

Abstract
Cholinesterase inhibitors (ChEIs) are the mainstay of treatment for AD but differ by secondary mechanisms of action. We determine the effects of sub-chronic dosing of ChEIs on α7 and non-α7 nAChRs and determine if differences can be observed between them. Sprague–Dawley rats were administered donepezil, galantamine; rivastigmine at two doses each, in saline SQ twice daily or with nicotine (0.4 mg/kg) as a positive control. After 14 days the animals were sacrificed, and the levels of nAChRs were measured using [3H]-EPI to measure non-α7 nAChRs and [3H]-MLA to measure α7 nAChRs. In the cortex, all compounds tested at the higher doses significantly increased the levels of both [3H]-EPI and [3H]-MLA. In the hippocampus all compounds significantly increased [3H]-EPI but had no effect on [3H]-MLA binding. No effects were observed in the striatum with treatment. There were no differences observed among the ChEIs. In cell cultures, none of the ChEIs increased non-α7 or α7 receptor binding. Treatment with ChEIs result in similar increases in receptor levels which suggest that the increases in nAChRs may be due simply to the increases in synaptic levels of acetylcholine.
Keywords: Donepezil, Rivastigmine, Galantamine, Nicotinic receptor, Alzheimer’s disease, PC-12 cells
Introduction
Alzheimer’s disease (AD) is a chronic neurodegenerative condition that is characterized by many hallmarks pathologically including a loss of cholinergic neurons. For this reason, one of the mainstays of AD pharmacotherapy remains the use of cholinesterase inhibitors (ChEIs) as a way to increase synaptic levels of acetylcholine. While the primary mode of action for these medications is the inhibition of the enzyme acetylcholinesterase, other possible mechanisms by which ChEIs exert biological effects on the brain in AD continue to be investigated (Sabbagh et al. 2006; Nordberg 2006). Among these mechanisms, much attention has been focused on the interaction between the nicotinic acetylcholine receptors (nAChRs) and ChEIs.
Nicotinic acetylcholine receptors are a family of ligand gated ion channels whose endogenous ligand is acetylcholine. They are pentameric in structure with 17 different subunits having been reported to date (α1-10, β1-4, γ, δ and ε) that can assemble to form a large family of receptors (Millar 2003). The nAChRs found in the brain are divided into two major subtypes, the α7 and non-α7. In the mammalian brain receptors of the α7 group are composed of homomers α7 subunits, and have a high affinity for the snake toxin α-bungarotoxin as well as the alkaloid methyllycaconitine (MLA; Davies and Feisullin 1981; Barrantes et al. 1995; Davies et al. 1999). In the rat brain, levels of this class of receptor are high in the hippocampus, moderate in the cortex and low in the striatum (Clarke et al. 1985; Davies et al. 1999; Mugnaini et al. 2002).
The second group of receptors, the non-α7 subtype, are composed of heteropentamers of α2-α4 and β2-β5 subunits in a 3:2 stoichiometry, with the predominant isoform being the (α4)3(β2)2 (Cooper et al. 1991). This class is often referred to as the α4β2 subtype. These receptors have a high affinity for nicotine as well as the frog toxin epibatidine and a low affinity for α-bungarotoxin. Previous studies of the rat brain show that the regional distribution of the non-α7 receptors is distinct from the α7 subtype, with high receptor levels in the cortex and striatum with lower levels in the hippocampus (Clarke et al. 1985; Whiteaker et al. 1999; Mugnaini et al. 2002).
Changes in nAChR levels have been studied extensively in AD (Albuquerque et al. 1996; Aubert et al. 1992; Banerjee et al. 2000; Flynn and Mash 1986; Nordberg 1994; Nordberg et al. 1992a; Perry et al. 1990; Sabbagh et al. 1998; Schroder et al. 1991; Sugaya et al. 1990; Martin-Ruiz et al. 1999) as well as other dementias (Perry et al. 1995; Court et al. 1999; Reid et al. 2000; Rinne et al. 1991; Sabbagh et al. 2001). Most show selective losses of the α4β2 subtype with relative sparing of the α7 subtype. Investigations have also shown that the α7 subtype interacts with Aβ (Wang et al. 2000; Kihara et al. 1997; Liu et al. 2001; Shimohama and Kihara 2001) and that Aβ acts as an antagonist for the α7 subtype expressed in oocytes (Lamb et al. 2005; Pym et al. 2005) although the data from animal hippocampal slices is equivocal (Spencer et al. 2006). While it is not clear if the interaction between Aβ and the α7 receptors is key to the pathogenesis of AD, it does suggest that there may be an important connection between the two.
Since ChEIs represent the mainstay of treatment in AD, a lot of research has focused on if and how ChEIs interact with cholinergic receptors such as the nAChR. (Barnes et al. 2000; Bhat et al. 1990; Maelicke et al. 2001; Nordberg et al. 1992b; Samochocki et al. 2000; Storch et al. 1995; Svensson and Nordberg 1996; Svensson and Nordberg 1998; Woodruff-Pak et al. 2001). One commonly reported finding is that chronic treatment with ChEIs increases nAChR levels in rodents (Bhat et al. 1990; Nilsson-Håkansson et al. 1990) and humans (Kadir et al. 2007). However, it is not known if this effect is due to a direct or indirect action at the nAChR.
While the primary mechanism of action for the ChEIs donepezil, rivastigmine, and galantamine is the inhibition of acetylcholinesterase, the pharmacological profiles and secondary mechanisms of action for the three primary drugs used in the treatment of AD are different. Galantamine is reported to be an allosteric modulator of nAChRs (Schrattenholz et al. 1996; Villarroya et al. 2007), while rivastigmine inhibits butrylcholinesterase in addition to acetylcholinesterase (Giacobini et al. 2002). Although not directly related to the cholinergic system, donepezil, a highly selective acetylcholinesterase inhibitor, has recently been shown to be a σ1 opiate receptor agonist (Maurice et al. 2006). Since differences in secondary pharmacological effects have been identified, it is important to discern whether these drugs differ in their effects on the cholinergic system. Indeed, recent studies have indicated that in okadaic acid and Aβ induced toxicity of SH-SY5Y cells, the neuroprotective active effect of all three drugs is not related to cholinesterase activity and in addition the action of galantamine and donepezil is distinct from that of rivastigmine (Arias et al. 2005).
Previously, we have shown that sub-chronic donepezil administration increases nAChR levels in vivo while having little effect in vitro (Reid and Sabbagh 2003). In the present study, we extend our earlier observations and look for possible differences in how the three most commonly prescribed ChEIs (donepezil, rivastigmine and galantamine) affect nAChR levels both in vivo and in vitro.
Methods
Materials
Donepezil was a generous gift from Eisai Inc. and all other compounds were purchased from standard commercial sources.
Animal studies
After approval for animal experimentation from the Sun Health IACUC in accordance with USPHS Policy on Human Care and Use of Laboratory Animals, Male Sprague– Dawley rats (160–180 g, n = 6 per group) were housed with a 12-h light dark cycle with free access to food and water. Compounds were administered twice daily by subcutaneous injection of saline, 2.6 μmol/kg nicotine ditartrate (RBI, Natick, MA, USA; equivalent to 0.4 mg/kg of the free base); 0.72 μmol/kg or 2.4 μmol/kg donepezil hydrochloride (equivalent to 0.3 and 1.0 mg/kg of the free base), 1.6 or 5.0 μmol/kg galantamine (equivalent to 0.3 and 0.9 mg/kg or 0.8 or 2.0 μmol/kg Rivastigmine (equivalent to 0.1 and 0.25 mg/kg of the free base) for 14 days. Doses were chosen based on literature references for pharmacologically active doses for the four compounds (Snape et al. 1999; Barnes et al. 2000; Scali et al. 2002; Reid and Sabbagh 2003; Geerts et al. 2005). On the morning of the 15th day, the animals were euthanized and the brains removed, regionally dissected and frozen on dry ice. Samples were stored at −80°C until assayed.
Membranes were prepared for ligand binding assays by homogenization in approximately 10 volumes of homogenization buffer (HB, 120 mM NaCl, 5 mM KCl, 1 mM EDTA, 0.1% phenylmethylsulfonyl chloride and 50 mM Tris HCl, pH 7.4) using a Polytron homogenizer (Brinkmann Instruments, Westbury, NY, USA) on setting 5 for 15 s. The resulting homogenate was centrifuged for 15 min at 19,000×g. The resulting pellet was resuspended in the original volume of HB, and the process repeated. The resulting pellet was used for binding. Protein content in the samples was determined using the BCA protein assay (Pierce Chemical Co., Rockford, IL, USA) with BSA as a standard.
Tissue culture
PC12 cells (ATCC, Rockville, MD, USA) were maintained on collagen type I coated dishes (Becton Dickinson, Bedford, MA, USA) with RPMI 1640 medium containing 10% fetal bovine serum and 5% horse serum in a humidified atmosphere containing 5% CO2 at 37°C. The culture media was changed thrice weekly and the cells subcultured once per week. To investigate the effects of nicotine and donepezil, cells were plated at 107 cells per dish in 100 mm culture dishes with the same media containing donepezil, galantamine or rivastigmine (10−7 to 10−4 M) and nicotine 10−4 M serving as a control. In these experiments the media, containing either the ChEIs or nicotine was replaced daily for 3 days. On the fourth day, the cells were scraped from the plates in media, centrifuged (2,000×g, 10 min), resuspended in 2.0 ml assay buffer (AB) and centrifuged a second time. The resulting pellet was resuspended in AB and used for the binding assays.
Ligand binding assays
Binding assays were performed as previously described (Reid and Sabbagh 2003). Briefly, [3H]-(±)epibatidine (45–60 Ci/mmol, NEN, Boston, MA, USA) ([3H]-EPI) binding was determined by incubating membrane preparations (0.25–0.5 mg protein/assay point) or cell pellets (0.1–0.2 mg protein/point) in 1 ml of a buffer containing 5 nM [3H]-EPI, 120 mM NaCl, 5 mM KCl, 2 mM CaCl2 1 mM MgCl2 and 50 mM Tris pH 7.4 (AB). Non-specific binding was determined in the presence of 1 μM EPI (RBI, Natick, MA, USA). Samples were incubated on ice for 2 h, and the assay terminated by rapid filtration through GF/C filters, presoaked in 0.5% polyethyleneimine for at least 1 h, using a Brandell Cell Harvester (Brandell Instruments, Gaithersburg, MD, USA). Filters were transferred to vials containing 5 ml Ecolume scintillation cocktail (ICN Biomedical, Costa Mesa, CA, USA) and radioactivity quantified by liquid scintillation spectrometry (LS 6500, Beckman Instruments, Torrance, CA, USA). There were three replicates per sample.
[3H]-Methyllyconitine binding ([3H]-MLA) was performed as described above for [3H]-EPI, with 10 nM [3H]-MLA in the AB. Non-specific binding was determined in the presence of 1 μMunlabelled MLA. In all experiments non-specific binding was less than 25% of the total binding.
The data was analyzed for statistical significance by ANOVA using commercially available software (Prizm, Instat Software, San Diego, CA, USA).
Results
The questions being addressed in this research involve how ChEIs may interact with nAChRs both in vivo and in vitro. The results from the in vivo phase are presented in Figs. 1a, b, c and 2a, b, c. In the cortical tissue samples, the [3H]-EPI binding for the saline control animals was 42.4 ± 3.1 fmol/mg protein (n = 6). The level of [3H]-EPI binding in response to nicotine, high dose donepezil and rivastigmine, as well as both galantamine doses treatment were significantly increased compared to saline control (Fig. 1a, P<0.05). In the hippocampus [3H]-EPI binding was 35.5 ± 0.8 fmol/mg protein (n = 6) in the saline control. Hippocampal [3H]-EPI binding was significantly increased for the animals treated with nicotine, donepezil, galantamine and rivastigmine. (Fig. 1b P<0.05). In the striatal tissue samples, the [3H]-EPI binding was 95.8 ± 3.2 fmol/mg (n = 6) for saline controls. There was no change in binding for any of the compounds tested (Fig. 1c P>0.05).
Fig. 1.

Effect of chronic treatment with nicotine or cholinesterase inhibitors on [3H]-EPI binding to rat brain membranes. Rats (n = 6 per group) were treated nicotine (0.4 mg/kg), donepezil (0.3 mg/kg, black bars or 1.0 mg/kg, checked bars), galantamine (0.3 mg/kg, black bars or 0.9 mg/kg, checked bars) or rivastigmine (0.1 mg/kg, black bars or 0.25 mg/kg, checked bars) twice daily for 14 days. Subsequent to the treatment, non-α7 nAChR levels three brain regions were determined by [3H]-EPI binding as described in “Methods.” a Cortex, b hippocampus, c striatum, *P<0.05
Fig. 2.

Effect of chronic treatment with nicotine or cholinesterase inhibitors on [3H]-MLA binding to rat brain membranes. Rats (n = 6 per group) were treated nicotine (0.4 mg/kg) or donepezil (0.3 mg/kg, black bars or 1.0 mg/kg, checked bars), galantamine (0.3 mg/kg, black bars or 0.9 mg/kg, checked bars) or rivastigmine (0.1 mg/kg, black bars or 0.25 mg/kg, checked bars) twice daily for 14 days. Subsequent to the treatment, α7 nAChR levels three brain regions were determined by[3H]-MLA binding as described in “Methods.” a Cortex, b hippocampus, c striatum, *P<0.05
When examining the results on the [3H]-MLA binding, the effects were less pronounced. In the cortex, the level of [3H]-MLA binding was 46.0 ± 4.2 fmol/mg protein for the saline control. Nicotine, high dose donepezil, high and low dose galantamine and rivastigmine significantly increased 3H-MLA binding in the cortex (Fig. 2a, P>0.05). Neither treatment had any significant effect in the hippocampus, for [3H]-MLA binding compared to saline control (Fig. 2b, P>0.1). There were also no significant differences for [3H]-MLA binding in the striatum (Fig. 2c, P>0.1).
Following the examination of the effects of donepezil in vivo, we also set out to determine how donepezil may regulate nAChR levels in cell culture models. The results of these experiments are shown in Figs. 3 and 4. Untreated PC12 cells measured 17.1 ± 2.4 fmol/mg protein for [3H]-EPI binding. When grown in the presence of 0–100 μM donepezil, galantamine or rivastigmine, no significant increases in the level of [3H]-EPI binding were seen, although there are slight but not significant increases at the 10 and 100 μM concentrations (P>0.1). Nicotine on the other hand, at a concentration of 10 μM for a 3 day treatment increased [3H]-EPI binding (P<0.01, Fig. 3).
Fig. 3.
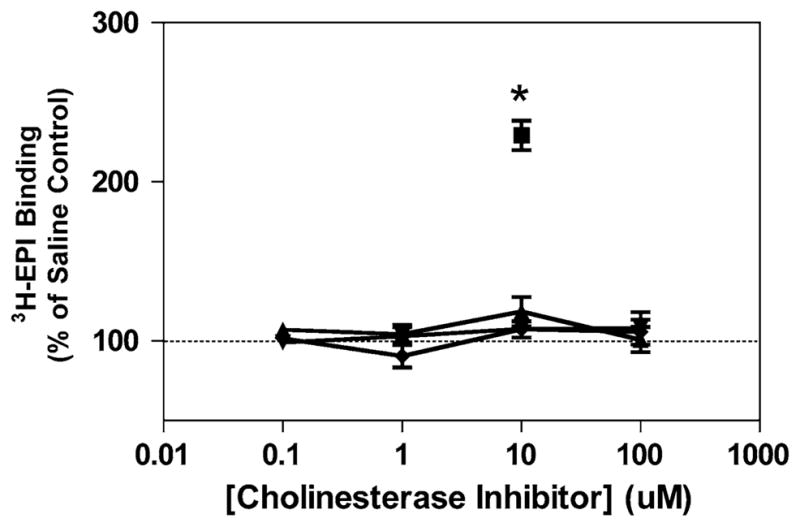
Effect of cholinesterase inhibitors on non-α7 nAChR levels in PC-12 cells. Cells were cultured in RPMI-1640 medium +10% FBS and 5% HS in the presence of 10 μM nicotine or 0–100 μM donepezil for 3 days, with the media replaced daily. Following the cell culture, nAChR levels were determined by [3H]-EPI binding as described in “Methods”. (dark filled square) nicotine, (dark filled triangle) donepezil (dark filled inverted triangle) galantamine and (dark filled diamond) rivastigmine
Fig. 4.
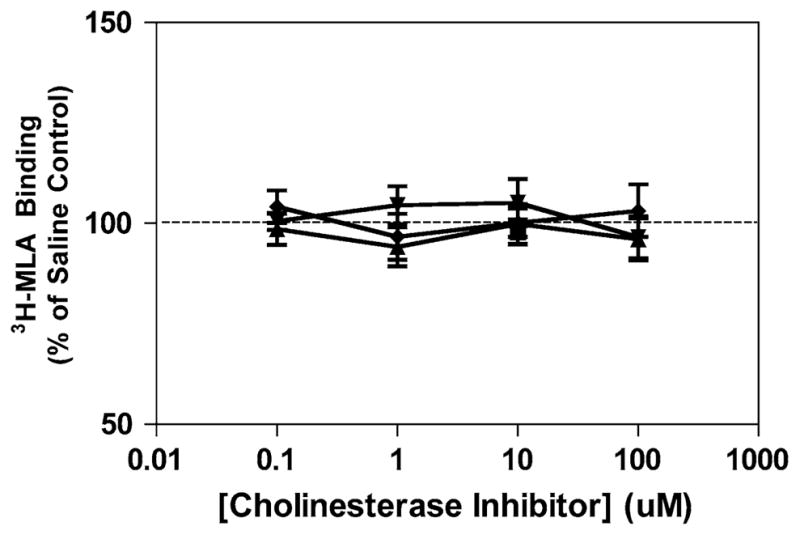
Effect of cholinesterase inhibitors on α7 nAChR levels in PC-12 cells. Cells were cultured in RPMI-1640 medium +10% FBS and 5% HS in the presence of 10 μM nicotine or 0–100 μM donepezil for 3 days, with the media replaced daily. Following the cell culture, nAChR levels were determined by[3H]-MLA binding as described in “Methods”. (dark filled square) nicotine, (dark filled triangle) donepezil (dark filled inverted triangle) galantamine and (dark filled diamond) rivastigmine
PC12 cells were also treated with nicotine, donepezil galantamine or rivastigmine and examined for changes in [3H]-MLA binding. Binding in the untreated control cells was 6.2 ± 0.9 fmol/mg protein. Figure 4 demonstrates that unlike the results obtained for [3H]-EPI binding, no increases [3H]-MLA binding were observed for either nicotine (10 μM) or with any of the ChEI (0.1–100 μM) treatments.
Discussion
In this report we present a side by side comparison of the effects on nAChR binding for three of the ChEIs commonly used in the treatment of AD. The most interesting finding to come out of this series of experiments is the result that although all three compounds have slightly different mechanisms of action, mainly relating to secondary effects of the compounds, they all up-regulate nAChRs in a similar way. Donepezil, rivastigmine and galantamine increase non-α7 and α7 receptor levels in the cortex of rats and increase non-α7 receptor levels in the hippocampus. The in vitro data suggests that the positive effects of ChEIs on nAChR binding activity are not related to a direct agonist action. Although the data does not allow us to make any statements on how secondary mechanisms such as allosteric potentiating effects may be involved in the observed nAChR up-regulation.
Without a full series of dose experiments we are unable to make quantitative statements about the relative potency, efficacy or duration of action of the three compounds. However, the in vivo results, along with the lack of any observed modulation of receptor levels in vitro, suggest that one possibility is that all of the tested ChEIs increase nAChR levels via a similar mechanism; by increasing synaptic acetylcholine (ACh) and not by any direct actions at the receptor itself. Additionally, our study only investigates the effects of ChEIs on total binding. Since we did not do single cell recording or other mechanistic studies, we cannot comment on the modulating properties reported for some ChEIs (Maelicke et al. 2001; Samochocki et al. 2000).
The observation that [3H]-EPI binding increases in vivo in the cortex and hippocampus but not the striatum may be related to receptor subtype differences in those three brain regions. It has recently shown that chronic nicotine administration decreased the level of conotoxin MII binding to α6 subunits in the striatum (Lai et al. 2005; Perry et al. 2007), and since nAChRs containing either α4 or α6 subunits are both found in this brain region and [3H]-EPI binds to both receptor subpopulations, the net effect of ChEIs on nAChRs in the striatum may be difficult to parse.
While we show no effects of the tested ChEIs on nAChRs in vitro, other researchers have shown that some of these compounds do increase nAChR levels in some cell lines. Hellstrom-Lindahl et al. (2000) demonstrated increases in [3H]-EPI binding in SH-SY5Y cells following 72 h incubations with donepezil or galantamine. One possible explanation for the difference between this work and ours is the choice of cell line with the former work using a human derived line while we used a rat derived line. There could be other explanations as Barik et al. (2005) also failed to see increases in nAChR levels in SH-SY5Y cells following incubation with galantamine.
The effects of ChEIs on nAChR levels in vivo are particularly interesting because they might lend additional explanations for secondary mechanism of action for ChEIs beyond simply inhibiting acetycholinesterase thereby increasing synaptic ACh (Nordberg 2006). ChEIs have been shown to have neuroprotective qualities. Tacrine and physostigmine have been shown to have interaction with the allosteric activator site of the α4β2 nAChR subtype (Svensson and Nordberg 1996; Smulders et al. 2005). Other studies suggest that galantamine might act in a similar manner, thus sensitizing the nicotinic receptor to ACh and nicotinic agonists. (Samochocki et al. 2000; Svensson and Nordberg 1997; Storch et al. 1995; Maelicke et al. 2001) Galantamine might even be a direct agonist of the nAChR (Maelicke et al. 2001). In another study both rivastigmine and galantamine have been shown to inhibit ion currents evoked by low concentrations of acetylcholine (Smulders et al. 2005). Both galantamine and donepezil, but not rivastigmine, have been shown to possess anti-apoptotic properties mediated through the α7 receptor (Arias et al. 2005). Donepezil and galantamine increase nAChR binding in a concentration dependent manner in neocortex (Reid and Sabbagh 2003; Woodruff-Pak et al. 2001; Barnes et al. 2000). Galantamine initially increases and then decreases nAChR binding in a concentration dependent manner and acts at the nAChR to decrease subsequent functional responses to acute stimulation with nicotine (Barik et al. 2005).
The findings on up-regulation presented here fit well with the currently accepted hypotheses on the mechanism of nicotinic receptor up-regulation. Whiteaker et al. (1998) propose an agonist mediated mechanism of up-regulation of nAChRs through a receptor state that is neither the activated nor the high affinity desensitized state. In their work, they also report little to no effect up-regulation by antagonists although they did not examine allosteric acting compounds in their study. Peng et al. (1994) proposed a similar mechanism of activation based up-regulation, although they also report up-regulation by the channel blocking antagonist mecamylamine. These reports support our hypothesis that since the ChEIs we tested are not direct agonists at the nAChR, they should not have a direct effect on the up-regulation of the receptor.
Further study will need to be focused on how ChEIs effect nAChR binding in transgenic mice and how the chronic cholinergic stimulation provided might affect AD pathology and whether it is mediated through nAChR activity. Further study will also need to elucidate the interaction between Aβ and nAChRs.
Acknowledgments
This work was funded by an unrestricted grant from Eisai Inc. and Pfizer Inc., the Erik and Ese Banck Clinical Research Center, NIA P30 AG 019610 and the Sun Health Research Institute.
Abbreviations
- AD
Alzheimer’s disease
- ChEI
Acetylcholinesterase inhibitor
- nAChR
Nicotinic acetylcholine receptor
- EPI
Epibatidine
- MLA
Methyllyconitine
Footnotes
Disclosures: Dr Reid has nothing to disclose. Dr Sabbagh is on the speaker’s bureau for Pfizer, Eisai, Forest and Novartis. He is a consultant for Lilly and Eisai. Dr Sabbagh receives clinical research funding from Eisai, Pfizer, GSK, Novartis, Avid, Wyeth, Lilly, Medivation, Abbott and Elan.
Contributor Information
Richard T. Reid, Email: richard.reid@thebanckcenter.org, Erik and Ese Banck Clinical Research Center, 8716 Production Avenue, San Diego, CA 92121, USA
Marwan N. Sabbagh, Email: Marwan.Sabbagh@sunhealth.org, The Cleo Roberts Center for Clinical Research, Sun Health Research Institute, 10515 W. Santa Fe Dr, Sun City, AZ 85351, USA
References
- Albuquerque E, Alkondon M, Pereira E, Castro N, Schrattenholz A, Barbosa C. Properties of neuronal nicotinic acetylcholine receptors, pharmacological characterization and modulation of synaptic function. J Pharmacol Exp Ther. 1996;280:1117–1136. [PubMed] [Google Scholar]
- Arias E, Gallego-Sandin S, Villarroya M, Garcia AG, Lopez MG. Unequal neuroprotection afforded by the acetylcholinesterase inhibitors galantamine, donepezil, and rivastigmine in SH-SY5Y neuroblastoma cells: role of nicotinic receptors. J Pharmacol Exp Ther. 2005;315(3):1346–1353. doi: 10.1124/jpet.105.090365. [DOI] [PubMed] [Google Scholar]
- Aubert D, Araujo M, Cecyre D, Robitaille Y, Gauthier S, Quirion E. Comparative alterations of nicotinic and muscarinic binding sites in Alzheimer’s and Parkinson’s diseases. J Neurochem. 1992;58:529–541. doi: 10.1111/j.1471-4159.1992.tb09752.x. [DOI] [PubMed] [Google Scholar]
- Banerjee C, Nyengaard JR, Wevers A, de Vos RA, Jansen-Steur EN, Lindstrom J, Pilz K, Nowacki S, Bloch W, Schroder H. Cellular expression of α7 nicotinic acetylcholine receptor protein in the temporal cortex in Alzheimer’s and Parkinson’s disease— a stereological approach. Neurobiol Dis. 2000;7:666–672. doi: 10.1006/nbdi.2000.0317. [DOI] [PubMed] [Google Scholar]
- Barik J, Dajas-Bailador F, Wonnacott S. Cellular responses to nicotinic receptor activation are decreased after prolonged exposure to galantamine in human neuroblastoma cells. Br J Pharmacol. 2005;145(8):1084–1092. doi: 10.1038/sj.bjp.0706278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes CA, Meltzer J, Houston F, Orr G, McGann K, Wenk GL. Chronic treatment of old rats with donepezil or galantamine: effects on memory, hippocampal plasticity and nicotinic receptors. Neuroscience. 2000;99:17–23. doi: 10.1016/s0306-4522(00)00180-9. [DOI] [PubMed] [Google Scholar]
- Barrantes GE, Rogers AT, Lindstrom J, Wonnacott S. α Bungarotoxin binding sites in rat hippocampal and cortical cultures: initial characterization, colocalization with α7 subunits and upregulation by chronic nicotine treatment. Brain Res. 1995;672:228–236. doi: 10.1016/0006-8993(94)01386-v. [DOI] [PubMed] [Google Scholar]
- Bhat RV, Turner SL, Marks MJ, Collins AC. Selective changes in sensitivity to cholinergic agonists and receptor changes elicited by continuous physostigmine infusion. J Pharmacol Exp Ther. 1990;255:187–196. [PubMed] [Google Scholar]
- Clarke PB, Schwartz RD, Paul SM, Pert CB, Pert A. Nicotinic binding in rat brain: autoradiographic comparison of [3H]acetylcholine, [3H]nicotine, and [125I]-alpha-bungarotoxin. J Neurosci. 1985;5(5):1307–1315. doi: 10.1523/JNEUROSCI.05-05-01307.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper E, Couturier S, Ballivet M. Pentameric structure and subunit stoichiometry of a neuronal nicotinic acetylcholine receptor. Nature. 1991;350(6315):235–238. doi: 10.1038/350235a0. [DOI] [PubMed] [Google Scholar]
- Court J, Spurden D, Lloyd S, McKeith I, Ballard C, Cairns N, Kerwin R, Perry R, Perry EK. Neuronal nicotinic receptors in dementia with lewy bodies and schizophrenia: α-bungarotoxin and nicotine binding in the thalamus. J Neurochemistry. 1999;73:1590–1597. doi: 10.1046/j.1471-4159.1999.0731590.x. [DOI] [PubMed] [Google Scholar]
- Davies P, Feisullin S. Postmortem stability of α-bungarotoxin binding sites in mouse and human brain. Brain Res. 1981;216:449–454. doi: 10.1016/0006-8993(81)90148-7. [DOI] [PubMed] [Google Scholar]
- Davies AR, Hardick DJ, Blagbrough IS, Potter BV, Wolstenholme AJ, Wonnacott S. Characterisation of the binding of [3H]methyllycaconitine: a new radioligand for labelling alpha 7-type neuronal nicotinic acetylcholine receptors. Neuropharmacology. 1999;38(5):679–690. doi: 10.1016/s0028-3908(98)00221-4. [DOI] [PubMed] [Google Scholar]
- Flynn DD, Mash DC. Characterization of L-[3H]Nicotine binding in human cerebral cortex: comparison between Alzheimer’s disease and the normal. J Neurochemistry. 1986;47:1948–1954. doi: 10.1111/j.1471-4159.1986.tb13113.x. [DOI] [PubMed] [Google Scholar]
- Geerts H, Guillaumat PO, Granthham C, Bode W, Anciaux K, Sachak S. Brain levels and acetylcholinesterase inhibition with galanthamine and donepezil in rats mice and rabits. Brain Res. 2005;1033:186–193. doi: 10.1016/j.brainres.2004.11.042. [DOI] [PubMed] [Google Scholar]
- Giacobini E, Spiegel R, Enz A, Veroff AE, Cutler NR. Inhibition of acetyl- and butyryl-cholinesterase in the cerebrospinal fluid of patients with Alzheimer’s disease by rivastigmine: correlation with cognitive benefit. J Neural Transm. 2002;109:1053–1065. doi: 10.1007/s007020200089. [DOI] [PubMed] [Google Scholar]
- Hellstrom-Lindahl E, Moore H, Nordberg A. Increased levels of tau protein in SH-SY5Y cells after treatment with cholinesterase inhibitors and nicotinic agonists. J Neurochem. 2000;74(2):777–784. doi: 10.1046/j.1471-4159.2000.740777.x. [DOI] [PubMed] [Google Scholar]
- Kadir A, Darreh-Shori T, Almkvist O, Wall A, Långström B, Nordberg A. Changes in brain 11C-nicotine binding sites in patients with mild Alzheimer’s disease following rivastigmine treatment as assessed by PET. Psychopharmacology (Berl) 2007;191(4):1005–1014. doi: 10.1007/s00213-007-0725-z. [DOI] [PubMed] [Google Scholar]
- Kihara T, Shimohama S, Sawada H, Kimura J, Kume T, Kochiyama H, Maeda T, Akaike A. Nicotinic receptor stimulation protects neurons against beta-amyloid toxicity. Ann Neurol. 1997;42:159–163. doi: 10.1002/ana.410420205. [DOI] [PubMed] [Google Scholar]
- Lai A, Parameswaran N, Khwaja M, Whiteaker P, Lindstrom JM, Fan H, McIntosh JM, Grady SR, Quik M. Long-term nicotine treatment decreases striatal alpha 6* nicotinic acetylcholine receptor sites and function in mice. Mol Pharmacol. 2005;67(5):1639–1647. doi: 10.1124/mol.104.006429. [DOI] [PubMed] [Google Scholar]
- Lamb PW, Melton MA, Yakel JL. Inhibition of neuronal nicotinic acetylcholine receptor channels expressed in Xenopus oocytes by beta-amyloid1–42 peptide. J Mol Neurosci. 2005;27(1):13–21. doi: 10.1385/JMN:27:1:013. [DOI] [PubMed] [Google Scholar]
- Liu Q, Kawai H, Berg DK. β-Amyloid peptide blocks the response of α7-containing nicotinic receptors on hippocampal neurons. Proc Natl Acad Sci USA. 2001;98:4734–4739. doi: 10.1073/pnas.081553598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maelicke A, Samochocki M, Jostock J, Fehrenbacher A, Ludwig J, Albuquerque EX, Zerlin M. Allosteric sensitization of nicotinic receptors by galanthamine, a new treatment strategy for Alzheimer’s disease. Biol Psychiatry. 2001;49:279–288. doi: 10.1016/s0006-3223(00)01109-4. [DOI] [PubMed] [Google Scholar]
- Martin-Ruiz CA, Court JA, Molnar E, Lee M, Gotti C, Mamalaki A, Tsouloufis T, Tzartos S, Ballard C, Perry RH, Perry EK. α4 but not α3 and α7 nicotinic etylcholine receptor subunits are lost from the temporal cortex in Alzheimer’s disease. J Neurochem. 1999;73:1635–1640. doi: 10.1046/j.1471-4159.1999.0731635.x. [DOI] [PubMed] [Google Scholar]
- Maurice T, Meunier J, Feng B, Ieni J, Monaghan DT. Interaction with sigma(1) protein, but not N-methyl-D-aspartate receptor, is involved in the pharmacological activity of donepezil. J Pharmacol Exp Ther. 2006;317(2):606–614. doi: 10.1124/jpet.105.097394. [DOI] [PubMed] [Google Scholar]
- Millar NS. Assembly and subunit diversity of nicotinic acetylcholine receptors. Biochem Soc Trans. 2003;31:869–874. doi: 10.1042/bst0310869. [DOI] [PubMed] [Google Scholar]
- Mugnaini M, Tessari M, Tarter G, Merlo Pich E, Chiamulera C, Bunnemann B. Upregulation of [3H]methyllycaconitine binding sites following continuous infusion of nicotine, without changes of alpha7 or alpha6 subunit mRNA: an autoradiography and in situ hybridization study in rat brain. Eur J Neurosci. 2002;16(9):1633–1646. doi: 10.1046/j.1460-9568.2002.02220.x. [DOI] [PubMed] [Google Scholar]
- Nilsson-Håkansson L, Lai Z, Nordberg A. Tetrahydroamino-acridine induces opposite changes in muscarinic and nicotinic receptors in rat brain. Eur J Pharmacol. 1990;186(2–3):301–305. doi: 10.1016/0014-2999(90)90448-f. [DOI] [PubMed] [Google Scholar]
- Nordberg A. Human nicotinic receptors—their role in aging and dementia. Neurochem Int. 1994;25:93–97. doi: 10.1016/0197-0186(94)90059-0. [DOI] [PubMed] [Google Scholar]
- Nordberg A. Mechanisms behind the neuroprotective actions of cholinesterase inhibitors in Alzheimer disease. Alzheimer Dis Assoc Disord. 2006;20(2 Suppl 1):S12–S18. doi: 10.1097/01.wad.0000213804.59187.2d. [DOI] [PubMed] [Google Scholar]
- Nordberg A, Alafuzoff I, Winblad B. Nicotinic and muscarinic subtypes in the human brain: changes with aging and dementia. J Neurosci Res. 1992a;31:103–111. doi: 10.1002/jnr.490310115. [DOI] [PubMed] [Google Scholar]
- Nordberg A, Lilja A, Lundqvist H, Hartvig P, Amberla P, Viitanen M, Warpman U, Johansson M, Hellstrom-Lindahl E, Bjurling P. Tacrine restores cholinergic nicotinic receptors and glucose metabolism in Alzheimer patients as visualized by positron emission tomography. Neurobiol Aging. 1992b;13:747–758. doi: 10.1016/0197-4580(92)90099-j. [DOI] [PubMed] [Google Scholar]
- Peng X, Gerzanich V, Anand R, Whiting PJ, Lindstrom J. Nicotine-induced increase in neuronal nicotinic receptors results from a decrease in the rate of receptor turnover. Mol Pharmacol. 1994;46(3):523–530. [PubMed] [Google Scholar]
- Perry EK, Smith CJ, Court JA, Perry RH. Cholinergic nicotinic and muscarinic receptors in dementia of Alzheimer, Parkinson, and Lewy body types. J Neural Transm. 1990;2:149–158. doi: 10.1007/BF02257646. [DOI] [PubMed] [Google Scholar]
- Perry EK, Morris CM, Court JA, Cheng A, Fairbairn AF, McKeith IG, Irving D, Brown A, Perry RH. Alteration in nicotine binding sites in Parkinson’s disease, Lewy body dementia and Alzheimer’s disease: possible index of early neuropathology. Neuroscience. 1995;64:385–395. doi: 10.1016/0306-4522(94)00410-7. [DOI] [PubMed] [Google Scholar]
- Perry DC, Mao D, Gold AB, McIntosh JM, Pezzullo JC, Kellar KJ. Chronic nicotine differentially regulates alpha6- and beta3-containing nicotinic cholinergic receptors in rat brain. J Pharmacol Exp Ther. 2007;322(1):306–315. doi: 10.1124/jpet.107.121228. [DOI] [PubMed] [Google Scholar]
- Pym L, Kemp M, Raymond-Delpech V, Buckingham S, Boyd CA, Sattelle D. Subtype-specific actions of beta-amyloid peptides on recombinant human neuronal nicotinic acetylcholine receptors (alpha7, alpha4beta2, alpha3beta4) expressed in Xenopus laevis oocytes. Br J Pharmacol. 2005;146(7):964–971. doi: 10.1038/sj.bjp.0706403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid RT, Sabbagh MN. Effects of donepezil treatment on rat nicotinic acetylcholine receptor levels in vivo and in vitro. J Alzheimer’s Dis. 2003;5(6):429–436. doi: 10.3233/jad-2003-5602. [DOI] [PubMed] [Google Scholar]
- Reid RT, Sabbagh MN, Corey-Bloom J, Tiraboschi P, Thal LJ. Nicotinic receptor losses in dementia with Lewy bodies: comparisons with Alzheimer’s disease. Neurobiol Aging. 2000;21:741–746. doi: 10.1016/s0197-4580(00)00168-8. [DOI] [PubMed] [Google Scholar]
- Rinne JO, Myllkyla T, Lonnberg P, Marjamaki P. A post mortem study of brain nicotinic receptors in Parkinson’s and Alzheimer’s disease. Brain Res. 1991;547:167–170. doi: 10.1016/0006-8993(91)90588-m. [DOI] [PubMed] [Google Scholar]
- Sabbagh MN, Reid RT, Corey-Bloom J, Rao TS, Hansen LA, Alford M, Masliah E, Adem A, Lloyd GK, Thal LJ. Correlation of nicotinic binding with neurochemical markers in Alzheimer’s disease. J Neural Transm. 1998;105:709–717. doi: 10.1007/s007020050090. [DOI] [PubMed] [Google Scholar]
- Sabbagh MN, Reid RT, Hansen LA, Alford M, Thal LJ. Correlation of nicotinic receptor binding with clinical and neuropathological changes in Alzheimer’s disease and dementia with Lewy bodies. J Neural Transm. 2001;108:1149–1157. doi: 10.1007/s007020170004. [DOI] [PubMed] [Google Scholar]
- Sabbagh MN, Farlow MR, Relkin NR, Beach TG. Do cholinergic therapies have disease modifying effects in Alzheimer’s disease? Alzheimers Dement. 2006;2(2):118–125. doi: 10.1016/j.jalz.2006.02.001. [DOI] [PubMed] [Google Scholar]
- Samochocki M, Zerlin M, Jostock R, Groot Kormelink PJ, Luyten WH, Albuquerque EX, Maelicke A. Galantamine is an allosterically potentiating ligand of the human alpha4/beta2 nAChR. Acta Neurol Scand Suppl. 2000;176:68–73. doi: 10.1034/j.1600-0404.2000.00310.x. [DOI] [PubMed] [Google Scholar]
- Scali C, Casamenti F, Bellucci A, Costagli C, Schmidt B, Pepu G. Effect of subchronic administration of metrifonate, rivastigmine and donepezil on brain acetylcholine on aged F344 rats. J Neural Transm. 2002;109:1067–1080. doi: 10.1007/s007020200090. [DOI] [PubMed] [Google Scholar]
- Schrattenholz A, Pereira EF, Roth U, Weber KH, Albuquerque EX, Maelicke A. Agonist responses of neuronal nicotinic acetylcholine receptors are potentiated by a novel class of allosterically acting ligands. Mol Pharmacol. 1996;49(1):1–6. [PubMed] [Google Scholar]
- Schroder H, Giacobini E, Struble RG, Zilles K, Maelicke A. Nicotinic cholinoceptive neurons of the frontal cortex are reduced in Alzheimer’s disease. Neurobiology. 1991;12:259–262. doi: 10.1016/0197-4580(91)90107-u. [DOI] [PubMed] [Google Scholar]
- Shimohama S, Kihara T. Nicotinic receptor-mediated protection against beta-amyloid neurotoxicity. Biol Psychiatry. 2001;49(3):233–239. doi: 10.1016/s0006-3223(00)01100-8. [DOI] [PubMed] [Google Scholar]
- Smulders CJ, Zwart R, Bermudez I, van Kleef RG, Groot-Kormelink PJ, Vijverberg HP. Cholinergic drugs potentiate human nicotinic alpha4beta2 acetylcholine receptors by a competitive mechanism. Eur J Pharmacol. 2005;509(2–3):97–108. doi: 10.1016/j.ejphar.2004.12.037. [DOI] [PubMed] [Google Scholar]
- Snape MF, Misra A, Murray TK, De Souza RJ, Williams JL, Cross AJ, Green AR. A comparative study in rats of the in vitro and in vivo pharmacology of the acetylcholinesterase inhibitors tacrine, donepezil and NXX-066. Neuropharmacology. 1999;38(1):181–93. doi: 10.1016/s0028-3908(98)00164-6. [DOI] [PubMed] [Google Scholar]
- Spencer JP, Weil A, Hill K, Hussain I, Richardson JC, Cusdin FS, Chen YH, Randall AD. Transgenic mice over-expressing human beta-amyloid have functional nicotinic alpha 7 receptors. Neuroscience. 2006;137(3):795–805. doi: 10.1016/j.neuroscience.2005.10.007. [DOI] [PubMed] [Google Scholar]
- Storch A, Schrattenholz A, Cooper JC, Abdel-Ghani EM, Gutbrod O, Weber KH, Reinhardt S, Lobron C, Hermsen B, Soskic V. Physostigmine, galanthamine and codeine act as ‘noncompetitive nicotinic receptor agonists’ on clonal rat pheochromocytoma cells. Eur J Pharmacol. 1995;290:207–219. doi: 10.1016/0922-4106(95)00080-1. [DOI] [PubMed] [Google Scholar]
- Sugaya K, Giacobini E, Chiappinelli VA. Nicotinic acetylcholine receptor subtypes in human frontal cortex: changes in Alzheimer’s disease. J Neurosci Res. 1990;27:349–359. doi: 10.1002/jnr.490270314. [DOI] [PubMed] [Google Scholar]
- Svensson AL, Nordberg A. Tacrine interacts with an allosteric activator site on alpha 4 beta 2 nAChRs in M10 cells. Neuroreport. 1996;7:2201–2205. doi: 10.1097/00001756-199609020-00029. [DOI] [PubMed] [Google Scholar]
- Svensson AL, Nordberg A. In: Alzheimer’s disease biology, diagnosis and therapeutics. Iqubal K, et al., editors. Wiley; Chichester: 1997. [Google Scholar]
- Svensson AL, Nordberg A. Tacrine and donepezil attenuate the neurotoxic effect of Aβ(25–35) in rat PC12 cells. Neuroreport. 1998;9:1519–1522. doi: 10.1097/00001756-199805110-00050. [DOI] [PubMed] [Google Scholar]
- Villarroya M, García AG, Marco-Contelles J, López MG. An update on the pharmacology of galantamine. Expert Opin Investig Drugs. 2007;16(12):1987–1998. doi: 10.1517/13543784.16.12.1987. [DOI] [PubMed] [Google Scholar]
- Wang HY, Lee DH, Davis CB, Shank RP. Amyloid peptide Aβ(1-42) binds selectively and with picomolar affinity to alpha7 nicotinic acetylcholine receptors. J Neurochemistry. 2000;75:1155–1161. doi: 10.1046/j.1471-4159.2000.0751155.x. [DOI] [PubMed] [Google Scholar]
- Whiteaker P, Sharples CG, Wonnacott S. Agonist-induced upregulation of alpha4beta2 nicotinic acetylcholine receptors in M10 cells: pharmacological and spatial definition. Mol Pharmacol. 1998;53(5):950–962. [PubMed] [Google Scholar]
- Whiteaker P, Davies RL, Marks MJ, Blackbrough IS, Potter BVL, Wolstenholme AJ, Collins AJ, Wonnacott S. An autoradiographic study of the distribution of binding sites for the novel a7 selective nicotinic ligand [3-H] methyllycaconitine in the mouse brain. Eur J Neurosci. 1999;11:2689–2696. doi: 10.1046/j.1460-9568.1999.00685.x. [DOI] [PubMed] [Google Scholar]
- Woodruff-Pak DS, Vogel RW, Wenk GL. Galantamine: effect on nicotinic receptor binding, acetylcholinesterase inhibition, and learning. Proc Natl Acad Sci USA. 2001;98:2089–2094. doi: 10.1073/pnas.031584398. [DOI] [PMC free article] [PubMed] [Google Scholar]