

Abstract
The title compound, C13H13O2P, crystallized as enantiomerically pure crystals; for the crystal measured, the P atom has R stereochemistry. The crystal structure displays O—H⋯O hydrogen bonding, which links individual molecules related by a 21 screw axis parallel to the crystallographic a-axis direction into continuous chains.
Related literature
For background to phosphinic acids, see: Beckmann et al. (2009 ▶); Burrow et al. (2000 ▶); Chen & Suslick (1993 ▶); Siqueira et al. (2006 ▶); Vioux et al. (2004 ▶). For a description of the Cambridge Structural Database, see: Allen (2002 ▶). Geometrical analysis was performed with Mogul (Bruno et al., 2004 ▶).
Experimental
Crystal data
C13H13O2P
M r = 232.20
Orthorhombic,

a = 5.7326 (2) Å
b = 12.3430 (3) Å
c = 16.7794 (4) Å
V = 1187.27 (6) Å3
Z = 4
Mo Kα radiation
μ = 0.21 mm−1
T = 295 K
0.65 × 0.34 × 0.22 mm
Data collection
Bruker X8 Kappa APEXII diffractometer
Absorption correction: gaussian (SADABS; Bruker, 2009 ▶) T min = 0.880, T max = 0.964
14337 measured reflections
3451 independent reflections
3119 reflections with I > 2σ(I)
R int = 0.028
Refinement
R[F 2 > 2σ(F 2)] = 0.039
wR(F 2) = 0.108
S = 1.05
3451 reflections
148 parameters
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.47 e Å−3
Δρmin = −0.23 e Å−3
Absolute structure: Flack (1983 ▶), 1447 Friedel pairs
Flack parameter: 0.00 (11)
Data collection: APEX2 (Bruker, 2009 ▶); cell refinement: SAINT (Bruker, 2009 ▶); data reduction: SAINT (Bruker, 2009 ▶); program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: DIAMOND (Brandenburg, 2009 ▶); software used to prepare material for publication: publCIF (Westrip, 2010 ▶).
Supplementary Material
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536811008245/zb2011sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536811008245/zb2011Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| O1—H1⋯O2i | 0.93 (3) | 1.58 (3) | 2.4838 (18) | 163 (3) |
Symmetry code: (i) 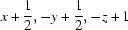 .
.
Acknowledgments
Financial support from the Conselho Nacional de Desenvolvimento Científico (CNPq, Brazil; grants 485245/2007–8 and 479747/2009–1) and the Fundação de Amparo à Pesquisa (FAPERGS, Rio Grande do Sul) is gratefully acknowledged, as are fellowships from CNPq (RAB) and the Coordenação de Aperfeiçoamento de Pessoas de Nível Superior (CAPES, Brazil; RMSS). The diffractometer was funded by a CT-INFRA grant from the Financiadora de Estrutos e Projetos (FINEP, Brazil).
supplementary crystallographic information
Comment
Phosphinic acids have found use for the construction of coordination polymers [Siqueira et al., 2006; Beckmann et al., 2009] for a wide range of applications [Vioux et al., 2004; Chen & Suslick,1993]. Continuing our research on phosphinic acids [Burrow et al., 2000], we report the synthesis and crystal structure of the title compound, (I).
The crystal structure of (I) is from an enantiomerically pure crystal (Flack parameter = 0.00 (11); 1447 Friedel pairs; Flack & Bernardinelli, 2000] with the P atom possessing R stereochemistry. An analysis of the geometry of (I) by Mogul [Bruno et al., 2004] using the CSD. [Allen, 2002] shows no unusual features for the benzyl and phenyl groups. However, an unusually long P═O bond [P1═O2 = 1.5104 (13) Å; average in Mogul:1.484 (17) Å for 16 observations, |z score| = 1.568] and a wider C—P—C angle [C11—P1—C21 angle = 109.493 (9)°; average in Mogul: 106.9(2.0) °, |z score| = 1.281] are found.
The individual molecules of (I) related by a 21 screw axis parallel to the crystallographic a direction are joined by hydrogen bonding of the type OH···O=P—OH···O=P to form continuous chains. The short P—O···O=P distance of 2.4838 (18) Å indicates a strong hydrogen bond. This is slightly shorter than the average O···O interaction distance in the CSD. [2.51 (5) Å, 45 observations] for other phosphinic acids.
Experimental
To a solution of phenylphosphinic acid (2.0 g, 14.1 mmol) in dichloromethane, 30 ml diisopropylethylamine (5.16 ml, 29.6 mmol) and trimethylsilyl chloride (3.74 ml, 29.6 mmol) were separately added at 0 °C under argon. The reaction mixture was stirred at room temperature for 2–3 h, cooled to 0 °C and 1-(bromomethyl)benzene (1.84 ml, 15.5 mmol) was added. After further stirring at room temperature for 48 h, the solvent was removed under vacuum. The residue was suspended in hydrochloric acid (2 M, 20 ml) and filtered on a glass frit. The white solid was washed with acetone and dried giving a yield of 1.70 g (65%) of pure product. IR: 1494 (m), 1439 (s), 1242 (m), 1132 (versus), 1069 (s), 969 (versus), 845 (s), 787 (s), 751 (s), 734 (s), 701 (s), 585 (m), 524 (s), 477 (s), 466 (m) cm-1. TGA: 310–361 °C: 99% loss. DTA: 181–193 °C & 310–361 °C endothermic peaks. Crystals suitable for single-crystal X-ray analysis were grown from an acetone solution in a desiccator with silical gel.
Refinement
The H atom on O1 was found in the difference Fourier map and its position was allowed to refine freely while its isotropic displacement factor was set to 1.5 times that of O1. The H atoms were positioned geometretically and allowed to ride on their parent atoms, with C—H bond lengths of 0.93 Å (aromatic CH) and 0.97 Å (methylene CH2) and isotropic displacement parameters equal to 1.2 times Ueq of the parent atom.
Figures
Fig. 1.

The molecular of (I) structure of the title compound, showing 30% probability ellipsoids.
Fig. 2.

The packing diagram of (I) in the crystallographic a direction with the crystallographic b axis pointing up. The O—H···O intermolecular hydrogen bond is shown dashed.
Crystal data
| C13H13O2P | Dx = 1.299 Mg m−3 |
| Mr = 232.20 | Melting point = 454–456 K |
| Orthorhombic, P212121 | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: P 2ac 2ab | Cell parameters from 4929 reflections |
| a = 5.7326 (2) Å | θ = 3.3–28.0° |
| b = 12.3430 (3) Å | µ = 0.21 mm−1 |
| c = 16.7794 (4) Å | T = 295 K |
| V = 1187.27 (6) Å3 | Block, colourless |
| Z = 4 | 0.65 × 0.34 × 0.22 mm |
| F(000) = 488 |
Data collection
| Bruker X8 Kappa APEXII diffractometer | 3451 independent reflections |
| Radiation source: fine-focus sealed tube | 3119 reflections with I > 2σ(I) |
| graphite | Rint = 0.028 |
| Detector resolution: 0.0833 pixels mm-1 | θmax = 30.0°, θmin = 3.5° |
| φ and ω scans | h = −8→8 |
| Absorption correction: gaussian (SADABS; Bruker, 2009) | k = −17→17 |
| Tmin = 0.880, Tmax = 0.964 | l = −20→23 |
| 14337 measured reflections |
Refinement
| Refinement on F2 | Secondary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: inferred from neighbouring sites |
| R[F2 > 2σ(F2)] = 0.039 | H atoms treated by a mixture of independent and constrained refinement |
| wR(F2) = 0.108 | w = 1/[σ2(Fo2) + (0.0579P)2 + 0.2054P] where P = (Fo2 + 2Fc2)/3 |
| S = 1.05 | (Δ/σ)max = 0.001 |
| 3451 reflections | Δρmax = 0.47 e Å−3 |
| 148 parameters | Δρmin = −0.23 e Å−3 |
| 0 restraints | Absolute structure: Flack & Bernardinelli (2000), 1447 Friedel pairs |
| Primary atom site location: structure-invariant direct methods | Flack parameter: 0.00 (11) |
Special details
| Experimental. SADABS (Bruker, 2009) was used to perform the numeric absorption correction based on the crystal dimensions determined by face indexing.The number of Friedel pairs measured is 1447. The crystal was not cut to size as it tended to fracture. |
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| C11 | 0.0173 (3) | 0.51865 (12) | 0.54568 (9) | 0.0333 (3) | |
| C12 | 0.2219 (4) | 0.53887 (16) | 0.58763 (12) | 0.0445 (4) | |
| H12 | 0.3342 | 0.4849 | 0.5926 | 0.053* | |
| C13 | 0.2585 (4) | 0.64005 (18) | 0.62216 (13) | 0.0530 (5) | |
| H13 | 0.3935 | 0.6530 | 0.6513 | 0.064* | |
| C14 | 0.0955 (4) | 0.72087 (17) | 0.61322 (14) | 0.0539 (5) | |
| H14 | 0.1215 | 0.7886 | 0.6359 | 0.065* | |
| C15 | −0.1073 (4) | 0.70186 (17) | 0.57065 (15) | 0.0539 (5) | |
| H15 | −0.2170 | 0.7567 | 0.5646 | 0.065* | |
| C16 | −0.1464 (3) | 0.60086 (15) | 0.53698 (12) | 0.0428 (4) | |
| H16 | −0.2828 | 0.5881 | 0.5085 | 0.051* | |
| C21 | 0.1121 (4) | 0.37609 (16) | 0.41002 (11) | 0.0463 (4) | |
| H21A | 0.2768 | 0.3904 | 0.4181 | 0.056* | |
| H21B | 0.0971 | 0.3022 | 0.3910 | 0.056* | |
| C22 | 0.0212 (3) | 0.45208 (15) | 0.34658 (10) | 0.0400 (4) | |
| C23 | 0.1432 (5) | 0.54488 (18) | 0.32764 (13) | 0.0561 (5) | |
| H23 | 0.2820 | 0.5606 | 0.3539 | 0.067* | |
| C24 | 0.0593 (6) | 0.6153 (2) | 0.26926 (16) | 0.0756 (8) | |
| H24 | 0.1425 | 0.6777 | 0.2566 | 0.091* | |
| C25 | −0.1438 (6) | 0.5929 (3) | 0.23093 (16) | 0.0769 (9) | |
| H25 | −0.2001 | 0.6403 | 0.1924 | 0.092* | |
| C26 | −0.2657 (6) | 0.5010 (3) | 0.24868 (14) | 0.0720 (7) | |
| H26 | −0.4033 | 0.4855 | 0.2216 | 0.086* | |
| C27 | −0.1855 (4) | 0.4311 (2) | 0.30663 (12) | 0.0535 (5) | |
| H27 | −0.2709 | 0.3693 | 0.3190 | 0.064* | |
| H1 | 0.101 (5) | 0.234 (3) | 0.5458 (19) | 0.080* | |
| O1 | 0.0790 (3) | 0.30554 (11) | 0.56144 (9) | 0.0480 (3) | |
| O2 | −0.2947 (2) | 0.37242 (9) | 0.49255 (9) | 0.0444 (3) | |
| P1 | −0.03542 (8) | 0.38743 (3) | 0.50410 (3) | 0.03482 (11) |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| C11 | 0.0375 (8) | 0.0272 (6) | 0.0350 (7) | −0.0003 (6) | 0.0014 (6) | −0.0005 (5) |
| C12 | 0.0399 (9) | 0.0415 (9) | 0.0520 (10) | 0.0028 (8) | −0.0048 (8) | −0.0043 (8) |
| C13 | 0.0490 (11) | 0.0526 (11) | 0.0574 (11) | −0.0089 (9) | −0.0107 (9) | −0.0098 (9) |
| C14 | 0.0639 (14) | 0.0378 (9) | 0.0600 (12) | −0.0056 (9) | −0.0009 (10) | −0.0138 (9) |
| C15 | 0.0590 (13) | 0.0342 (9) | 0.0684 (13) | 0.0105 (9) | −0.0095 (10) | −0.0101 (9) |
| C16 | 0.0420 (9) | 0.0341 (8) | 0.0525 (10) | 0.0031 (7) | −0.0078 (8) | −0.0058 (7) |
| C21 | 0.0504 (10) | 0.0418 (9) | 0.0465 (9) | 0.0155 (8) | 0.0003 (8) | −0.0070 (8) |
| C22 | 0.0468 (10) | 0.0405 (8) | 0.0327 (7) | 0.0060 (7) | 0.0035 (7) | −0.0051 (6) |
| C23 | 0.0652 (14) | 0.0509 (11) | 0.0523 (11) | −0.0075 (10) | 0.0080 (10) | −0.0044 (9) |
| C24 | 0.111 (2) | 0.0522 (13) | 0.0638 (14) | −0.0030 (16) | 0.0222 (15) | 0.0105 (12) |
| C25 | 0.109 (2) | 0.0749 (18) | 0.0464 (12) | 0.0311 (18) | 0.0081 (14) | 0.0147 (12) |
| C26 | 0.0729 (15) | 0.103 (2) | 0.0404 (10) | 0.0214 (15) | −0.0098 (11) | −0.0021 (12) |
| C27 | 0.0559 (12) | 0.0641 (13) | 0.0405 (10) | −0.0047 (10) | −0.0024 (9) | −0.0037 (9) |
| O1 | 0.0649 (10) | 0.0328 (6) | 0.0461 (7) | 0.0112 (6) | −0.0006 (6) | 0.0035 (5) |
| O2 | 0.0413 (6) | 0.0326 (5) | 0.0592 (8) | −0.0041 (5) | 0.0011 (6) | −0.0015 (6) |
| P1 | 0.0409 (2) | 0.02614 (17) | 0.03746 (19) | 0.00195 (14) | 0.00110 (17) | −0.00116 (16) |
Geometric parameters (Å, °)
| C11—C16 | 1.390 (2) | C21—H21B | 0.9700 |
| C11—C12 | 1.391 (3) | C22—C23 | 1.379 (3) |
| C11—P1 | 1.7893 (16) | C22—C27 | 1.386 (3) |
| C12—C13 | 1.393 (3) | C23—C24 | 1.395 (4) |
| C12—H12 | 0.9300 | C23—H23 | 0.9300 |
| C13—C14 | 1.375 (3) | C24—C25 | 1.359 (5) |
| C13—H13 | 0.9300 | C24—H24 | 0.9300 |
| C14—C15 | 1.384 (3) | C25—C26 | 1.365 (5) |
| C14—H14 | 0.9300 | C25—H25 | 0.9300 |
| C15—C16 | 1.387 (3) | C26—C27 | 1.379 (4) |
| C15—H15 | 0.9300 | C26—H26 | 0.9300 |
| C16—H16 | 0.9300 | C27—H27 | 0.9300 |
| C21—C22 | 1.511 (3) | O1—P1 | 1.5420 (14) |
| C21—P1 | 1.7962 (19) | O1—H1 | 0.93 (3) |
| C21—H21A | 0.9700 | O2—P1 | 1.5104 (13) |
| C16—C11—C12 | 119.46 (16) | C23—C22—C21 | 120.2 (2) |
| C16—C11—P1 | 120.38 (13) | C27—C22—C21 | 121.29 (19) |
| C12—C11—P1 | 120.15 (13) | C22—C23—C24 | 120.3 (3) |
| C11—C12—C13 | 119.89 (18) | C22—C23—H23 | 119.9 |
| C11—C12—H12 | 120.1 | C24—C23—H23 | 119.9 |
| C13—C12—H12 | 120.1 | C25—C24—C23 | 120.0 (3) |
| C14—C13—C12 | 120.20 (19) | C25—C24—H24 | 120.0 |
| C14—C13—H13 | 119.9 | C23—C24—H24 | 120.0 |
| C12—C13—H13 | 119.9 | C24—C25—C26 | 120.3 (2) |
| C13—C14—C15 | 120.26 (18) | C24—C25—H25 | 119.8 |
| C13—C14—H14 | 119.9 | C26—C25—H25 | 119.8 |
| C15—C14—H14 | 119.9 | C25—C26—C27 | 120.2 (3) |
| C14—C15—C16 | 119.90 (19) | C25—C26—H26 | 119.9 |
| C14—C15—H15 | 120.0 | C27—C26—H26 | 119.9 |
| C16—C15—H15 | 120.0 | C26—C27—C22 | 120.6 (2) |
| C15—C16—C11 | 120.27 (18) | C26—C27—H27 | 119.7 |
| C15—C16—H16 | 119.9 | C22—C27—H27 | 119.7 |
| C11—C16—H16 | 119.9 | P1—O1—H1 | 120 (2) |
| C22—C21—P1 | 114.11 (12) | O2—P1—O1 | 114.73 (8) |
| C22—C21—H21A | 108.7 | O2—P1—C11 | 109.11 (8) |
| P1—C21—H21A | 108.7 | O1—P1—C11 | 106.16 (8) |
| C22—C21—H21B | 108.7 | O2—P1—C21 | 109.93 (9) |
| P1—C21—H21B | 108.7 | O1—P1—C21 | 107.28 (8) |
| H21A—C21—H21B | 107.6 | C11—P1—C21 | 109.49 (9) |
| C23—C22—C27 | 118.5 (2) | ||
| C16—C11—C12—C13 | 1.6 (3) | C24—C25—C26—C27 | 1.1 (4) |
| P1—C11—C12—C13 | −177.83 (16) | C25—C26—C27—C22 | −1.1 (4) |
| C11—C12—C13—C14 | −1.6 (3) | C23—C22—C27—C26 | 0.7 (3) |
| C12—C13—C14—C15 | 0.7 (4) | C21—C22—C27—C26 | −179.8 (2) |
| C13—C14—C15—C16 | 0.1 (4) | C16—C11—P1—O2 | −22.31 (17) |
| C14—C15—C16—C11 | −0.1 (3) | C12—C11—P1—O2 | 157.10 (14) |
| C12—C11—C16—C15 | −0.7 (3) | C16—C11—P1—O1 | −146.45 (15) |
| P1—C11—C16—C15 | 178.69 (17) | C12—C11—P1—O1 | 32.96 (17) |
| P1—C21—C22—C23 | 102.5 (2) | C16—C11—P1—C21 | 98.04 (16) |
| P1—C21—C22—C27 | −77.0 (2) | C12—C11—P1—C21 | −82.55 (16) |
| C27—C22—C23—C24 | −0.2 (3) | C22—C21—P1—O2 | 54.98 (17) |
| C21—C22—C23—C24 | −179.8 (2) | C22—C21—P1—O1 | −179.66 (14) |
| C22—C23—C24—C25 | 0.2 (4) | C22—C21—P1—C11 | −64.87 (17) |
| C23—C24—C25—C26 | −0.6 (4) |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| O1—H1···O2i | 0.93 (3) | 1.58 (3) | 2.4838 (18) | 163 (3) |
Symmetry codes: (i) x+1/2, −y+1/2, −z+1.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: ZB2011).
References
- Allen, F. H. (2002). Acta Cryst. B58, 380–388. [DOI] [PubMed]
- Beckmann, J., Duthie, A., Rüttinger, R. & Schwich, T. (2009). Z. Anorg. Allg. Chem. 635, 1412–1419.
- Brandenburg, K. (2009). DIAMOND Crystal Impact GbR, Bonn, Germany.
- Bruker (2009). APEX2, SAINT and SADABS Bruker AXS Inc., Madison, Wisconsin, USA.
- Bruno, I. J., Cole, J. C., Kessler, M., Luo, J., Motherwell, W. D. S., Purkis, L. H., Smith, B. R., Taylor, R., Cooper, R. I., Harris, S. E. & Orpen, A. G. (2004). J. Chem. Inf. Comput. Sci. 44, 2133–2144. [DOI] [PubMed]
- Burrow, R. A., Farrar, D. H., Lough, A. J., Siqueira, M. R. & Squizani, F. (2000). Acta Cryst. C56, e357–e358.
- Chen, C.-T. & Suslick, K. S. (1993). Coord. Chem. Rev. 128, 293–322.
- Flack, H. D. (1983). Acta Cryst A39, 876–881.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Siqueira, M. R., Tonetto, T. C., Rizzatti, M. R., Lang, E. S., Ellena, J. & Burrow, R. A. (2006). Inorg. Chem. Commun. 9, 536–540.
- Vioux, A., Le Bideau, J., Hubert Mutin, P. & Leclerq, D. (2004). Top. Curr. Chem. 232, 145–174.
- Westrip, S. P. (2010). J. Appl. Cryst. 43, 920–925.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536811008245/zb2011sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536811008245/zb2011Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report