosphorylation of the N-terminal transactivation domain of the c-Jun transcription factor.1–4 Three JNK isoforms (JNK1, 2 and 3) share more than 90% amino acid sequence identity and the ATP pocket is highly conserved (>98% identities). These proteins are often activated in response to a large variety of cellular stresses including irradiation, hypoxia, peroxides, heat shock, and chemotoxins as well as various cytokines, thus participating in the onset of apoptosis.5,6 It has been clearly established that excessive up-regulation of JNK activity results or is associated with a number of human disorders including type-2 diabetes and obesity, neurodegeneration and stroke, cancer and inflammation.1–3 Hence, JNK inhibitors are expected to be viable agents to devise novel therapies against these diseases, and there have been large efforts in identifying small molecule JNK inhibitors targeting its ATP binding site.7–13
Peculiar to JNKs substrates and scaffold proteins, is a JNK interacting conserved consensus sequence R/KXXXXLXL termed the D-domain.14,15 A short peptide corresponding to the D-domain of the scaffolding protein JIP-1 (aa 153–163; pep-JIP1) has been shown to inhibit JNK activity in vitro, displaying noteworthy selectivity with little inhibition of the closely related Erk and p38 MAPKs.16–19 Furthermore, recent in vivo data, generated for studies focusing on pep-JIP1 fused to the cell permeable HIV-TAT peptide, show that its administration in various mice models of insulin resistance and type-2 diabetes restores normoglycemia without causing hypoglycemia.20 Despite these encouraging data, peptide’s instability in vivo may hamper the development of novel JNK-related therapies based on such peptides.16–20
Based on these premises, a drug discovery program in our laboratory was initiated with the aim of identifying and characterizing small molecule JNK inhibitors as novel chemical entities targeting its JIP binding site rather than the highly conserved ATP binding site of the protein. Very recently, we have reported the identification of 5-(5-nitrothiazol-2-ylthio)-1,3,4-thiadiazol-2-amine series21 related to compound BI-78D322 (Figure 1), as initial JIP mimetic inhibitors. These compounds were discovered using a displacement assay with a biotinylated-pepJIP1 peptide and employing a DELFIA assay platform in a medium size screening campaign.22 In our continued interest in the development of JNK inhibitors21–23 we now report further structure-activity relationship studies describing novel small molecules thiophene-carboxamide derivatives as JNK inhibitors targeting its JIP/substrate docking site. Intriguingly, we believe that the compounds are also able to function as ATP mimetics for JNK, which makes them particularly interesting. The 4,5-dimethyl-2-(2-(naphthalen-1-yl)acetamido)thiophene-3-carboxamide (1, Figure 1) was qualified as a hit and became the starting point of our medicinal chemistry efforts, with an IC50 value for the displacement of pepJIP1 in the DELFIA assay of 15.8 μM, inhibiting JNK1 kinase activity in the Lantha assay platform with an IC50 value of 26.0 μM. To investigate the effects on potency induced by small changes in the structure of 1, we developed the general synthetic route for the preparation of this series. A variety of commercially available 2-aryl acetic acids were treated with aryl 2-amino-3-carboxamides in the presence of EDC at room temperature to give 5a–5g and 11–74 (Schemes 1, 2, and 3) in moderate to good yields. Replacement of the thiophene moiety with a phenyl ring led to compound 3 that showed a drastic drop in activity (IC50 > 100 μM), similarly replacing the 3-carboxamide group on the thiophene with an acid, resulting in compound 5a, or an ester, resulting in compound 5b, or a cyano group, as in compound 5c, also resulted in a significant loss of JNK1 inhibitory activity (Table 1). The position of carboxamide is also important for JNK1 inhibitory activity as the analogue with the carboxamide at the 5-position on the thiophene (compound 5f) was completely inactive. The 4-methyl (5d) or 5-methyl (5e) or 4,5-dimethyl substitutions on the thiophene of compound 1 also resulted in less active compounds (IC50 > 25 μM), compared to the un-substituted compound (5g, IC50 = 5.4 μM). Therefore, we retained 4 and 5-positions unsubstituted and carboxamide on the 3-position on the thiophene, and explored modifications at the 2-position. We observed that introducing substituents with one carbon linker did not affect the inhibitory properties of the series (i.e. compound 7, IC50 = 3.6 μM versus compound 8, no linker, IC50 = 5.9 μM), while longer chains (i.e. compound 9 with a 2-carbon linker, IC50 > 100 μM, or compound 10 with a trans-2-carbon linker, IC50 > 100 μM) are not tolerated (Table 1). Based on these observations, we synthesized additional analogs of compound 7 with a variety of aryl or heteroaryl substitutions (Scheme 3). The mono fluoro or difluoro substitutions (compounds 29, 30, 31, 52, 53, 54, 55, 56, and 71) on the benzene ring were well tolerated (IC50 = 8.3 μM, 9.4 μM, 5.1 μM, 8.2 μM, 10.2 μM, 9.7 μM, 7.4 μM, 5.8 μM, 4.9 μM respectively) but the penta-fluoro benzene, compound 60, was inactive (IC50 > 50 μM). Similarly, a chlorine on positions 2 and 3 on the benzene ring resulted in compounds with good inhibitory activities (compound 26, IC50 = 1.4 μM and compound 27, IC50 = 2.6 μM). However a chlorine (28, IC50 = 18.7 μM) or larger substituents such as bromine (34, IC50 > 25 μM), isopropyl (50, IC50 > 25 μM), phenyl (62, IC50 > 50 μM), and isobutyl (20, IC50 > 25 μM) in the 4-position on the benzene ring were not tolerated, whereas similar substitutions in positions 2 or 3 resulted in compounds with good inhibitory properties against JNK1. Furthermore, introducing potential hydrogen bonding acceptors such as methoxy (compound 37, IC50 = 11.9 μM, compound 38, IC50 = 2.6 μM and compound 39, IC50 = 5.7 μM), methylene dioxy (compound 19, IC50 = 1.8 μM), and benzodioxane (compound 18 IC50 = 2.7 μM) groups on the benzene ring resulted in compounds with significant JNK1 inhibition while other polar groups, such as a morpholine or a piperazine (compounds 64, 65, 67, and 68) resulted in compounds with poor inhibitory activities (IC50 > 50 μM), except for compound 66 in which some activity was retained (IC50 = 7.6 μM). Finally, we found that replacing the benzene with a benzothiophene (compound 25, Figure 1) resulted in a significant improvement with respect to both JNK1 kinase activity inhibition and JNK1/pepJIP displacement.
Figure 1.

Previously reported key JIP1 site binding JNK inhibitor (BI-78D3) and thiophene 3-carboxamide inhibitors indentified via HTS (1 and 25).
Scheme 1.

Reagents and conditions: (a) 2-(1-Naphthyl) acetic acid, EDC, HOBt, DIEA, DMF, room temp.
Scheme 2.

Reagents and conditions: (a) phenyl acetic acid, EDC, HOBt, DIEA, DMF, room temp; (b) benzoic acid, EDC, HOBt, DIEA, DMF, room temp.; (c) 3-phenylpropanoic acid, EDC, HOBt, DIEA, DMF, room temp.; (d) trans cinnamic acid, EDC, HOBt, DIEA, DMF, room temp.
Scheme 3.

Reagents and conditions: (a) 2-aryl-acetic acids, EDC, HOBT, DIEA, DMF, room temp.
Table 1.
Inhibition results for thiophene carboxamide series against JNK-1.
| Compd. | Kinase activity assay IC50a(μM) | Compd. | Kinase activity assay IC50a(μM) | Compd. | Kinase activity assay IC50a(μM) |
|---|---|---|---|---|---|
| 1 | 26.0 | 24 | >25 | 50 | >25 |
| 3 | >100 | 25 | 1.3 | 51 | 1.9 |
| 5a | >100 | 26 | 1.4 | 52 | 8.2 |
| 5b | >100 | 27 | 2.6 | 53 | 10.2 |
| 5c | >100 | 28 | 18.7 | 54 | 9.7 |
| 5d | >25 | 29 | 8.3 | 55 | 7.4 |
| 5e | >25 | 30 | 9.4 | 56 | 5.8 |
| 5f | >100 | 31 | 5.1 | 57 | 12.5 |
| 5g | 5.4 | 32 | 2.9 | 58 | >25 |
| 7 | 3.6 | 33 | 1.8 | 59 | 3.1 |
| 8 | 5.9 | 34 | >25 | 60 | >50 |
| 9 | >100 | 35 | 3.4 | 61 | 8.1 |
| 10 | >100 | 36 | 1.8 | 62 | >50 |
| 11 | >25 | 37 | 11.9 | 63 | 9.4 |
| 12 | >25 | 38 | 2.6 | 64 | >50 |
| 13 | >25 | 39 | 5.7 | 65 | 65.3 |
| 14 | 3.2 | 40 | 6.0 | 66 | 7.6 |
| 15 | 2.7 | 41 | 5.4 | 67 | 83.5 |
| 16 | >50 | 42 | 4.3 | 68 | 75.1 |
| 17 | >25 | 43 | >25 | 69 | 3.6 |
| 18 | 2.7 | 44 | 2.9 | 70 | 2.5 |
| 19 | 1.8 | 45 | 7.6 | 71 | 4.9 |
| 20 | >25 | 46 | 6.4 | 72 | 2.5 |
| 21 | >25 | 47 | 5.8 | 73 | 1.9 |
| 22 | 2.30 | 48 | 20.9 | 74 | 2.5 |
| 23 | >25 | 49 | 3.4 |
Values are means of at least three or more experiments. Standard deviation generally within ±25%
Subsequently, we performed dose response measurements and enzyme kinetics assays with compound 25. Compound 25 showed an IC50 of 1.32 μM in the kinase assay (Figure 2) and an IC50 of 4.62 μM in displacing pepJIP1, as measured by DELFIA assay, that indicates that the compound may bind directly at JIP site (Figure 2). However, enzyme kinetics experiments suggested that compound 25 is an ATP competitive inhibitor as well as a mixed substrate competitive inhibitor (Figure 2). This is quite intriguing, suggesting that the compound may bind independently to both the ATP and the JIP binding sites on JNK. Interestingly, our findings are similar to what recently reported by Chen et al.24 that have identified a similar dual JIP site and ATP site JNK inhibitor based on kinetic experiments.
Figure 2.

Dose response curves for the displacement of pepJIP1 from JNK1 by compound 25 in the DELFIA assay (A), and for the inhibition of JNK1 kinase activity in the LANTHA assay by compound 25 (B). Lineweaver-Burk analyses for the inhibition of JNK1 kinase activity (LANTHA assay) by compound 25 with increasing substrate ATF2 (C) or ATP (D) concentrations. Data were collected in triplicates.
Hence, to determine the selectivity, compound 25 was tested against a panel of 26 kinases. Compound 25 was found to be nearly inactive (Table 2) against p38α (2% inhibition at 25 μM), a member of the MAPK family with high structural similarity to JNK (52% sequence identity, considering the kinase domains), and inactive against other MAP kinases and lipid kinases (Table 2), further corroborating that these compounds may elicit their activity by binding not only to the highly conserved ATP site but also the JNK docking site. This compound showed a preferential inhibition of JNK1 and JNK2 over JNK3 (Table 2), in agreement with our previous findings with compound BI-78D322 (Figure 1) and with the reported data on pepJIP1.16–20
Table 2.
| Kinase | % inhibition at 25 μM | Kinase | % inhibition at 25 μM | Kinase | % inhibition at 25 μM |
|---|---|---|---|---|---|
| JNK1 | 79%c | BRAF | 2% | MAPK14 (p-38α) | 11% |
| JNK2 | 72%c | CAMK1 | 2% | NEK1 | 0% |
| JNK3 | 18%c | CDK1 | 3% | PDK1 direct | 0% |
| MAPK14 (p-38α, direct) | 2% | CHEK1 (CHK1) | 0% | PI3Kα | 4% |
| MAPK3 (ERK1) | 3% | CHUK (IIKα) | 0% | PIM1 | 0% |
| MAPK1 (ERK2) | 4% | FRAP1 (mTOR) | 0% | PRKCA (PKCα) | 0% |
| AKT1 | 5% | GSK3α | 16% | ROCK1 | 0% |
| AMPK A1 | 0% | MAK2K1 (MEK1) | 0% | SPHK1 | 1% |
| AURK A1 | 0% | MAPK3K9 (MLK1) | 0% | MAP4K2 (GCK) | 1% |
From Invitrogen (Life Technologies) kinase profiling service;
Values are means of at least two independent experiments;
Z’-LYTE assay.
Finally, to assess the activity of the compounds in cell we employed a cell-based LanthaScreen™ kinase assay. In this assay, compound 25 is able to inhibit TNF-α stimulated phosphorylation of c-Jun with an IC50 value of 7.5 μM (Supporting Information), which parallels well the in vitro activity and previous findings with this type of assay and other JNK inhibitors.22
To shed light into the possible binding mode of these compounds, we performed molecular modeling studies,25–27 using the ATP and JIP binding pockets derived from the X-ray crystal structure of the ternary complex including JNK1, pepJIP1, and the ATP mimic SP600125 (PDB ID 1UKI). From the docked binding pose, compounds of this series, represented by compound 25 (Figure 3) appeared to be deeply inserted in the ATP binding site and further stabilized by hydrogen-bonding interactions involving its carboxamide NH2 and side-chain atoms of the residue Gln 37. The docked poses of compounds 18, 19 and 38 suggest the presence of additional hydrogen bonding interactions with the backbone amide of Met 111 in the ATP binding site, providing a justification for the increased inhibitory properties for these compounds (Supporting Information).
Figure 3.
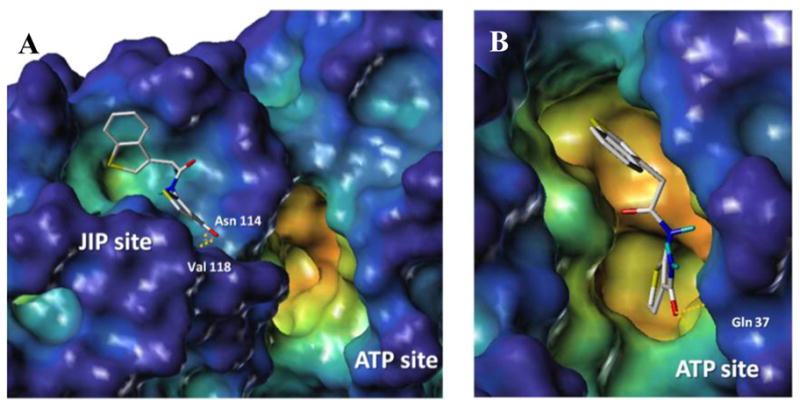
Docking studies of compound 25 within the JIP (A) and ATP (B) binding sites of JNK1 (PDB ID 1UKI). The surface was generated with MOLCAD27 and color coded according to cavity depth (blue, shallow; yellow, deep). Predicted hydrogen bonding interactions are highlighted with yellow dashed lines.
Furthermore, the JIP1 docking site seemed also able to potentially accommodate compounds of this series, as shown for compound 25 (Figure 3) through hydrophobic contacts and hydrogen bonding interactions between the carboxamide –CO– and –NH2 groups with the backbone NH of Val 118 and the backbone carbonyl of Asn 114, respectively. Hence, both binding modes would reflect the observed SAR including the direct ability of the compounds to displace the pepJIP1/JNK interactions in the DELFIA assay, suggesting that the dual inhibition is possible also on a theoretical basis.
To further corroborate this hypothesis, we conducted isothermal titration calorimetry experiments to monitor the thermodynamics of bindings of compound 25 to JNK2 in presence and absence of saturating concentrations of either a non-hydrolizable ATP or of pepJIP1. These studies revealed indeed that compound 25 binds to JNK2 with a Kd value of 640 nM with a 2:1 ligand to protein stoichiometry, and that the binding decreases significantly in presence of either the non-hydrolizable ATP or pepJIP1. The measured Kd values for compound 25 to JNK1 under these saturating conditions were 1.1 μM (in presence of ATP) and 3.1 μM (in presence of pepJIP1), both with a 1:1 ligand to protein stoichiometry.
Finally, to assess basic pharmacological properties of the proposed thiophene carboxamide derivatives, we determined in vitro plasma and microsomal stability for selected compounds. Compounds 25, 33, and 26 remained basically unaltered after 60 min of incubation in rat plasma while were degraded 85% (T1/2 18 min), 74% (T1/2 24 min), and 70% (T1/2 27 min) respectively, after 60 min of incubation in rat microsomal preparations, data suggesting that the molecules might be suitable for further optimizations and in vivo studies. Thiophenes are considered potential toxicophores due to their metabolites that can causehepatotoxicity, immunotoxicity and nephrotoxicity. 28, 29,30 However, di-substitutions or ring deactivation by addition of adjacent functionalities, have been proven to minimize metabolic oxidation hence reducing toxicity. 28,30 Hence, the observed relatively long microsomal half-lives of these compounds can be attributed to the presence of di-substitutions on the thiophene ring. Finally, two very recent reports, describing the pharmacology of other thiophene-based JNK inhibitors, albeit with rather different structures form those reported herein, corroborated the present observations 31,32.
In summary, we report data on a series of novel JNK inhibitors that resulted in the selection of compound 25, that apparently inhibits JNK by possibly binding to both the ATP and JIP binding site, independently. Because the compound possesses a good balance of potency, selectivity, solubility, metabolic stability, and cellular activity, we anticipate that this series may hold promise for further development of novel JNK inhibitors as chemical probes and eventually for therapeutic purposes.
Methods
Synthetic chemistry
Unless otherwise indicated, all anhydrous solvents were commercially obtained and stored in Sure-seal bottles under nitrogen. All other reagents and solvents were purchased as the highest grade available and used without further purification. Thin-layer chromatography (TLC) analysis of reaction mixtures was performed using Merck silica gel 60 F254 TLC plates, and visualized using ultraviolet light. NMR spectra were recorded on Varian 300 or 500 MHz instruments. Chemical shifts (δ) are reported in parts per million (ppm) referenced to Me4Si. Coupling constants (J) are reported in Hz throughout. Mass spectral data were acquired on Shimadzu LCMS-2010EV for low resolution, and on an Agilent ESI-TOF for either high or low resolution. Purity of key compounds was obtained in a HPLC Breeze from Waters Co. using an Atlantis T3 3μm 4.6×150 mm reverse phase column. The eluant was a linear gradient with a flow rate of 1 ml/min from 95% A and 5% B to 5% A and 95% B in 15 min followed by 5 min at 100% B (Solvent A: H2O with 0.1% TFA; Solvent B: ACN with 0.1% TFA). The compounds were detected at λ=254 nm. Purity of key compounds was established by elemental analysis as performed on a Perkin Elmer series II-2400. Combustion analysis was performed by NuMega Resonance labs, Inc., San Diego, CA. Details on the experimental procedures and key analytical data for each of the reported compound are available as supplementary material.
Plasma Stability Assay
Test compound solution was incubated (1 μM, 2.5% final DMSO concentration) with fresh rat plasma at 37 °C. The reactions were terminated at 0, 30, and 60 min by the addition of two volumes of methanol containing internal standard. Following protein precipitation and centrifugation, the samples were analyzed by LC-MS. The percentage of parent compound remaining at each time point relative to the 0 min sample is calculated from peak area ratios in relation to the internal standard. Compounds were run in duplicate with a positive control known to be degraded in plasma.
Microsomal Stability Assay (RLM assay)
Solutions of three compounds were incubated with RAT liver microsomes (RLM) for 60 minutes at 37.5 °C. The final incubation solutions contained 4 μM test compound, 2 mM NADPH, 1 mg/ml (total protein) microsomes, and 50 mM phosphate (pH 7.2). Compound solutions, protein, and phosphate were pre-incubated at 37.5 °C for 5 min and the reactions were initiated by the addition of NADPH and incubated for 1 hour at 37.5 °C. Aliquots were taken at 15 minute time-points and quenched with the addition of methanol containing internal standard. Following protein precipitation and centrifugation, the samples were analyzed by LC-MS. Test compounds were run in duplicate with 2 control compounds of known half life.
DELFIA Assay (dissociation enhanced lanthanide fluoro-immuno assay)
To each well of 96-well streptavidin-coated plates (Perkin-Elmer) 100 μL of a 100 ng/ml solution of biotin-labeled pep-JIP11 (Biotin-lc-KRPKRPTTLNLF, where lc indicates a hydrocarbon chain of 6 methylene groups) was added. After 1 hr incubation and elimination of unbound biotin-pep-JIP11 by 3 washing steps, 87 μL of Eu-labeled anti-GST antibody solution (300ng/ml; 1.9 nM), 2.5 μL DMSO solution containing test compound, and 10 μL solution of GST-JNK1 for a final protein concentration of 10 nM was added. After 1 hr incubation at 0 °C, each well was washed 5 times to eliminate unbound protein and the Eu-antibody if displaced by a test compound. Subsequently, 200 μL of enhancement solution (Perkin-Elmer) was added to each well and fluorescence measured after 10 min incubation (excitation wavelength, 340 nm; emission wavelength, 615 nm). Controls include unlabeled peptide and blanks receiving no compounds. Protein and peptide solutions were prepared in DELFIA buffer (Perkin-Elmer).
In vitro Kinase Assay
The LanthaScreen™ assay platform from Invitrogen was utilized. The time-resolved fluorescence resonance energy transfer assay (TR-FRET) was performed in 384 well plates. Each well received JNK1 (35 ng/mL), ATF2 (400 nM), and ATP (0.2 μM) in 50 mM HEPES, 10mM MgCl 2, 1mM EGTA and 0.01% Brij-35, pH 7.5 and test compounds. The kinase reaction was performed at room temperature for 1 hr. After which, the terbium labeled antibody and EDTA were added into each well. After an additional hour incubation, the signal was measured at 520/495 nm emission ratio on a fluorescence plate reader (Victor 2, Perkin-Elmer).
Isothermal Titration Calorimetry
Titrations were done using a VP-ITC calorimeter from Microcal (Northampton, MA). Depending upon the solubility of the titrants, JNK2 was used at 25–50 μM in 20 mM sodium phosphate buffer (pH 7.4), 5% DMSO, and 0.01% triton X-100. Titrants were used at 15× protein concentration (375–750 μM) in the same buffer. For competition titrations, 350 μM 25 was titrated to 25μM JNK2 in the presence of 250 μM ATPγS (Biomol International, Plymouth Meeting, PA) or pepJIP1 (ie. 250 μM ATPγS or pepJIP1 was present in both the syringe and cell of the ITC). Titrations of compound alone into buffer were done to determine the heats of dilution of compounds and were negligible compared to compound to protein titrations. Compound heats of dilution were subtracted from compound/protein titrations. Titrations were carried out at 25 °C. Data were analyzed using Microcal Origin software provided by the ITC manufacturer.
Molecular Modeling
Molecular modeling studies were conducted on a Linux workstation and a 64 3.2-GHz CPUs Linux cluster. Docking studies were performed using the X-ray coordinates of JNK1 (PDB code 1UKI). The complexed JIP peptide was extracted from the protein structure and was used to define the binding site for docking of small molecules. The genetic algorithm (GA) procedure in the GOLD25 docking software performed flexible docking of small molecules whereas the protein structure was static and compound poses minimized according to the GOLD scoring function.25 For each compound, 20 geometries were generated and the solution with the best fit according to the scoring function25 was used to represent the docked pose. The protein surface was prepared with the program MOLCAD as implemented in Sybyl and was used to analyze the binding poses for studied small molecules.
Cell based assays for c-Jun phosphorylation
The cell based kinase assays for c-Jun and ATF2 phosphorylation were carried out using the LanthaScreen c-Jun (1–79) Hela (Invitrogen, Carlsbad, CA) which stably express GFP-c-Jun 1–79. Phosphorylation was determined by measuring the time resolved FRET (TR-FRET) between a terbium labeled phospho-specific antibody and the GFP-fusion protein. The cells were plated in white tissue culture treated 384 well plates at a density of 10000 cell per well in 32 μl assay medium (Opti-MEM®, supplemented with 1% charcoal/dextran-treated FBS, 100 U/mL penicillin and 100 μg/mL streptomycin, 0.1 mM non-essential amino acids, 1 mM sodium pyruvate, 25 mM HEPES pH 7.3, and lacking phenol red). After overnight incubation, cells were pretreated for 60 min with compound (indicated concentration) followed by 30 min of stimulation with 2 ng/ml of TNF-alpha which stimulates both JNK and p38. The medium was then removed by aspiration and the cells were lysed by adding 20 μl of lysis buffer (20 mM TRIS-HCl pH 7.6, 5 mM EDTA, 1% NP-40 substitute, 5 mM NaF, 150 mM NaCl, 1:100 protease and phosphatase inhibitor mix, SIGMA P8340 and P2850 respectively). The lysis buffer included 2 nM of the terbium labeled anti-pc-Jun (pSer73) detection antibodies (Invitrogen). After allowing the assay to equilibrate for 1 h at room temperature, TR-FRET emission ratios were determined on a BMG Pherastar fluorescence plate reader (excitation at 340 nm, emission 520 nm and 490 nm; 100 μs lag time, 200 μs integration time, emission ratio = Em520 / Em 490).
Supplementary Material
Acknowledgments
We gratefully acknowledge financial support from the NIH (grant # DK080263 to MP).
Footnotes
Supporting Information Available
Detailed synthetic procedures and analytical data are available for the reported compounds. A dose response curve for the inhibition of JNK in cell is also reported. Docking studies with compounds 19 and 39 are also included.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. Science. 2002;298:1912. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 2.Manning AM, Davis RJ. Nat Rev Drug Discov. 2003;2:554. doi: 10.1038/nrd1132. [DOI] [PubMed] [Google Scholar]
- 3.Bogoyevitch MA, Arthur PG. Biochim Biophys Acta. 2008;1784:76. doi: 10.1016/j.bbapap.2007.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gupta S, Barrett T, Whitmarsh AJ, Cavanagh J, Sluss HK, Derijard B, Davis RJ. Embo J. 1996;15:2760. [PMC free article] [PubMed] [Google Scholar]
- 5.Kyriakis JM, Avruch J. Physiol Rev. 2001;81:807. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- 6.Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH. Endocr Rev. 2001;22:153. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- 7.Shin Y, Chen W, Habel J, Duckett D, Ling YY, Koenig M, He Y, Vojkovsky T, LoGrasso P, Kamenecka TM. Bioorg Med Chem Lett. 2009;19:3344. doi: 10.1016/j.bmcl.2009.03.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gaillard P, Jeanclaude-Etter I, Ardissone V, Arkinstall S, Cambet Y, Camps M, Chabert C, Church D, Cirillo R, Gretener D, Halazy S, Nichols A, Szyndralewiez C, Vitte PA, Gotteland JP. J Med Chem. 2005;48:4596. doi: 10.1021/jm0310986. [DOI] [PubMed] [Google Scholar]
- 9.Ruckle T, Biamonte M, Grippi-Vallotton T, Arkinstall S, Cambet Y, Camps M, Chabert C, Church DJ, Halazy S, Jiang X, Martinou I, Nichols A, Sauer W, Gotteland JP. J Med Chem. 2004;47:6921. doi: 10.1021/jm031112e. [DOI] [PubMed] [Google Scholar]
- 10.Bennett BL, Sasaki DT, Murray BW, O’Leary EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, Bhagwat SS, Manning AM, Anderson DW. Proc Natl Acad Sci U S A. 2001;98:13681. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao H, Serby MD, Xin Z, Szczepankiewicz BG, Liu M, Kosogof C, Liu B, Nelson LT, Johnson EF, Wang S, Pederson T, Gum RJ, Clampit JE, Haasch DL, Abad-Zapatero C, Fry EH, Rondinone C, Trevillyan JM, Sham HL, Liu G. J Med Chem. 2006;49:4455. doi: 10.1021/jm060465l. [DOI] [PubMed] [Google Scholar]
- 12.Angell RM, Atkinson FL, Brown MJ, Chuang TT, Christopher JA, Cichy-Knight M, Dunn AK, Hightower KE, Malkakorpi S, Musgrave JR, Neu M, Rowland P, Shea RL, Smith JL, Somers DO, Thomas SA, Thompson G, Wang R. Bioorg Med Chem Lett. 2007;17:1296. doi: 10.1016/j.bmcl.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 13.Kamenecka T, Jiang R, Song X, Duckett D, Chen W, Ling YY, Habel J, Laughlin JD, Chambers J, Figuera-Losada M, Cameron MD, Lin L, Ruiz CH, LoGrasso PV. J Med Chem. 53:419. doi: 10.1021/jm901351f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kallunki T, Deng T, Hibi M, Karin M. Cell. 1996;87:929. doi: 10.1016/s0092-8674(00)81999-6. [DOI] [PubMed] [Google Scholar]
- 15.Yang SH, Whitmarsh AJ, Davis RJ, Sharrocks AD. Embo J. 1998;17:1740. doi: 10.1093/emboj/17.6.1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barr RK, Kendrick TS, Bogoyevitch MA. J Biol Chem. 2002;277:10987. doi: 10.1074/jbc.M107565200. [DOI] [PubMed] [Google Scholar]
- 17.Bonny C, Oberson A, Negri S, Sauser C, Schorderet DF. Diabetes. 2001;50:77. doi: 10.2337/diabetes.50.1.77. [DOI] [PubMed] [Google Scholar]
- 18.Dickens M, Rogers JS, Cavanagh J, Raitano A, Xia Z, Halpern JR, Greenberg ME, Sawyers CL, Davis RJ. Science. 1997;277:693. doi: 10.1126/science.277.5326.693. [DOI] [PubMed] [Google Scholar]
- 19.Heo YS, Kim SK, Seo CI, Kim YK, Sung BJ, Lee HS, Lee JI, Park SY, Kim JH, Hwang KY, Hyun YL, Jeon YH, Ro S, Cho JM, Lee TG, Yang CH. Embo J. 2004;23:2185. doi: 10.1038/sj.emboj.7600212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaneto H, Nakatani Y, Miyatsuka T, Kawamori D, Matsuoka TA, Matsuhisa M, Kajimoto Y, Ichijo H, Yamasaki Y, Hori M. Nat Med. 2004;10:1128. doi: 10.1038/nm1111. [DOI] [PubMed] [Google Scholar]
- 21.De SK, Stebbins JL, Chen LH, Riel-Mehan M, Machleidt T, Dahl R, Yuan H, Emdadi A, Barile E, Chen V, Murphy R, Pellecchia M. J Med Chem. 2009;52:1943. doi: 10.1021/jm801503n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stebbins JL, De SK, Machleidt T, Becattini B, Vazquez J, Kuntzen C, Chen LH, Cellitti JF, Riel-Mehan M, Emdadi A, Solinas G, Karin M, Pellecchia M. Proc Natl Acad Sci U S A. 2008;105:16809. doi: 10.1073/pnas.0805677105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vazquez J, De SK, Chen LH, Riel-Mehan M, Emdadi A, Cellitti J, Stebbins JL, Rega MF, Pellecchia M. J Med Chem. 2008;51:3460. doi: 10.1021/jm800068w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen T, Kablaoui N, Little J, Timofeevski S, Tschantz WR, Chen P, Feng J, Charlton M, Stanton R, Bauer P. Biochem J. 2009;420:283. doi: 10.1042/BJ20081899. [DOI] [PubMed] [Google Scholar]
- 25.Jones G, Willett P, Glen RC, Leach AR, Taylor R. J Mol Biol. 1997;267:727. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 26.Eldridge MD, Murray CW, Auton TR, Paolini GV, Mee RP. J Comput Aided Mol Des. 1997;11:425. doi: 10.1023/a:1007996124545. [DOI] [PubMed] [Google Scholar]
- 27.Teschner M, Henn C, Vollhardt H, Reiling S, Brickmann J. J Mol Graph. 1994;12:98. doi: 10.1016/0263-7855(94)80074-x. [DOI] [PubMed] [Google Scholar]
- 28.Dalvie DK, Kalgutkar AS, Khojasteh-Bakht SC, Obach RS, O’Donnell JP. Chem Res Toxicol. 2002;15:269. doi: 10.1021/tx015574b. [DOI] [PubMed] [Google Scholar]
- 29.Dansette PM, Bonierbale E, Minoletti C, Beaune PH, Pessayre D, Mansuy D. Eur J Drug Metab Pharmacokinet. 1998;23:443. doi: 10.1007/BF03189993. [DOI] [PubMed] [Google Scholar]
- 30.Gardner I, Zahid N, MacCrimmon D, Uetrecht JP. Mol Pharmacol. 1998;53:991. [PubMed] [Google Scholar]
- 31.Hom RK, Bowers S, Sealy JM, Truong AP, Probst GD, Neitzel ML, Neitz RJ, Fang L, Brogley L, Wu J, Konradi AW, Sham HL, Toth G, Pan H, Yao N, Artis DR, Quinn K, Sauer JM, Powell K, Ren Z, Bard F, Yednock TA, Griswold-Prenner I. Bioorg Med Chem Lett. 2010;20:7303. doi: 10.1016/j.bmcl.2010.10.066. [DOI] [PubMed] [Google Scholar]
- 32.Probst GD, Bowers S, Sealy JM, Truong AP, Hom RK, Galemmo RA, Jr, Konradi AW, Sham HL, Quincy DA, Pan H, Yao N, Lin M, Toth G, Artis DR, Zmolek W, Wong K, Qin A, Lorentzen C, Nakamura DF, Quinn KP, Sauer JM, Powell K, Ruslim L, Wright S, Chereau D, Ren Z, Anderson JP, Bard F, Yednock TA, Griswold-Prenner I. Bioorg Med Chem Lett. 2011;21:315. doi: 10.1016/j.bmcl.2010.11.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.