

Abstract
Pneumococcal surface protein A (PspA) is an immunogenic protein expressed on the surface of all strains of Streptococcus pneumoniae (pneumococcus) and induces antibodies which protect against invasive infection in mice. Pneumococci used for infectious challenge in protection studies are typically collected from cultures grown in semisynthetic medium in vitro. The purpose of these studies is to confirm that PspA is expressed by pneumococci during growth in vivo at a level sufficient for antibodies to PspA to be protective. Mice were actively immunized with purified PspA or by passive transfer of monoclonal antibody (MAb) and challenged with a capsular type 3 strain in diluted whole blood from bacteremic mice. All were protected against challenge with 10 times the 50% lethal dose (LD50), and mice challenged with 1,000 times the LD50 had increased survival compared with controls. Additionally, nonimmune mice treated with MAbs to PspA or PspA immune serum at 6 and 12 h after infection with 10 times the LD50 also showed increased survival. Northern blot analysis of RNA from pneumococci grown either in vitro or in vivo showed similar levels of PspA mRNA. These results demonstrate that PspA is expressed in vivo in a mouse model and that immunization with PspA induces antibodies to an antigen which is expressed during the course of invasive infection. Immunotherapy with antibodies to PspA may have some utility in treating pneumococcal infections in humans.
Bacterial infectious diseases continue to cause significant morbidity and mortality worldwide, and Streptococcus pneumoniae (pneumococcus) is among the most common etiologies of lower respiratory tract infections, meningitis, and septicemia in humans of all age groups (11, 20). Currently, two formulations of pneumococcal vaccines are available, a 23-valent polysaccharide vaccine and a heptavalent protein-polysaccharide conjugate vaccine; these vaccines have proven useful in preventing invasive disease in adults and children, respectively. However, each has limitations that may reduce long-term effectiveness. The 23-valent vaccine is poorly immunogenic in certain high-risk groups and fails to induce a significant memory immune response (7, 17, 25). These characteristics have resulted in recommendations for the reimmunization of some groups at 5 years after the primary immunization (8). The conjugate vaccine contains protein-conjugated polysaccharides of the seven most common pneumococcal serotypes which cause invasive disease in children less than 5 years of age. Although this vaccine is highly effective in preventing invasive disease (29) it is relatively expensive to synthesize, and there has been some concern that serotypes not included in the vaccine may increase in frequency (12, 15). Proteins which are antigenically conserved across clinically relevant serotypes would be more effective immunogens and, potentially, be more cost effective. Recent reviews have described protein virulence factors of pneumococci which are being investigated as components of improved vaccines (6, 14, 22).
Among the most well-characterized protein antigens is pneumococcal surface protein A (PspA). This protein is present on all strains of pneumococci and can induce an antibody response which protects against an otherwise lethal challenge dose in an animal model (4, 5, 9). Although serologically variable, heterologous PspA molecules are fairly cross-reactive and immunization with one PspA family can protect against pneumococci expressing PspA of separate families (3, 6). This protective immunity can be induced by either systemic or mucosal immunization (1, 30). Although PspA has proven to be a potent immunogen, the studies reported to date have used pneumococci grown in vitro in synthetic media as the sourceof both infectious organisms for challenge and native PspA for characterization and immunization. These prior studies were somewhat artificial, as human infections result from translocation of pneumococci colonizing mucosal surfaces of the upper respiratory tract. It is now evident that bacterial pathogens such as pneumococci regulate expression of virulence factors in response to the unique environmental stimuli of diverse anatomical locations in hosts (27, 28). For any vaccine candidate, it is important to demonstrate that the antigen is expressed by the pathogen during growth in vivo. Both pneumococcal carriage and otitis media caused by pneumococci induce antibodies to PspA in children, suggesting that PspA is expressed by pneumococci replicating on mucosal surfaces (24). The purpose of the present study is to test the hypothesis that PspA is expressed by pneumococci which are multiplying in blood in vivo and that immunization studies using infectious challenge with organisms grown in vitro are valid models for predicting the natural history of invasive infection in an intact host. In the present studies, this test was performed in two ways. The first approach was to challenge actively or passively immunized animals with pneumococci grown in vivo. The second approach was to administer PspA antibodies 6 to 24 h after animals had been infected with pneumococci grown in vitro.
Immunization.
PspA used for immunization was isolated from strain R36A, an unencapsulated derivative of the capsule type 2 laboratory strain D39 (26). Cells were grown in chemically defined medium with ethanolamine and the supernatant was passed over a Sepharose column conjugated with choline as previously described (5). This purification method yields a single detectable band on Coomassie blue-stained sodium dodecyl sulfate-polyacrylamide gels which reacts with anti-PspA antibodies on immunoblotting. Total protein was quantitated with a protein assay (Bio-Rad, Hercules, Calif.). Mice used in this study were obtained from Jackson Laboratories (Bar Harbor, Maine), and all procedures were approved by the local Animal Care and Use Committee. BALB/cByJ mice were immunized subcutaneously on days 0, 14, and 28 with 0.5 μg of purified PspA in sterile 0.9% NaCl containing 1 mg of alum (Pierce Chemicals, Rockford, Ill.)/ml. Control mice received column-purified culture supernatants from strain WG44.1 in the same formulation and schedule. WG44.1 is a PspA-deficient mutant derived from the functionally unencapsulated strain Rx1 (31). Blood was collected for antibody measurement from each actively immunized animal prior to the initial immunization and on day 35 immediately prior to infectious challenge. Anti-PspA antibody concentrations were determined by an enzyme-linked immunosorbent assay technique as previously described (5).
Infection model.
Infectious challenge of animals was performed with strain A66.1 or WU2 as noted.Both of these are mouse-virulent capsule type 3 pneumococci (3) and express family 1, clade 2, PspA serologically similar to the family 1, clade 2, PspA of strain R36A (13). Immunization with R36A PspA (or with the identical PspA from strain Rx1) has been shown previously to protect mice from in vitro-grown WU2 or A66.1 (4). To prepare in vivo-grown pneumococci, naive BALB/cByJ mice were infected by intravenous (i.v.) or intraperitoneal (i.p.) inoculation of A66.1 and blood was collected in sterile nonheparinized tubes 24 h later, the time at which the bacterial density was approximately 106 to 107 CFU/ml (as determined by preliminary studies). Whole blood was diluted with sterile saline and used immediately to infect the experimental animals. An aliquot of diluted blood was saved for serial dilution plate counts to determine the actual number of organisms used for infectious challenge and to assure the identity and purity of the bacteria collected from septic animals. Animals were inoculated with 100 μl of diluted blood via the tail vein, and survival was observed for 5 days.
Immunotherapy.
Passive immunization was performed by i.p. injection of 5 μg of XiR278, an anti-PspA immunoglobulin G1 monoclonal antibody. This antibody was made using a PspA with 100% sequence identity to that of strains R36A and Rx1 (19). CBA/CaHN-Btk(xid)/J mice were inoculated with strain A66.1 or WU2 by an i.p. or i.v. route as indicated, and 20 μg of XiR278 antibody or 4.3 μg of pooled anti-PspA antibody was administered i.p. at 6 or 12 h postinfection. The pooled antibody used was from CBA/N mice hyperimmunized with recombinant Rx1 M1 PspA (18). Prior studies have shown that antibody given in this manner equilibrates with the blood within 1 h (19).
RNA procedure.
Total RNA from pneumococci growing in the logarithmic phase in Todd-Hewitt broth was isolated by pelleting cells, washing twice in diethyl polycarbonate-treated water, and resuspending the washed pellets in a 1/10 volume of lysis buffer (0.05% deoxycholate and 0.1% sodium dodecyl sulfate). The cell suspension was incubated at 37°C for 30 min to lyse the pneumococcal cells and clear the suspension. A High Pure RNA isolation system (Roche, Indianapolis, Ind.) was used to isolate and purify the RNA from the lysate. Total RNA from pneumococci growing in vivo was isolated from strain D39 samples which had been collected from bacteremic mice. Animals were infected i.p. with 103 to 105 CFU of strain D39 and bled under anesthesia after 24 to 48 h. Strain D39 replicates to high numbers in mouse blood (≥109 CFU/ml) after this period of time, and small amounts of blood from bacteremic mice can yield a significant quantity of bacterial cells. Blood was collected in 5 mM EDTA on ice and was immediately processed. Whole blood was centrifuged for 15 s at 5,000 × g at 4°C to separate plasma from cellular elements. The plasma was collected and centrifuged at 10,000 × g at 4°C for 1 min to pellet bacterial cells. The cells were then immediately processed for RNA purification as described above. Total RNA was separated on MOPS (morpholinepropanesulfonic acid)/formaldehyde agarose gels and vacuum blotted onto positively charged nylon membranes as described previously (2). A cloned fragment of pspA representing the first 864 bases of the coding region was labeled with digoxigenin and used as a probe on the blotted membranes according to the manufacturer's protocol (Genius System; Roche).
Results and discussion.
The relative amounts of PspA mRNA were observed to be similar in pneumococci growing in vitro and pneumococci collected directly from bacteremic animals when measured by Northern blotting (Fig. 1). This confirms earlier reports of pspA transcription in vivo (21). The data regarding phenotypic expression of PspA are supported by challenge experiments with pneumococci which were collected from bacteremic animals and used immediately for infection in actively or passively immunized mice.
FIG. 1.
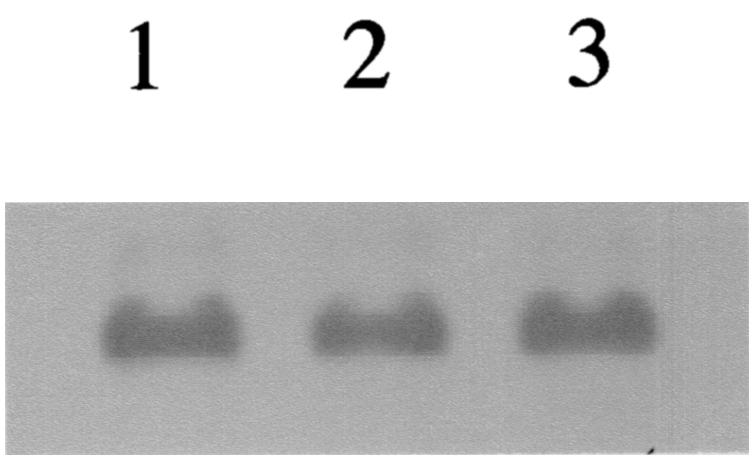
Northern blot of total RNA isolated from pneumococci grown in THY medium (lane 1), pneumococci collected from bacteremic mice infected with strain A66.1 (lane 2), and pneumococci collected from bacteremic mice infected with strain D39 (lane 3). Each lane represents 10 μg of total RNA that was hybridized with a digoxigenin-labeled pspA DNA probe as described in the text.
To confirm that the quantity of PspA expressed in vivo is sufficient to bind protective antibodies, two experiments were performed. In the first, mice were actively immunized with purified PspA or passively immunized with a monoclonal antibody to PspA and infected with in vivo-grown pneumococci. Mice actively immunized with PspA responded with various antibody concentrations. In 20 animals actively immunized in this study, anti-PspA total immunoglobulin levels ranged from 13 to 1,350 μg/ml (geometric mean, 92 μg/ml; upper and lower limits of standard error, 66 to 129 μg/ml). All actively and passively immunized animals challenged i.v. with 10 times the 50% lethal dose of a mouse-virulent pneumococcal strain survived. When the inoculum was increased to 1,000 times the 50% lethal dose only 40% of the immunized mice survived, but survival for those immunized mice that had a lethal outcome was extended by just over 2 days (Fig. 2). In this study pneumococcal cells were transferred immediately from one host to another, so the full complement of virulence factors expressed during bacteremia should have been present. If PspA expression were repressed or down-regulated in vivo, then preexisting antibodies would not be expected to have such a significant protective effect.
FIG. 2.

(A) Survival of animals inoculated i.v. with 4.5 × 104 CFU of strain A66.1. Passively immunized animals received 5 μg of anti-PspA monoclonal antibody XiR278 at either 1 h before or 1 h after infection. Actively immunized mice received PspA isolated from strain R36A. Control mice received an equal volume of column eluate of WG44.1, a strain that does not express PspA. Each group of actively or passively immunized mice differed from the controls at P = 0.0068 by the Mann-Whitney two-sample rank test. (B) Survival of immunized animals inoculated i.v. with 106 CFU of strain A66.1. The groups were immunized as described for panel A except that no group received XiR278 monoclonal antibody before infection. Each group of actively or passively immunized mice differed from the controls at P < 0.0001 by the Mann-Whitney two-sample rank test.
An alternative method used to study the in vivo expression of PspA was to transfer anti-PspA antibodies to animals with established bacteremia with strain WU2 or A66. Pneumococci replicating in vivo for ≥6 h will have gone through multiple cell divisions and should differentially express all genes required for growth in the host blood. Table 1 shows that PspA-specific antibodies can, in fact, clear preestablished pneumococcal bacteremia with strain WU2 when given at 6 and 12 h postinfection (Table 1). Identical results were obtained for i.p. infection with WU2 (data not shown). For strain A66.1, which is more virulent in animal models of infection (3), antibodies given at 6 and 12 h delayed mortality but only those given at 6 h postinfection rapidly cleared bacteria from the blood. All mice that survived infection following treatment had sterile blood at 8 or 16 days following infection. No survival benefit was observed when antibodies were used to treat strain A66.1 infections at 24 h after challenge (data not shown). Although PspA antibodies were not able to reverse the course of disease once sepsis had become established for 24 h, these data provide strong support for the hypothesis that PspA is expressed at sufficient levels in vivo to be an effective immunogen during the course of bacteremia. The failure of antibody to PspA to protect against sepsis after some critical time point may be related, in part, to the small amount of antibody used and to the mechanism of action of PspA. PspA inhibits complement deposition on the pneumococcal cell surface, and antibody to PspA appears to block this inhibition (23, 23a). Complement levels are generally depleted during sepsis, and thus, a potentially beneficial effect of antibody to PspA is possibly reduced in septic animals. It is possible that antibodies to PspA are more effective at preventing infections than at eradicating them, especially as infection progresses to high-level bacteremia.
TABLE 1.
Use of PspA antibody to treat established pneumococcal bacteremia with capsular type 3 strains
| Challengea | Antibodyb
|
Log
CFU ± SE (% bacteremic) at the indicated time
(h)c
|
% Survivald | Median days to death | P vs control (days to death)e | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Strainf | Log CFU | Source | Time of treatment (h) | 6 | 12 | 30-36 | 54-60 | 144 | |||
| WU2 | 2.36 | XiR278 | 6 | <1.7 (0) | 100 | >8 | <0.0001 | ||||
| WU2 | 2.36 | XiR278 | 12 | <1.7 (0) | 100 | >8 | <0.0001 | ||||
| WU2 | 2.36 | None | 5.9 ± 0.4 (100) | 0 | 2 | ||||||
| A66.1 | 2.38 | Anti-PspA | 6 | 3.0 ± 0.2 (86) | 2.4 ± 0.4 (43) | 2.5 ± 0.4 (43) | <1.7 (0) | 71 | >16 | 0.0006 | |
| A66.1 | 2.38 | Anti-PspA | 12 | 4.1 ± 0.5 (86) | 3.4 ± 0.5 (71) | 4.2 ± 0.8 (83) | 4.4 ± 0.3 (100) | 29 | 8 | 0.0012 | |
| A66.1 | 2.38 | None | 4.1 ± 0.4 (100) | >6.7 | None surviving | None surviving | 0 | 1 | |||
Bacteria were injected i.v. at time 0.
XiR278 monoclonal antibody was given at 20 μg/animal. Anti-PspA serum raised in CBA/N mice by immunization with recombinant strain Rx1 PspA was injected at a dilution to give each animal 4.3 μg in 100 μl.
Blood was collected at 6 or 12 h immediately prior to the administration of antibody. Bacteremia was scored as the percentage of surviving animals with at least 50 CFU (log = 1.7), the limit of detection by plating.
Percentage of inoculated mice surviving at 8 or 16 days. No mice surviving at the time of euthanasia had detectable pneumococci in their blood.
P values were calculated by a two-tailed Mann-Whitney two-sample rank test to compare time until death of control mice with that for mice given antibody.
Each group given strain WU2 contained 10 mice; groups receiving strain A66.1 contained 7 animals.
In the past, standard approaches for the identification of virulence factors or vaccine components of bacterial pathogens relied on manipulations of bacterial cultures grown in vitro and protection studies typically used bacterial cells growing in laboratory medium as the infectious challenge. This was true for PspA when it was originally characterized (19). This same type of approach led to the development of a human vaccine for Borrelia burgdorferi in which the antigen, OspA, was expressed in the insect vector but not in the human host (10). It is now clear that bacterial pathogens respond to environmental signals, and phenotypes characterized from growth in vitro cannot be extrapolated to the in vivo environment (16). In this study, transcription of PspA in vivo was confirmed by Northern blotting of total RNA with a pspA probe and protein expression was demonstrated by the protective efficacy of PspA-specific antibodies against pneumococci replicating in the blood of an intact host. We now know that as determined on the basis of these results, previous protection studies of immune responses to PspA and pneumococci grown in vitro are models which accurately predict the natural history of pneumococcal infection in hosts with preexisting PspA antibodies. The results of these present studies also suggest that antibodies to PspA can be useful for immunotherapy in some circumstances. Immunotherapy for invasive pneumococcal disease may be an effective treatment modality which is not compromised by the growing prevalence of antimicrobial resistance.
Acknowledgments
These studies were supported in part by NIH grant AI121548.
Editor: J. N. Weiser
REFERENCES
- 1.Arulanandam, B. P., J. M. Lynch, D. E. Briles, S. Hollingshead, and D. W. Metzger. 2001. Intranasal vaccination with pneumococcal surface protein A and interleukin-12 augments antibody-mediated opsonization and protective immunity against Streptococcus pneumoniae infection. Infect. Immun. 69:6718-6724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ausubel, F., R. Brent, R. E. Kingston, et al. (ed.). 1993. Current protocols in molecular biology. John Wiley & Sons, Inc., New York, N.Y.
- 3.Briles, D. E., M. J. Crain, B. M. Gray, C. Forman, and J. Yother. 1992. Strong association between capsular type and virulence for mice among human isolates of Streptococcus pneumoniae. Infect. Immun. 60:111-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Briles, D. E., S. K. Hollingshead, J. King, A. Swift, P. A. Braun, M. K. Park, L. M. Ferguson, M. H. Nahm, and G. S. Nabors. 2000. Immunization of humans with recombinant pneumococcal surface protein A (rPspA) elicits antibodies that passively protect mice from fatal infection with Streptococcus pneumoniae bearing heterologous PspA. J. Infect. Dis. 182:1694-1701. [DOI] [PubMed] [Google Scholar]
- 5.Briles, D. E., J. D. King, M. A. Gray, L. S. McDaniel, E. Swiatlo, and K. A. Benton. 1996. PspA, a protection-eliciting pneumococcal protein: immunogenicity of isolated native PspA in mice. Vaccine 14:858-867. [DOI] [PubMed] [Google Scholar]
- 6.Briles, D. E., R. C. Tart, E. Swiatlo, J. Dillard, P. Smith, K. A. Benton, B. A. Ralph, A. Brooks-Walter, M. J. Crain, S. K. Hollingshead, and L. S. McDaniel. 1998. Pneumococcal diversity: considerations for new vaccine strategies with emphasis on pneumococcal surface protein A (PspA). Clin. Microbiol. Rev. 11:645-657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Butler, J. C., E. D. Shapiro, and G. M. Carlone. 1999. Pneumococcal vaccines: history, current status, and future directions. Am. J. Med. 107(Suppl. 1A):69S-76S. [DOI] [PubMed] [Google Scholar]
- 8.Centers for Disease Control and Prevention. 1997. Prevention of pneumococcal disease: recommendations of the Advisory Committee on Immunization Practices (ACIP). Morb. Mortal. Wkly. Rep. 46:1-24. [Google Scholar]
- 9.Crain, M. J., W. D. Waltman, J. S. Turner, J. Yother, D. F. Talkington, L. S. McDaniel, B. M. Gray, and D. E. Briles. 1990. Pneumococcal surface protein A (PspA) is serologically highly variable and is expressed by all clinically important capsular serotypes of Streptococcus pneumoniae. Infect. Immun. 58:3293-3299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Silva, A., S. Telford III, L. Brunet, S. Barthold, and E. Fikrig. 1996. Borrelia burgdorferi OspA is an arthropod-specific transmission-blocking Lyme disease vaccine. J. Exp. Med. 183:271-275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fedson, D. S., J. Anthony, and G. Scott. 1999. The burden of pneumococcal disease among adults in developed and developing countries: what is known and what is not known. Vaccine 17:S11-S18. [DOI] [PubMed] [Google Scholar]
- 12.Hausdorff, W. P., J. Bryant, P. R. Paradiso, and G. R. Siber. 2000. Which pneumococcal serogroups cause the most invasive disease: implications for conjugate vaccine formulation and use, part I. Clin. Infect. Dis. 30:100-121. [DOI] [PubMed] [Google Scholar]
- 13.Hollingshead, S. K., R. Becker, and D. E. Briles. 2000. Diversity of PspA: mosaic genes and evidence for past recombination in Streptococcus pneumoniae. Infect. Immun. 68:5889-5900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jedrzejas, M. J. 2001. Pneumococcal virulence factors: structure and function. Microbiol. Mol. Biol. Rev. 65:187-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klein, D. L. 1999. Pneumococcal disease and the role of conjugate vaccines. Microb. Drug Resist. 5:147-157. [DOI] [PubMed] [Google Scholar]
- 16.Marra, A., J. Asundi, M. Bartilson, S. Lawson, F. Fang, J. Christine, C. Wiesner, D. Brigham, W. P. Schneider, and A. E. Hromockyj. 2002. Differential fluorescence induction analysis of Streptococcus pneumoniae identifies genes involved in pathogenesis. Infect. Immun. 70:1422-1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McCashland, T. M., L. C. Preheim, and M. J. Gentry-Nielsen. 2000. Pneumococcal vaccine response in cirrhosis and liver transplantation. J. Infect. Dis. 181:757-760. [DOI] [PubMed] [Google Scholar]
- 18.McDaniel, L. S., B. A. Ralph, D. O. McDaniel, and D. E. Briles. 1994. Localization of protection-eliciting epitopes on PspA of Streptococcus pneumoniae between amino acid residues 192 and 260. Microb. Pathog. 17:323-337. [DOI] [PubMed] [Google Scholar]
- 19.McDaniel, L. S., G. Scott, J. F. Kearney, and D. E. Briles. 1984. Monoclonal antibodies against protease-sensitive pneumococcal antigens can protect mice from fatal infection with Streptococcus pneumoniae. J. Exp. Med. 160:386-397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mulholland, K. 1999. Strategies for the control of pneumococcal diseases. Vaccine 17:S79-S84. [DOI] [PubMed] [Google Scholar]
- 21.Ogunniyi, A. D., P. Giammarinaro, and J. C. Paton. 2002. The genes encoding virulence-associated proteins and the capsule of Streptococcus pneumoniae are upregulated and differentially expressed in vivo. Microbiology 148:2045-2053. [DOI] [PubMed] [Google Scholar]
- 22.Paton, J. C., A. M. Berry, and R. A. Lock. 1997. Molecular analysis of putative pneumococcal virulence proteins. Microb. Drug Resist. 3:1-10. [DOI] [PubMed] [Google Scholar]
- 23.Ren, B., A. Szalai, J., O. Thomas, S. K. Hollingshead, and D. E. Briles. 2003. Both family 1 and family 2 PspA proteins can inhibit complement deposition and confer virulence to a capsular serotype 3 strain of Streptococcus pneumoniae. Infect. Immun. 71:75-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23a. Ren, B., A. J. Szalai, S. K. Hollingshead, and D. E. Briles. Effects of PspA and antibodies to PspA on activation and deposition of complement on the pneumococcal surface. Infect. Immun., in press. [DOI] [PMC free article] [PubMed]
- 24.Simell, B., M. Korkeila, H. Pursiainen, T. M. Kilpi, and H. Kayhty. 2001. Pneumococcal carriage and otitis media induce salivary antibodies to pneumococcal surface adhesin A, pneumolysin, and pneumococcal surface protein A. J. Infect. Dis. 183:887-896. [DOI] [PubMed] [Google Scholar]
- 25.Tasker, S. A., M. R. Wallace, J. B. Rubins, W. B. Paxton, J. O'Brien, and E. N. Janoff. 2002. Reimmunization with 23-valent pneumococcal vaccine for patients infected with human immunodeficiency virus type 1: clinical, immunologic, and virologic responses. Clin. Infect. Dis. 34:813-821. [DOI] [PubMed] [Google Scholar]
- 26.Tiraby, G., M. S. Fox, and H. Bernheimer. 1975. Marker discrimination in deoxyribonucleic acid-mediated transformation of various pneumococcus strains. J. Bacteriol. 121:608-618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weiser, J. N., R. Austrian, P. K. Sreenivasan, and H. R. Masure. 1994. Phase variation in pneumococcal opacity: relationship between colonial morphology and nasopharyngeal colonization. Infect. Immun. 62:2582-2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weiser, J. N., Z. Markiewicz, E. I. Tuomanen, and J. H. Wani. 1996. Relationship between phase variation in colony morphology, intrastrain variation in cell wall physiology, and nasopharyngeal colonization by Streptococcus pneumoniae. Infect. Immun. 64:2240-2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Whitney, C. G., M. M. Farley, J. Hadler, L. H. Harrison, N. M. Bennett, R. Lynfield, A. Reingold, P. Cieslak, T. Pilishvili, D. Jackson, R. R. Facklam, J. H. Jorgensen, and A. Schuchat. 2003. Decline in invasive pneumococcal disease after the introduction of protein-polysaccharide conjugate vaccine. N. Engl. J. Med. 348:1737-1746. [DOI] [PubMed] [Google Scholar]
- 30.Wu, H.-Y., M. H. Nahm, Y. Guo, M. W. Russell, and D. E. Briles. 1997. Intranasal immunization of mice with PspA (pneumococcal surface protein A) can prevent intranasal carriage, pulmonary infection, and sepsis with Streptococcus pneumoniae. J. Infect. Dis. 175:839-846. [DOI] [PubMed] [Google Scholar]
- 31.Yother, J., G. L. Handsome, and D. E. Briles. 1992. Truncated forms of PspA that are secreted from Streptococcus pneumoniae and their use in functional studies and cloning of the pspA gene. J. Bacteriol. 174:610-618. [DOI] [PMC free article] [PubMed] [Google Scholar]