

Abstract
DNA methylation plays critical roles in transcriptional regulation and chromatin remodeling. Differentially methylated regions (DMRs) have important implications for development, aging and diseases. Therefore, genome-wide mapping of DMRs across various temporal and spatial methylomes is important in revealing the impact of epigenetic modifications on heritable phenotypic variation. We present a quantitative approach, quantitative differentially methylated regions (QDMRs), to quantify methylation difference and identify DMRs from genome-wide methylation profiles by adapting Shannon entropy. QDMR was applied to synthetic methylation patterns and methylation profiles detected by methylated DNA immunoprecipitation microarray (MeDIP-chip) in human tissues/cells. This approach can give a reasonable quantitative measure of methylation difference across multiple samples. Then DMR threshold was determined from methylation probability model. Using this threshold, QDMR identified 10 651 tissue DMRs which are related to the genes enriched for cell differentiation, including 4740 DMRs not identified by the method developed by Rakyan et al. QDMR can also measure the sample specificity of each DMR. Finally, the application to methylation profiles detected by reduced representation bisulphite sequencing (RRBS) in mouse showed the platform-free and species-free nature of QDMR. This approach provides an effective tool for the high-throughput identification of potential functional regions involved in epigenetic regulation.
INTRODUCTION
DNA methylation, as a natural and inheritable epigenetic event, affects biological phenotype by inhibiting gene expression without changing the DNA sequence (1). The quantification of methylation difference across large numbers of samples and the identification of sample-specificity are important in genomic function analysis, and may provide an important reference for identifying specific drug targets. Differentially methylated regions (DMRs), as genomic regions with different methylation statuses among multiple samples (tissues, cells, individuals or others), are regarded as possible functional regions involved in gene transcriptional regulation. The identification of DMRs among multiple tissues (T-DMRs) provides a comprehensive survey of epigenetic differences among human tissues (2). DMRs between cancer and normal samples (C-DMRs) demonstrate the aberrant methylation in cancers (3). It is well known that DNA methylation is associated with cell differentiation and proliferation (4). Many DMRs have been found in the development stages (D-DMRs) (5) and in the reprogrammed progress (R-DMRs) (6). In addition, there are intra-individual DMRs (Intra-DMRs) with longitudinal changes in global DNA methylation along with the increase of age in a given individual (7). There are also inter-individual DMRs (Inter-DMRs) with different methylation patterns among multiple individuals (8).
With the progress of DNA sequencing technologies, DNA methylation profiling techniques have undergone a veritable revolution over the past decade (9). Several techniques have been developed for profiling DNA methylation patterns across various cells or tissues. In the earliest studies, tissue-specific DNA methylation in a few genes was detected by restriction enzymes (10) or restriction landmark genomic scanning (RLGS) method (11). But these methods are subject to restriction enzyme sites and not suitable for complete whole-genome scan. The discovery of pre-treatment with sodium bisulphite chemically spurred a revolution in high sensitivity mapping of methylated cytosines (12). This approach has been widely applied in various methylation mapping projects including human epigenome project (HEP) (13). However, it is prohibitively expensive for genome-wide applications. To circumvent these limitations, Weber et al. (14) developed methylated DNA immunoprecipitation (MeDIP), which utilizes antibody against 5-methylcytosine to enrich methylated DNA. MeDIP, in combination with oligonucleotide arrays (MeDIP-chip) becomes a powerful approach for DNA methylation profiling (2,15). With the recent advances of next-generation sequencing techniques, several sequencing-based techniques, including bisulphite-based techniques MethylC-Seq (16) and reduced representation bisulphite sequencing (RRBS) (17), and enrichment-based methods MeDIP sequencing (MeDIP-seq) (18) and MBD-isolated Genome Sequencing (MiGS) (19) and enzyme-based techniques methyl-sensitive cut counting (MSCC) (20) and methylation mapping analysis by paired-end sequencing (Methyl-MAPS) (21), have been developed for the genome-wide study of DNA methylation. In most of current methylation mapping techniques, the original or pretreated DNA methylation status is represented by continuous values with measurement scale from 0 to 1 (22). The unprecedented scale and precision of data have enabled the quantitative analysis of differential DNA methylation status in gene regulation across cells/tissues (23).
With high throughput technologies over recent years, there have been considerable efforts in identifying DMRs from experimental profiles produced by specific methylation profiling techniques. Bibikova et al. compared the difference in mean methylation level between two cells, and selected the regions with P < 0.001 (t-test) as DMRs (24). In another study, the statistical significance of DMRs was defined by permutation test and the empirical Bayes approach (3). In the case of identifying DMRs from three or more samples, analysis of variance (ANOVA) and Kruskall–Wallis test were used respectively by Byun et al. (25) and Eckhardt et al. (13). The use of ANOVA assumes that the data follows a normal distribution, but this assumption is likely to be invalid with methylation data which follows bimodal distribution (5,26). Kruskall–Wallis test, as a non-parameter test, is more suitable for methylation data. However, since this method utilizes the ranks of the data rather than their original values to calculate the statistic, it may lose numeric information of the original data such as relative methylation degree among samples and the maximum fluctuation range for all samples. In addition to these statistic approaches, two non-statistical methods were proposed. Fan et al. identified DMRs as the regions with both hypermethylation (>50%) and hypomethylation (<50%) among various samples (27). It is obvious that the cut-off value 50% may induce some false DMRs in which the methylation levels are close to 50% in all samples. Another method derived by Rakyan et al. identifies a region as a hypermethylated T-DMR if the methylation level of this region in a tissue is >60% and methylation levels of this region in at least other three somatic tissues are <40%. This method identifies a region as a hypomethylated T-DMR if the methylation level of this region in a tissue is <40% and methylation levels of this region in at least other three somatic tissues are >60%. (2). In principle, the determination of tissue-specific DMRs by this mentioned method is supposed to be influenced by sample number, that is, the threshold should be redefined along with the sample number. In brief, the development of DNA methylation measurement proposes big challenges for the DMR calling methods.
Shannon entropy (28), as a quantitative measure of difference and uncertainty in a data set, has been widely applied in quantitative biology, such as identification of potential drug targets (29) and tissue-specific genes (30). To quantify methylation difference and further identify DMRs across multiple samples, we adapted the Shannon entropy and developed an improved approach, quantitative differentially methylated region (QDMR). Based on the Shannon entropy, two optimizations, preprocessing of methylation data and adjustment of entropy, were performed to quantify methylation difference. The application of QDMR on synthetic and biological methylation data demonstrated that QDMR can give a reasonable quantitative measure of methylation difference across multiple samples. In order to identify DMRs, the threshold of DMRs was determined according to a methylation probability model which was used to control for a degree of the random biological variability among samples. By the threshold, QDMR can identify T-DMRs with better performance than pervious methods. To further determine the sample-specificity of DMR, the categorical sample-specificity was also pre-defined according to entropy difference. To facilitate biomedical researchers, we developed a standalone and a web-based version of QDMR software, which is available at http://bioinfo.hrbmu.edu.cn/qdmr. In summary, QDMR can be used as an effective tool for the quantification of methylation difference and identification of DMRs across multiple samples.
MATERIALS AND METHODS
Synthetic methylation data
We generated methylation levels, which are scaled from 0 to 1 (0 = unmethylated, 1 = 100% methylation) across 10 samples in eight regions representing eight potential methylation patterns (Supplementary Table S1).
Human methylation data
The genome-wide methylation data in human was downloaded from ftp://ftp.ebi.ac.uk/pub/software/ensembl/efg/MeDIP-chip/ (2). This data set consists of human genome-wide methylation profiles processed by Batman (18) for 16 tissues/cells including 13 normal somatic tissues, placenta, sperm and the GM06990 immortalized cell line. Each region of interest (ROI) in this data set contains 5 × 50-mer probes typically. For each ROI, the methylation level in a tissue was the mean methylation level of the probes. We selected 40 437 ROIs whose methylation levels have been detected in all 16 tissues/cells. These ROIs were used to examine the capability of QDMR in quantification of methylation difference and identification of DMRs across different tissues.
Genomic annotations
The 40 437 ROIs in human were classified into seven categories (Up2kb, 5′-UTR, CodingExon, Intron, 3′-UTR, Down2kb and Intergenic regions) according to their relative positions with six gene elements based on the Refseq gene annotation in UCSC table browser (http://genome.ucsc.edu/cgi-bin/hgTables) (31). Seven categories were defined according to the following rules: (i) if the centre of a ROI is located in a gene element, the ROI is then classified into the category related to the gene element; (ii) if the centre of a ROI is located in more than one gene related categories, it is classified into a category according to the following priority: Up2kb→5′-UTR→CodingExon→Intron→3′-UTR→Down2kb and (iii) the ROIs that cannot be assigned into any gene related category are classified into Intergenic category. The distribution of ROIs in seven categories is shown in Supplementary Table S2.
CpG islands in human (hg18) were predicted by CpG_MI approach, which was developed by Su et al. (32). CpG island shores were defined as the regions located within 2 kb of CpG islands as described in Irizarry et al.’s work (3). A total of 40 437 ROIs are classified into three categories (CGIsland, CGIshore and Other) according to their relative position to CpG islands. CGIsland category consists of the ROIs whose centres are located within a CpG island. CGIshore category refers to the ROIs whose centres are located in the CpG island shores as defined above, and the remaining regions are classified into the Other category.
Gene Ontology annotations
In order to analyse the potential roles of T-DMRs in seven genomic categories, seven gene sets were obtained according to the following rules: (i) the genes related to the T-DMRs in the genomic category are classified into the same gene set; (ii) if a gene is related to T-DMRs in different genomic categories, it is classified into a gene set according to the following priority: Up2kb→5′-UTR→CodingExon→Intron→3′-UTR→Down2kb and (iii) if a gene can’t be assigned into any gene set above and there is a Intergenic T-DMR whose centre is located within 5 kb upstream or downstream of this gene, it is classified into Intergenic gene set. we investigated the functional relevance of each gene set using g:GOst in the g:profiler web service (33) for the genes related to each of the seven genome categories above. For the comparison of T-DMRs identified by QDMR and Rakyan’s method, we used another annotation tool to avoid preexisting bias in the ontology terms in g:GOst. We obtained Gene Ontology annotations for the category of ‘biological process’ using functional annotation tools in the DAVID Bioinformatics Resources 6.7 website (34). A GO term is considered significantly enriched if the Bonferroni corrected P < 0.05.
Gene expression data
Gene expression data used in this study was downloaded from the Gene Expression Omnibus (GSE1133) (35,36). There were only 11 tissues (B cells, CD4+ T cells, CD8+ T cells, liver, lung, pancreas, prostate, placenta, skeletal muscle, uterus and whole blood) with gene expression profiles that can be used for expression analysis. Annotations of probes for hg18 were downloaded from UCSC Genome Browser (http://genome.ucsc.edu/) (37), and then assigned to human refseq genes (hg18). For each probe, the expression value is the mean of MAS5-condensed fluorescence intensities in three replicates per tissue. The normalized expression data using the GCRMA algorithm were also analysed to avoid the bias of normalization algorithm. Mean expression value was used when multiple probes were available for a single gene. We used the linear expression values to quantify expression difference and identify tissue-specific differentially expressed genes (T-DEGs) by ROKU method which works on the linear expression values (38).
Histone modification data
In this work, 20 histone methylations and 18 acetylation modifications detected by ChIP-Seq experiments in human CD4+ T cells were used. These data were obtained from Human Histone Modification Database (HHMD, http://bioinfo.hrbmu.edu.cn/hhmd) (39). The histone modification tags were mapped to tissue-specific DMRs. And the tag count was normalized by the total number of bases in the region and the total read number of the given library to obtain normalized tag density (40).
Mouse methylation data
DNA methylation data of mouse (mm8) was downloaded from ftp://ftp.broad.mit.edu/pub/papers/rrbs/Meissner2008/ (5). This data set consists of mouse genome-wide methylation profiles on approximately 1 million distinct CpG dinucleotides detected by RRBS (17) for 18 tissues/cells. Seven adult tissues/cells (Brain, Liver, Lung, Spleen, B cells, CD4+ T cells and CD8+ T cells) were selected for this study. CpG islands of mouse (mm8) were also predicted by CpG_MI. The methylation level of a CpG island was estimated as the mean methylation level across all CpG dinucleotides with ≥5-fold coverage overlapping the same CpG island, requiring at least five such CpG dinucleotides. We selected 9636 CpG islands with sufficient methylation data in all the seven adult tissues/cells. The genomic categories of CpG islands are the same as described in the human methylation data.
Quantifying methylation difference using entropy
In order to quantify methylation difference across samples, we proposed a new method based on Shannon entropy. Although entropy has been used previously to identify tissue-specific genes from gene expression data (30), we first apply entropy to quantify methylation difference. As far as we know, methylation data has two unique characteristics. First, in most of methylation mapping techniques, DNA methylation status can be represented by continuous values with measurement scale from 0 to 1, or 0 to 100%, where 0 represents that the particular locus is unmethylated and 1 or 100% indicates that the particular locus is fully methylated (22). Second, methylation data follows bimodal distribution, which is different from other biological profiles such as gene expression profiles (26). Thus, the original entropy used to quantify gene expression difference from gene expression data could not be directly used to quantify DNA methylation difference from DNA methylation data. Therefore, we used two steps of optimization in order to adapt the original entropy.
The methylation vector mr of region r across  samples was defined as
samples was defined as  , where
, where 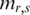 represents the methylation level in sample s. The sum of methylation levels of region
represents the methylation level in sample s. The sum of methylation levels of region  in
in 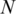 samples (
samples (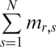 ) was treated as a total methylation value. The ratio of methylation level of region
) was treated as a total methylation value. The ratio of methylation level of region  in sample
in sample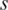 relative to the total value was defined as the relative methylation probability
relative to the total value was defined as the relative methylation probability 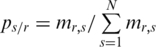 . The original Shannon entropy
. The original Shannon entropy 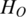 of the region
of the region  can be calculated as
can be calculated as
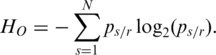 |
(1) |
According to this formula, methylation levels 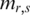 in vector mr determine the distribution of
in vector mr determine the distribution of  which further determines the value of
which further determines the value of 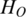 . The lower
. The lower  is the greater the methylation difference is represented across samples. The regions with consistent methylation among all samples were assigned high entropy. The regions specific methylation in minor samples should be assigned lower entropy. However, as described by Kadota et al. in the development of ROKU method (38), the original Shannon entropy is biased towards specific high values in minor samples. Thus, the specific hypermethylation in minor samples can bring about low entropy while specific hypomethylation would not. Such situations could be observed frequently in a number of promoters that were hypomethylated only in minor tissues, cancers or development stages. In order to equally quantify the methylation difference of the regions with hyper- or hypomethylation in minor samples, we calculated a one-step Tukey biweight (
is the greater the methylation difference is represented across samples. The regions with consistent methylation among all samples were assigned high entropy. The regions specific methylation in minor samples should be assigned lower entropy. However, as described by Kadota et al. in the development of ROKU method (38), the original Shannon entropy is biased towards specific high values in minor samples. Thus, the specific hypermethylation in minor samples can bring about low entropy while specific hypomethylation would not. Such situations could be observed frequently in a number of promoters that were hypomethylated only in minor tissues, cancers or development stages. In order to equally quantify the methylation difference of the regions with hyper- or hypomethylation in minor samples, we calculated a one-step Tukey biweight (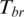 ) for region
) for region  as Kadota et al. did in the development of ROKU method (38). One-step Tukey biweight provides a robust weighted mean that is relatively insensitive to outliers (41). The median
as Kadota et al. did in the development of ROKU method (38). One-step Tukey biweight provides a robust weighted mean that is relatively insensitive to outliers (41). The median 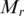 for methylation levels in
for methylation levels in 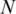 samples of region
samples of region  was first computed. Then, the absolute distance for each
was first computed. Then, the absolute distance for each 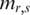 from the median was calculated as
from the median was calculated as 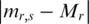 . Third, the median of the absolute distance (
. Third, the median of the absolute distance (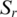 ) from Mr was determined. For each sample
) from Mr was determined. For each sample 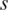 , a uniform measure of distance from the centre was defined as
, a uniform measure of distance from the centre was defined as
| (2) |
where 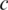 is a tuning constant (default c = 5) and
is a tuning constant (default c = 5) and 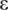 is a very small value used to avoid zero values from happening in the denominator (default
is a very small value used to avoid zero values from happening in the denominator (default  = 0.0001). A weight in each sample was then calculated by the bisquare function:
= 0.0001). A weight in each sample was then calculated by the bisquare function:
| (3) |
For each sample 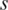 , the weight was reduced by a function of its distance from the median Mr. Thus outliers can be effectively discounted by a smooth function. When methylation levels are very far from the median, their weights are reduced to zero. Finally, the one-step Tukey biweight (
, the weight was reduced by a function of its distance from the median Mr. Thus outliers can be effectively discounted by a smooth function. When methylation levels are very far from the median, their weights are reduced to zero. Finally, the one-step Tukey biweight (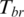 ) for region
) for region  was calculated as
was calculated as
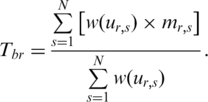 |
(4) |
The processed methylation level 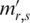 for sample
for sample 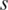 then can be calculated by using Tbr (a weighted mean)
then can be calculated by using Tbr (a weighted mean)
| (5) |
The processed methylation vector 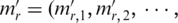
 of region
of region  was then used to calculate the region’s entropy as
was then used to calculate the region’s entropy as
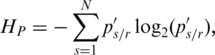 |
(6) |
where 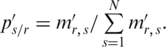
However, the range of variation was considered in neither the original Shannon entropy nor ROKU method when they were designed for expression arrays. Therefore, these two methods may not be appropriate for the analysis of the methylation arrays in which methylation level ranges from 0 to 1 (or from 0 to 100%). For example, if two genome regions A and B exhibit the same relative methylation ps/r for each 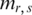 , these two regions will be assigned to the same entropy whether they have the same methylation range or not. It is possible that region A and B have different function in affecting biological process as they are very different in terms of methylation status. To overcome the shortcoming of these two methods, the entropy for each region was adjusted by a methylation weight which was defined as
, these two regions will be assigned to the same entropy whether they have the same methylation range or not. It is possible that region A and B have different function in affecting biological process as they are very different in terms of methylation status. To overcome the shortcoming of these two methods, the entropy for each region was adjusted by a methylation weight which was defined as
| (7) |
where 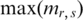 and
and 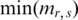 are the max and min methylation level of region r in all samples respectively, and the
are the max and min methylation level of region r in all samples respectively, and the 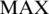 and
and 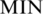 are defined as the highest methylation level 1 (or 100%, while the methylation level ranges from 0 to 100%) and the lowest methylation level 0, respectively, and
are defined as the highest methylation level 1 (or 100%, while the methylation level ranges from 0 to 100%) and the lowest methylation level 0, respectively, and  is a small value used to avoid zero values in the logarithm (default
is a small value used to avoid zero values in the logarithm (default 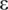 = 0.0001). Then the entropy calculated by processed methylation vector was adjusted by weight
= 0.0001). Then the entropy calculated by processed methylation vector was adjusted by weight 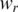 as
as
| (8) |
where 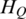 represents the extent of methylation difference across multiple samples. It ranges from zero for regions differentially methylated in a single sample with the biggest range to
represents the extent of methylation difference across multiple samples. It ranges from zero for regions differentially methylated in a single sample with the biggest range to 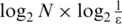 for regions with uniform methylation level in all samples considered. The maximum value of
for regions with uniform methylation level in all samples considered. The maximum value of 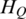 depends on the number of samples and value
depends on the number of samples and value  .
.
Determination of threshold for identification of DMRs
Since the methylation difference of a region can be represented by 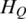 , this region can be defined as a DMR if
, this region can be defined as a DMR if 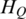 is lower than an appropriate threshold, otherwise, this regions can be defined as the N-DMR. In this study, we determined the threshold for DMRs from the methylation probability model as Schug et al. did in selecting tissue-specifically expressed genes from gene expression profiles (30). To model the effect of experimental variability, we simulated distribution of entropy from uniformly methylated regions. We computed the fold change between replicate-dependent difference from the average level across replicates and the theoretical maximum range of methylation. The fold change follows a normal distribution with mean equal to zero and some unknown, but ‘small’, standard deviation (SD) (Supplementary Figure S1). Therefore, the experimental variability will be estimated by appropriate methylation levels. To model a uniformly methylated region, we assumed that a region exhibits an average methylation level across all samples and then allow the methylation levels in individual samples to follow a narrow distribution of random fold changes from the mean level. Compared with Schug’s method, there were two major differences in this method. First, the entropy in current work is independent of the average methylation across all samples because it is derived from the methylation value processed by
is lower than an appropriate threshold, otherwise, this regions can be defined as the N-DMR. In this study, we determined the threshold for DMRs from the methylation probability model as Schug et al. did in selecting tissue-specifically expressed genes from gene expression profiles (30). To model the effect of experimental variability, we simulated distribution of entropy from uniformly methylated regions. We computed the fold change between replicate-dependent difference from the average level across replicates and the theoretical maximum range of methylation. The fold change follows a normal distribution with mean equal to zero and some unknown, but ‘small’, standard deviation (SD) (Supplementary Figure S1). Therefore, the experimental variability will be estimated by appropriate methylation levels. To model a uniformly methylated region, we assumed that a region exhibits an average methylation level across all samples and then allow the methylation levels in individual samples to follow a narrow distribution of random fold changes from the mean level. Compared with Schug’s method, there were two major differences in this method. First, the entropy in current work is independent of the average methylation across all samples because it is derived from the methylation value processed by 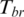 . Therefore, the biological variability modeled in this approach exhibited the average methylation level
. Therefore, the biological variability modeled in this approach exhibited the average methylation level 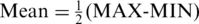 across all samples. Second, the fold change between sample-dependent difference from the average level and the theoretical maximum range of methylation was defined as
across all samples. Second, the fold change between sample-dependent difference from the average level and the theoretical maximum range of methylation was defined as 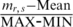 . It was assumed in this study that the fold change follows a normal distribution with mean equal to zero and some unknown, but ‘small’ SD. Thus, SD can be used to indicate the degree of the biological variation. If SD equals to zero, the methylation levels in all samples will be the same, and equal to the
. It was assumed in this study that the fold change follows a normal distribution with mean equal to zero and some unknown, but ‘small’ SD. Thus, SD can be used to indicate the degree of the biological variation. If SD equals to zero, the methylation levels in all samples will be the same, and equal to the 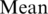 . The larger the SD is, the greater the methylation difference across multiple samples is. Setting SD = 0.07 means a relatively small amount of variation with methylation levels between 43 and 57 in 68% of the samples, between 36 and 64 in 95% of the samples, between 29 and 71 in 99% of the samples.
. The larger the SD is, the greater the methylation difference across multiple samples is. Setting SD = 0.07 means a relatively small amount of variation with methylation levels between 43 and 57 in 68% of the samples, between 36 and 64 in 95% of the samples, between 29 and 71 in 99% of the samples.
Take the determination of DMR threshold for 16 samples as an example. In total 80 000 (5000 × 16) random values were generated from the normal distribution model with mean = 0 and SD = 0.07. And 5000 uniformly methylated regions across 16 samples were modeled. Then entropy 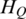 for each of these regions was calculated. The entropy value at P = 0.05 (one-sided) from the distribution of 5000 entropies, which was normal, was determined as a threshold. This process was repeated 10 times, and therefore 10 thresholds with mean (SD) equals to 5.326 (0.022) were produced. This mean was determined as the threshold
for each of these regions was calculated. The entropy value at P = 0.05 (one-sided) from the distribution of 5000 entropies, which was normal, was determined as a threshold. This process was repeated 10 times, and therefore 10 thresholds with mean (SD) equals to 5.326 (0.022) were produced. This mean was determined as the threshold 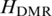 for DMR identification. Regions with entropy that is lower than
for DMR identification. Regions with entropy that is lower than  are defined as DMRs while remaining regions are not differentially methylated regions (N-DMRs). With this method, the
are defined as DMRs while remaining regions are not differentially methylated regions (N-DMRs). With this method, the 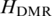 thresholds were produced for samples that vary in number from 2 to 100 (Supplementary Table S3).
thresholds were produced for samples that vary in number from 2 to 100 (Supplementary Table S3).
Measurement of sample specificity for DMRs
Based on Shannon entropy theory, the increase of variable number would reduce uncertainty, while significant changes in the individual variables would result in a substantial increase of uncertainty. The sample-specific methylation levels were considered as the main individual factors that determine the methylation differences across samples. For the region  , the entropy
, the entropy 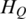 represents the methylation difference across all samples. For each sample
represents the methylation difference across all samples. For each sample 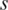 , the entropy
, the entropy 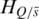 for the methylation difference across the samples that do not include sample
for the methylation difference across the samples that do not include sample 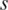 can also be calculated. Thus, the contribution of sample
can also be calculated. Thus, the contribution of sample 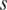 to the whole methylation difference can be reflected by the entropy difference
to the whole methylation difference can be reflected by the entropy difference 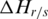 between
between  and
and 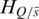 which was defined as
which was defined as
| (9) |
When region  is specifically methylated in sample
is specifically methylated in sample 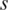 ,
, 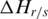 is greater than 0. To further identify hypermethylation or hypomethylation in a region, the categorical sample-specificity (
is greater than 0. To further identify hypermethylation or hypomethylation in a region, the categorical sample-specificity ( ) was presented as
) was presented as
| (10) |
where 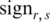 was the sign of the difference between methylation level
was the sign of the difference between methylation level 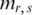 in sample
in sample  and the median methylation level of vector
and the median methylation level of vector 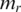 in region
in region  . Thus, the absolute value of
. Thus, the absolute value of 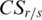 is then associated with
is then associated with 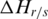 , and the sign of
, and the sign of 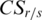 is the same as
is the same as 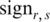 . When value in the sample
. When value in the sample 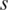 is very close to the median,
is very close to the median, 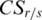 equals to zero. Specific hyper-methylation in sample
equals to zero. Specific hyper-methylation in sample 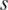 will have
will have 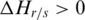 , and since
, and since 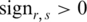 , so
, so 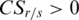 .
. 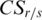 reaches its maximum when a region is relatively high-methylated in the sample
reaches its maximum when a region is relatively high-methylated in the sample 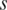 , and decreases as either the number of samples high-methylated in the region
, and decreases as either the number of samples high-methylated in the region  increases, or as the relative contribution of sample
increases, or as the relative contribution of sample  to the region’s overall pattern decreases. Similarly specific hypo-methylation in sample
to the region’s overall pattern decreases. Similarly specific hypo-methylation in sample 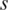 will have
will have 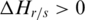 , and since
, and since  , so
, so 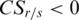 .
. 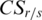 reaches its minimum when a region is relatively low-methylated in the sample
reaches its minimum when a region is relatively low-methylated in the sample 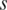 , and increases as either the number of samples low-methylated in the region
, and increases as either the number of samples low-methylated in the region  increases, or as the relative contribution of sample
increases, or as the relative contribution of sample  to the region’s overall pattern decreases.
to the region’s overall pattern decreases.
RESULTS
QDMR overview
We have developed QDMR, a bioinformatic tool for genome-wide quantitative comparisons of DNA methylation among multiple samples based on Shannon entropy (28) (details in methods). QDMR starts from the imported methylation data across a number of samples. It then performs the following steps, including quantification of methylation difference, identification of DMRs and measurement of sample-specificity (Figure 1 and Supplementary Figure S2). In the following sections, we applied QDMR to synthetic data and experimental data respectively to evaluate the performance of QDMR in quantification of methylation difference, identification of DMRs and measurement of sample-specificity for DMRs.
Figure 1.

Overview of QDMR. In step 1, the methylation data of the regions to be analysed should be detected in the lab or processed by bioinformatics methods. In step 2, methylation difference across multiple samples for each region is quantified by the adapted entropy. In step 3, based on the quantified methylation difference, DMRs are identified by the threshold for the corresponding sample number. In step 4, for each DMR identified above, the sample-specificity is measured by the categorical sample-specificity defined according to entropy difference. Finally, all results in QDMR can be exported for further analysis.
Evaluation of QDMR in quantification of methylation difference by synthetic data
To evaluate the performance of QDMR, we generated eight possible synthetic methylation patterns across ten samples which are represented by red points in Figure 2 (Supplementary Table S1). For each pattern, the methylation difference was quantified by the entropy derived from three different entropy methods, the original entropy (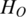 ), the entropy calculated from processed methylation vector (
), the entropy calculated from processed methylation vector (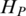 ) and the entropy derived by QDMR (
) and the entropy derived by QDMR (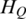 ). For each of the three calculation methods, the entropy ranges from zero to the maximum [Max(
). For each of the three calculation methods, the entropy ranges from zero to the maximum [Max( ) = Max(
) = Max(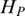 ) = log2(10) = 3.32 and Max(
) = log2(10) = 3.32 and Max(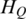 ) = log2(10) × log2(1/0.0001) = 44.1]. The lower the entropy is, the greater the methylation difference across samples is. Thus the fold change between entropy to the maximum was used to compare the performance of three entropy methods in quantification of methylation difference.
) = log2(10) × log2(1/0.0001) = 44.1]. The lower the entropy is, the greater the methylation difference across samples is. Thus the fold change between entropy to the maximum was used to compare the performance of three entropy methods in quantification of methylation difference.
Figure 2.

Quantification of methylation difference for various synthetic methylation patterns. Eight subgraphs (A–H) represent eight synthetic methylation patterns, respectively. In each subgraph, original methylation data are shown as connected red dots, and the processed data by Tukey biweight as green dots, respectively. Two blue lines represent the highest and the lowest methylation values of each region across samples, and the distance between them represents the methylation range. HO, HP and HQ represent the original entropy, the entropy calculated from processed methylation vector and the entropy derived by QDMR, respectively. The pattern identified as DMR by QDMR is tagged as ‘DMR’, while that identified as N-DMR is tagged as ‘N-DMR’, above the corresponding subgraph.
The comparative analysis demonstrated that QDMR can provide a reasonable quantitative measure of methylation difference, which is intuitive, for each of the eight methylation difference patterns. For the pattern with consistent methylation levels across samples in Figure 2A, the entropy by each of these three methods reaches its maximum indicating no methylation difference. This pattern was identified as an N-DMR by QDMR. For the pattern with specific high methylation level in one sample and low levels in others (Figure 2B), the fold change by QDMR was close to 0 while those by the other two methods were close to 0.5 and 0.3, respectively. This observation indicated that QDMR was superior to other methods in quantifying methylation difference for the region with most specific methylation pattern. The similar result is shown in the third pattern (Figure 2C). When the number of specific samples increases, QDMR can also quantify methylation difference for regions with large methylation fluctuation across samples (Figure 2D and E). QDMR identified the four patterns in Figure 2B–E as DMRs. Moreover, hypermethylation or hypomethylation in a small fluctuation range across samples, as shown in Figures 2F and G, are considered as N-DMRs in most methylation studies. High entropy was obtained from all three methods. QDMR identified this pattern as an N-DMR. However, the fold change by QDMR was near to 0.1 while those by other two methods reached 1 and 0.9, respectively, indicating QDMR may be more appropriate for the regions with small but potentially functional methylation difference. Furthermore, for the pattern with consistently high methylation in half of the samples and low methylation in the other half as shown in Figure 2H, the fold change by QDMR was close to 0, while those by other two methods were close to 0.8 and 1, respectively. This methylation pattern becomes more frequent when the number of samples decreases. The regions with this methylation pattern would be identified as DMRs by QDMR. These results indicated the importance of two-step optimization and the usability of QDMR in quantifying the difference from various methylation patterns across two or more samples.
Quantification of methylation difference from methylation profiles in 16 human tissues
The QDMR method was then applied to the human genome-wide methylation profile including 40 437 regions of interesting (ROIs) with methylation value in all 16 tissues (2). Each region was assigned an entropy value by QDMR based on the methylation levels for all the tissues. Then these regions were ranked by the entropy from low to high as shown in Figure 3A. ROIs with larger methylation difference were in the upper region while the consistently methylated or unmethylated regions were in the lower region. The top 100 and bottom 100 regions were selected for a clearer visualization and comparison (Supplementary Tables S4 and S5). All the top 100 regions exhibited different methylation status across all tissues or tissue specificity (Figure 3B). Most of them were differentially hypomethylated specifically in sperm. In contrast, all the bottom 100 regions showed relatively invariant methylation state, especially consistent hypomethylation across tissues (Figure 3C). In addition, entropy scores differed significantly among different genomic categories (Supplementary Figure S3A). Especially, CpG islands showed higher entropy than that of CpG island shores and other genome regions (Supplementary Figure S3B and C). And there is a significant positive correlation between entropy and ObsCpG/ExpCpG ratio (Supplementary Figure S3D). The entropy of a genome region can reflect the methylation difference across multiple tissues, while ObsCpG/ExpCpG ratio can reflect the enrichment of CpG dinucleotides in the same region. It is indicated that the genome regions with higher CpG density may possess more stable methylation status among tissues, which is consistent with the results from the other studies in DNA methylation and CpG density (2,5,15). Overall, QDMR can provide a precise approach to quantify methylation difference in genome regions among different tissues.
Figure 3.

Methylation heat map across 16 human tissues of regions ranked by entropy derived from QDMR. (A). Methylation heat map of all 40 437 regions ranked from top to bottom by ascending entropy. The methylation levels range from 0 (green) to 100 (red). (B) Clearer methylation heat map of the top 100 regions with the lowest entropy. The number in the last column is the entropy derived from QDMR for the region in the same row. (C) Clearer methylation heat map of the bottom 100 regions with highest entropy.
Identification of T-DMRs from genome-wide methylation profiles by QDMR
To identify T-DMRs based on the quantified methylation difference across 16 tissues/cells mentioned above, the threshold 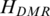 = 5.326 for identification of DMRs for the 16 samples was obtained based on methylation probability profiles (details in ‘Materials and Methods’ section). Among 40 437 regions, 10 651 (26%) with lower entropy value than
= 5.326 for identification of DMRs for the 16 samples was obtained based on methylation probability profiles (details in ‘Materials and Methods’ section). Among 40 437 regions, 10 651 (26%) with lower entropy value than 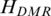 were classified as T-DMRs (Supplementary Table S6), while the remaining 29 786 (74%) regions were defined as N-DMRs. The distribution of T-DMRs and N-DMRs in seven genome categories showed that T-DMRs were present in all genome categories although they were less frequent than N-DMRs (Figure 4). There were a smaller proportion of T-DMRs in promoter than other categories, which was consistent with the previous findings (13). Interestingly, T-DMRs had a preference for certain chromosomes or regions (Supplementary Figure S4). In addition, the number of T-DMRs overlapping with CpG island shores was 1.6-fold as that with CpG islands (36 versus 22%, P < 0.0001), which was consistent with a previous finding that most tissue-specific DNA methylation occurs at CpG island shores rather than CpG islands (3).
were classified as T-DMRs (Supplementary Table S6), while the remaining 29 786 (74%) regions were defined as N-DMRs. The distribution of T-DMRs and N-DMRs in seven genome categories showed that T-DMRs were present in all genome categories although they were less frequent than N-DMRs (Figure 4). There were a smaller proportion of T-DMRs in promoter than other categories, which was consistent with the previous findings (13). Interestingly, T-DMRs had a preference for certain chromosomes or regions (Supplementary Figure S4). In addition, the number of T-DMRs overlapping with CpG island shores was 1.6-fold as that with CpG islands (36 versus 22%, P < 0.0001), which was consistent with a previous finding that most tissue-specific DNA methylation occurs at CpG island shores rather than CpG islands (3).
Figure 4.

Distribution of T-DMRs and N-DMRs in seven genome categories.
Previous studies found that promoter T-DMRs are associated with genes that are thought to function in a tissue-specific manner (2,42). However, the role of intragenic and intergenic T-DMRs is still not clear. We analysed the functions of the genes related to the T-DMRs identified by QDMR in seven genome categories. To this end, seven non-overlapping gene sets, each of which consists of the genes related to T-DMRs in the same categories, was obtained (details in ‘Materials and Methods’ section). Then the functional relevance of each gene set was investigated using g:GOst in the g:profiler web service (33) (Table 1 and Supplementary Table S7). In addition to the genes related to promoter T-DMRs, the genes related to intragenic T-DMRs also exhibited enrichment for multicellular organismal process and cell differentiation functions. The possible interpretation for this observation could be that methylation difference in gene-body may be related to alternative splicing (43) which involves in transcription regulation in the development of multicellular organisms (44). The genes related to T-DMRs in coding exons also tend to be targeted by miRNAs. It is well known that miRNAs also show tissue specificity (45) and participate in determination of cell fate (46). This unexpected finding hinted at an epigenetic control of gene function involving miRNAs and DNA methylation for which supporting evidence has been obtained by the latest study (47). Moreover, the genes close to Intergenic T-DMRs also showed enrichment in tissue-specific functions, for example cell fate specification, organ morphogenesis, and cell differentiation. It has been verified by previous finding that methylation in Intergenic T-DMRs regulates gene functions in association with multiple distal regulatory elements (48), such as enhancer (49), silencer (50). It suggests that T-DMRs identified by QDMR may have influence on those genes that participate in multicellular organismal development and cell differentiation.
Table 1.
Functional enrichment of genes related to T-DMRs by g:GOst
| Term type | Term name | P-value | Term type | Term name | P-value |
|---|---|---|---|---|---|
| Up 2 kb | |||||
| GOBP | Multicellular organismal process | 1.62E-17 | GOBP | System development | 3.02E-06 |
| GOBP | Signal transduction | 2.52E-11 | GOBP | Biological regulation | 6.59E-06 |
| GOBP | Cell surface receptor linked signalling pathway | 4.04E-09 | GOCC | Intermediate filament cytoskeleton | 1.82E-14 |
| 5′-UTR | |||||
| KEGG | Retinol metabolism | 2.63E-04 | KEGG | Leukocyte transendothelial migration | 5.74E-04 |
| Coding exon | |||||
| GOBP | Homophilic cell adhesion | 3.82E-17 | KEGG | Calcium signalling pathway | 8.03E-07 |
| GOBP | Multicellular organismal development | 9.49E-12 | REAC | Signalling by GPCR | 2.07E-11 |
| GOBP | Cell–cell adhesion | 6.91E-09 | MiRNA | MI:hsa-miR-886-5p | 1.83E-08 |
| GOBP | Calcium-dependent cell–cell adhesion | 7.48E-08 | MiRNA | MI:hsa-miR-663 | 1.96E-08 |
| GOBP | Cell morphogenesis involved in differentiation | 5.89E-07 | MiRNA | MI:hsa-miR-339-3p | 2.67E-08 |
| GOBP | Cell differentiation | 1.29E-06 | MiRNA | MI:hsa-miR-324-3p | 8.65E-08 |
| GOMF | Transcription factor activity | 6.16E-06 | MiRNA | MI:hsa-miR-638 | 5.95E-06 |
| Intron | |||||
| GOBP | Multicellular organismal development | 2.46E-12 | GOBP | Regulation of cell communication | 2.24E-06 |
| GOBP | Anatomical structure development | 4.27E-11 | GOBP | Cell adhesion | 2.65E-06 |
| GOBP | Multicellular organismal process | 4.52E-08 | GOCC | Cell projection | 9.83E-09 |
| GOBP | Cell differentiation | 4.74E-08 | GOMF | Cytoskeletal protein binding | 2.24E-12 |
| GOBP | Organ development | 1.87E-07 | GOMF | Calcium ion binding | 3.22E-07 |
| 3′-UTR | |||||
| GOCC | Intracellular | 1.69E-05 | |||
| Down 2 kb | |||||
| None | |||||
| Intergenic | |||||
| GOBP | Regulation of transcription | 1.40E-21 | GOBP | Regulation of developmental process | 2.19E-05 |
| GOBP | Regulation of gene expression | GOCC | Nucleus | 3.70E-12 | |
| GOBP | Cell fate specification | 8.96E-12 | GOMF | DNA binding | 5.10E-21 |
| GOBP | Multicellular organismal development | 2.00E-08 | GOMF | Nucleic acid binding | 6.13E-16 |
| GOBP | Organ development | 2.64E-08 | GOMF | Transcription regulator activity | 3.58E-13 |
| GOBP | Cell fate commitment | 1.11E-07 | GOMF | Transcription factor activity | 3.73E-11 |
| GOBP | Anatomical structure development | 1.35E-07 | MiRNA | MI:hsa-miR-615-5p | 3.66E-10 |
| GOBP | Organ morphogenesis | 2.97E-07 | MiRNA | MI:hsa-miR-339-3p | 6.62E-06 |
| GOBP | Regulation of transcription from RNA polymerase II promoter | 9.34E-07 | MiRNA | MI:hsa-miR-663 | 1.05E-05 |
| GOBP | Embryonic morphogenesis | 7.70E-06 | MiRNA | MI:hsa-miR-423-3p | 4.13E-05 |
| GOBP | Cell differentiation | 8.52E-06 | MiRNA | MI:hsa-miR-296-5p | 7.11E-05 |
| GOBP | Regulation of cell differentiation | 1.53E-05 | MiRNA | MI:hsa-miR-886-5p | 1.17E-04 |
Term type: Annotation database and gradation. GOBP: biological process in GO; GOCC: cellular component in GO; GOMF: molecular function in GO; KEGG: KEGG pathway; REAC: reactome pathway; MiRNA: microRNA target.
Comparison with Rakyan’s method in identification of DMRs
In order to evaluate the performance of QDMR in identification of DMRs, we compared it with a counting method developed for 16 samples by Rakyan et al. For the same regions used in this work, Rakyan’s method identified 6541 T-DMRs and 33 896 N-DMRs (Figure 5A). Thus, these two methods classed all ROIs into four groups (I, II, III and IV) as shown in Figure 5A. It was shown that the two methods were common in identification of most of T-DMRs and N-DMRs for the 16 tissues. More than half (5911/10 651) of T-DMRs identified by QDMR were also defined as T-DMRs by Rakyan’s method (Figure 5A-II), while nearly 98% (29 156/29 786) of the N-DMRs identified by QDMR were also assorted as N-DMRs by Rakyan’s method (Figure 5A-III).
Figure 5.

Performance of QDMR in identification of T-DMRs and N-DMRs. (A) Different categories of regions by Rakyan’s method and QDMR. X-axis are the ROIs which are divided into two groups according Rakyan’s method, T-DMRs represented by orange dots (I and II), and N-DMRs represented by blue dots (III and IV). Y-axis is the entropy for each ROI derived from QDMR. All 40 437 regions are classified as N-DMRs (I and III) and T-DMRs (II and IV) by DMR threshold represented by the red line. The number indicates the amount of regions in the corresponding category. (B) The mean methylation levels across 16 tissues of the regions in I and IV. (C) The range of methylation levels in 16 tissues of the regions in I and IV. (D) The standard deviation of methylation levels in 16 tissues of the regions in I and IV.
However, there were also some differences in classification of the regions in groups I and IV. Group I consisted of 630 regions defined as T-DMRs by Rakyan’s method but as N-DMR by QDMR (Figure 5A–I). The example for this group showed little methylation difference among 16 tissues (Supplementary Figure S5–I). Group IV consisted of 4740 regions as T-DMRs by QDMR but as N-DMRs by Rakyan’s method (Figure 5A–IV). The example for this group showed large methylation difference across 16 tissues, and specific hypermethylation and hypomethylation in CM06990 and Sperm, respectively (Supplementary Figure 5–IV).
To examine the methylation properties of these two groups, we calculated the mean, range and standard deviation for each region in these two groups. The mean methylation levels of ROIs in group I were close to 50, while those in group IV ranged from 10 to 90 (Figure 5B). Furthermore, ROIs in group IV showed wider range and greater standard deviation than those in group I (Figure 5C and D). To further analyse the functional properties of these two groups, Gene Ontology annotation was performed at the genes related to each group. The T-DMRs identified only by QDMR were located within or nearby 2697 genes. These genes showed enrichment for cellular developmental process, cell differentiation and cell–cell adhesion in higher specificity (Table 2 and Supplementary Table S8). Cell–cell adhesion, especially calcium-dependent cell–cell adhesion, has a key role in the organization of tissues comprising multiple cell types (51). However, 388 genes related to T-DMR identified only by Rakyan’s method did not show any functional enrichment under the same P-value threshold (Supplementary Table S8). Therefore, the T-DMRs identified by QDMR may possess more unstable methylation patterns across multiple tissues. Further enrichment analysis of the genes related with these T-DMRs suggests that these T-DMRs may participate in biological functions. Overall, QDMR provides a good performance approach for identifying DMRs across multiple samples.
Table 2.
Functional enrichment of genes related to T-DMRs identified only by QDMR based on biological process (BP)
| BP term | Gene number | Bonferroni P-value |
|---|---|---|
| Multicellular organismal process | 789 | 7.56E-15 |
| Homophilic cell adhesion | 59 | 4.80E-13 |
| Biological adhesion | 174 | 7.53E-10 |
| Cell–cell adhesion | 88 | 1.29E-09 |
| Cell adhesion | 173 | 1.33E-09 |
| Anatomical structure development | 485 | 2.25E-09 |
| Nervous system development | 243 | 2.27E-09 |
| System development | 449 | 1.38E-08 |
| Developmental process | 575 | 8.33E-08 |
| Multicellular organismal development | 527 | 2.78E-07 |
| System process | 299 | 5.24E-06 |
| Neurological system process | 249 | 7.04E-06 |
| Calcium-dependent cell–cell adhesion | 17 | 1.36E-05 |
| Cellular developmental process | 314 | 6.18E-03 |
| Cell differentiation | 303 | 6.22E-03 |
| Cell–cell signalling | 128 | 1.67E-02 |
| Anatomical structure morphogenesis | 228 | 2.12E-02 |
| Cell communication | 161 | 2.19E-02 |
| Synaptic transmission | 73 | 2.25E-02 |
| Cognition | 179 | 3.56E-02 |
| Neurogenesis | 126 | 4.85E-02 |
Only annotations with Bonferroni P < 0.05 for GO in all levels are listed here. Full lists and more details are provided in Supplementary Table S7.
Measurement of sample specificity for DMRs by QDMR
As mentioned above, QDMR can identify functional T-DMRs from genome-wide methylation data. Some T-DMRs may exhibit not only methylation difference across tissues but also specificity in a particular tissue. To measure the tissue specificity for T-DMRs identified by QDMR, categorical sample-specificity 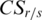 was defined based on the entropy difference
was defined based on the entropy difference  (details in ‘Materials and Methods’ section). For each tissue, specific hypermethylated T-DMRs (Hyper-T-DMRs) and specific hypomethylated T-DMRs (Hypo-T-DMRs) were selected from 10 651 regions by positive and negative
(details in ‘Materials and Methods’ section). For each tissue, specific hypermethylated T-DMRs (Hyper-T-DMRs) and specific hypomethylated T-DMRs (Hypo-T-DMRs) were selected from 10 651 regions by positive and negative  , respectively (Table 3 and Supplementary Table S9). The number of tissue-specific methylated regions varied widely among the tissues (Supplementary Figure S6). Especially, there were more specific T-DMRs in Uterus and Sperm suggesting the distinctive methylation patterns in germ cell-related tissues (52). In addition, the proportion of Hyper-T-DMRs and Hypo-T-DMRs was also different among different tissues. For example, the number of Hypo-T-DMRs was 2.5 times more than that of Hyper-T-DMRs in CD4+ T cells. Further analysis on gene EPYC, which involves in female pregnancy, demonstrated that the specific hypomethylation of the T-DMR in the promoter of EPYC may account for its specific high expression in placenta (Supplementary Figure S7). Therefore, it is speculated that specific tissue may possess its unique methylation patterns which determine its development and differentiation.
, respectively (Table 3 and Supplementary Table S9). The number of tissue-specific methylated regions varied widely among the tissues (Supplementary Figure S6). Especially, there were more specific T-DMRs in Uterus and Sperm suggesting the distinctive methylation patterns in germ cell-related tissues (52). In addition, the proportion of Hyper-T-DMRs and Hypo-T-DMRs was also different among different tissues. For example, the number of Hypo-T-DMRs was 2.5 times more than that of Hyper-T-DMRs in CD4+ T cells. Further analysis on gene EPYC, which involves in female pregnancy, demonstrated that the specific hypomethylation of the T-DMR in the promoter of EPYC may account for its specific high expression in placenta (Supplementary Figure S7). Therefore, it is speculated that specific tissue may possess its unique methylation patterns which determine its development and differentiation.
Table 3.
Specifically hyper- and hypomethylated T-DMRs across 16 human tissues
| Tissue | T-DMR | Hyper-T-DMR (%) | Hypo-T-DMR (%) |
|---|---|---|---|
| B cell | 461 | 180 (52.7) | 281 (47.3) |
| CD4+ T cell | 522 | 117 (22.4) | 405 (77.6) |
| CD8+ T cell | 840 | 619 (73.7) | 221 (26.3) |
| Cervix | 478 | 304 (63.6) | 174 (36.4) |
| Colon | 375 | 181 (50.9) | 194 (49.1) |
| Liver | 1174 | 851 (72.5) | 323 (27.5) |
| Lung | 412 | 224 (54.4) | 188 (45.6) |
| Pancreas | 657 | 321 (48.9) | 336 (51.1) |
| Prostate | 419 | 220 (52.5) | 199 (47.5) |
| Rectum | 628 | 280 (44.6) | 348 (55.4) |
| Skeletal muscle | 1865 | 1614 (86.5) | 251 (13.5) |
| Uterus | 4876 | 2057 (42.2) | 2819 (57.8) |
| Whole blood | 926 | 552 (59.6) | 374 (40.4) |
| Placenta | 1170 | 722 (61.7) | 448 (38.3) |
| Sperm | 4079 | 585 (14.3) | 3494 (85.7) |
| Gm06990 | 2415 | 1606 (66.5) | 809 (33.5) |
The relationship between histone modification and specific T-DMRs
Histone modifications play important roles in stem cell maintenance and tissue differentiation (53,54). A recent study reported the specificity of histone modifications in lineage fate determination of differentiating CD4+ T cells (55). We investigated the normalized tag density of each histone modification in Hyper-T-DMRs and Hypo-T-DMRs in CD4+ T cells. The ratio between the mean tag density in Hypo-T-DMRs and that in Hyper-T-DMRs was defined as the relative modification intensity. We compared the tag densities of each histone modification between Hyper- and Hypo-T-DMRs. As shown in Figure 6, Hypo-T-DMRs were more likely to be overlapped with active chromatin marks, such as H4K20me1, H3K79me3, H2BK5me1, H3K79me2, H3K79me1, H3K4me1, H3R2me2, H3K9me1, H4K16ac and H4K19ac, most of which correlates with active transcription of genes (40) and play critical role in mammal development (56). On the contrary, Hyper-T-DMRs were in preference in suppressive histone modifications, such as H3K9me3, H3K27me3 and H3K27me2, most of which are involved in pluripotency maintenance and cell fate decisions (53). These results suggested that Hyper-T-DMRs and Hypo-T-DMRs may correlate with histone modifications that have different activities and functions.
Figure 6.

Relative modification intensity between CD4+ T cell-specific Hyper-T-DMRs and Hypo-T-DMRs. X-axis is 38 histone modifications in CD4+ T cell. Y-axis is the relative modification intensity of histone modification between hypermethylated and hypomethylated T-DMRs. The horizontal line represents the same modification intensity between CD4+ T cell-specific Hyper-T-DMRs and Hypo-T-DMRs. ‘Asterisk’ represents those histone modifications with significantly different modification intensity between CD4+ T cell-specific Hyper-T-DMRs and Hypo-T-DMRs.
The correlation between DNA methylation difference and gene expression difference
Irizarry et al. found differential methylation across transcription start sites exhibited a strong inverse relationship with differential gene expression (3). To examine whether this relationship is still hold genome-widely, we obtained 16 258 ROIs related to 10 220 Refseq genes for 11 tissues whose corresponding gene expressions were available in the gene atlas data (36). We investigated the correlation between methylation difference and expression difference of associated genes in seven genome categories. The methylation difference of each ROI was quantified by the entropy derived from QDMR and the expression difference of each gene by the entropy was derived from ROKU (38) for average expression status of related genes of the same ROI. For ROIs in Up2kb, 5′-UTR, CodingExon and Intron, the Pearson correlation coefficient (PCC) between methylation entropy and expression entropy shows that methylation difference is positively correlated with gene expression difference (Supplementary Figure S8). This observation is consistent with a recent study which demonstrated that differential DNA methylation correlates with differential expression of angiogenic factors in human heart failure (57).
Recent studies demonstrated that T-DMRs are associated with differences in gene expression (3,42). We studied the locations of T-DMRs identified by QDMR and tissue-specific differentially expressed genes (T-DEGs) in 11 tissues. Based on the quantified methylation difference, QDMR identified 2391 T-DMRs and 13 867 N-DMRs using the threshold  = 4.637 for 11 samples. And based on the quantified expression difference, we selected 2965 T-DEGs and 7255 N-DEGs using the threshold
= 4.637 for 11 samples. And based on the quantified expression difference, we selected 2965 T-DEGs and 7255 N-DEGs using the threshold 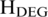 = 2.326 which was estimated from probability model of gene expression as described in Schug’s work (30). About 35.1% (840/2391) of T-DMRs located from upstream 2000 bp to downstream 2000 bp of a T-DEG (Supplementary Table S10), while only 27.5% (3813/13 867) N-DMRs located from upstream 2000 bp to downstream 2000 bp of a T-DEG. Thus T-DMRs overlapped with T-DEGs much more than expected (Chi-square test, P < 0.0001, Supplementary Table S10), which was consistent with the finding of previous studies (3,57). For example, there was a T-DMR in the first intron of gene IL7R (Supplementary Figure S9A). The hypomethylation of this T-DMR in CD4+ T cells and CD8+ T cells may involve in regulating the high expression of this gene (Supplementary Figure S9B–D), which has been demonstrated by Kim et al. (58). These results indicated that T-DMRs may involve in regulation of cell/tissue-specific gene expression which is considered as a natural event in the tissue formation process (43).
= 2.326 which was estimated from probability model of gene expression as described in Schug’s work (30). About 35.1% (840/2391) of T-DMRs located from upstream 2000 bp to downstream 2000 bp of a T-DEG (Supplementary Table S10), while only 27.5% (3813/13 867) N-DMRs located from upstream 2000 bp to downstream 2000 bp of a T-DEG. Thus T-DMRs overlapped with T-DEGs much more than expected (Chi-square test, P < 0.0001, Supplementary Table S10), which was consistent with the finding of previous studies (3,57). For example, there was a T-DMR in the first intron of gene IL7R (Supplementary Figure S9A). The hypomethylation of this T-DMR in CD4+ T cells and CD8+ T cells may involve in regulating the high expression of this gene (Supplementary Figure S9B–D), which has been demonstrated by Kim et al. (58). These results indicated that T-DMRs may involve in regulation of cell/tissue-specific gene expression which is considered as a natural event in the tissue formation process (43).
The software package
The results have demonstrated that QDMR is useful in quantification of methylation difference, identification of DMRs and measurement of sample specificity for each DMR. To facilitate its use in analysis of DMRs, we developed stand-alone and web-based software packages using Java (Supplementary Figure S10). This software includes all the features discussed in this article. It can process data files with at least two samples with the following steps: data import, differentiation quantization, DMR identification, specificity measurement and methylation visualization. Two output formats are available: tabular and graphical. The tabular output is a table of DMRs entries with columns representing region information, entropy, sample specificity and raw methylation data. The graphical output allows the user to inspect the raw methylation data pattern, DMR distribution on chromosomes and genome information at UCSC Genome Browser. The standalone and online version of QDMR is provided at http://bioinfo.hrbmu.edu.cn/qdmr. In addition, the source code is also open to the public.
Application of QDMR to the methylation profiles in mouse
Finally, QDMR software was applied to analyse the methylation data set which was detected by RRBS in seven adult mouse tissues/cells of 9636 CpG islands (5). The heat map of methylation in seven tissues/cells of mouse demonstrated that QDMR also can quantify methylation difference for mouse methylation data (Supplementary Figure S11A). Most CpG islands exhibited consistent hypomethylation across seven tissues/cells, which is consistent with a previous finding that CpG islands are often free of methylation in normal somatic tissues (59). According to the threshold 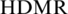 = 3.636 for seven samples, only 4% (397/9636) of CpG islands were identified as T-DMRs (Supplementary Table S11). It is implied that CpG islands possess less tissues/cells differential methylation which is consistent with the finding in human genome in this article. There were less T-DMRs than N-DMRs in each genome category, while the CpG islands in Up2kb, 5′-UTR and Intron exhibited a smaller proportion of T-DMRs than other genome categories (Supplementary Figure S11B). The distribution of all the T-DMRs identified by QDMR on mouse chromosomes was also shown in the visualization module in QDMR software (Supplementary Figure S11C). The total 326 genes that are related to these T-DMRs showed enrichment for organ development (Supplementary Table S11). For example, there is a T-DMR in the promoter of gene HOXA5 encoding a transcription factor which plays key roles in differentiation of adult cells (60). Previous studies have demonstrated that the methylation of this T-DMR is involved in regulation of cell-type-specific expression of gene HOXA5 (61,62), which was also shown in our analysis (Supplementary Figure S12).
= 3.636 for seven samples, only 4% (397/9636) of CpG islands were identified as T-DMRs (Supplementary Table S11). It is implied that CpG islands possess less tissues/cells differential methylation which is consistent with the finding in human genome in this article. There were less T-DMRs than N-DMRs in each genome category, while the CpG islands in Up2kb, 5′-UTR and Intron exhibited a smaller proportion of T-DMRs than other genome categories (Supplementary Figure S11B). The distribution of all the T-DMRs identified by QDMR on mouse chromosomes was also shown in the visualization module in QDMR software (Supplementary Figure S11C). The total 326 genes that are related to these T-DMRs showed enrichment for organ development (Supplementary Table S11). For example, there is a T-DMR in the promoter of gene HOXA5 encoding a transcription factor which plays key roles in differentiation of adult cells (60). Previous studies have demonstrated that the methylation of this T-DMR is involved in regulation of cell-type-specific expression of gene HOXA5 (61,62), which was also shown in our analysis (Supplementary Figure S12).
DISCUSSION
Shannon entropy, as a measure of the uncertainty associated with a random variable, has been previously used to carry out biological research, such as to identify potential drug targets (29), to prioritize promoter activity (63) and to measure tissue specificity of gene expression in many tissues (30). Due to the unique characteristics of the methylation data, a two-step optimization was performed based on Shannon entropy. The main difference between QDMR and Shannon entropy is that QDMR introduces a weight to adjust the entropy, which makes significant improvement in quantification of methylation difference. In order to show the impact of weighting, we selected three methylation patterns as shown in Figure 7A–C from the data in Figure 2B divided by 2, 10 and 100, respectively. With this process, the three new patterns in Figure 7 have smaller fluctuation range compared with region in Figure 2B. As shown in Figure 7, when the fluctuation range of methylation becomes smaller, 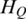 from QDMR becomes bigger, while both
from QDMR becomes bigger, while both  and
and 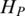 have no change. Therefore, the entropy adjustment process by weight plays an important role in quantifying the methylation difference for regions with small methylation fluctuation range. This methylation pattern is very common in genomes with numerous CpG islands which have constitutive hypomethylation among all samples.
have no change. Therefore, the entropy adjustment process by weight plays an important role in quantifying the methylation difference for regions with small methylation fluctuation range. This methylation pattern is very common in genomes with numerous CpG islands which have constitutive hypomethylation among all samples.
Figure 7.

Importance of methylation range across samples. Three regions with different methylation range across samples are artificially synthesized based on the methylation values in Figure 2B. (A) Methylation values are produced from Figure 2B divided by 2. (B) Methylation values are produced from Figure 2B divided by 10. (C) Methylation values are produced from Figure 2B divided by 100.
There are two major differences between QDMR and the previous methods in identification of DMRs. The first difference is that QDMR identifies DMRs based on quantified methylation difference, while previous methods based on statistics or counting. The entropy derived from QDMR can quantify methylation difference reasonably, and can reflect the biological characteristics of methylation difference, such as methylation difference distribution, the relationship between methylation difference and CpG density, and the association between methylation difference and gene expression difference. QDMR can be used to quantify methylation difference among various numbers of samples, which benefits from the mathematical properties of Shannon entropy. Moreover, the thresholds determined from methylation probability model can be used to identify DMRs based on the quantified methylation difference. The second difference of QDMR is its adaptability to the number of samples. The previous methods were designed for the particular data set with the given numbers of samples in their works. Instead, QDMR was developed for identifying DMRs for variable sample numbers. Therefore, QDMR may be a more suitable method for identification of DMRs from methylation profiles with multiple samples.
QDMR is independent of specific methylation mapping technique. Currently, nearly all of these techniques need the pre-treatment of DNA before amplification or hybridization by three main approaches, including endonuclease digestion, affinity enrichment and bisulphite conversion as reviewed by Laird (22). For biological and historical reasons, the methylation data is with measurement scale from 0 to 1 (0 = unmethylated, 1 = 100% methylated) in most of methylation mapping techniques, especially some sequencing-based techniques MethylC-Seq, RRBS, MeDIP-seq and MSCC. QDMR works on the fraction or percentage methylation across multiple samples, and identifies DMRs in a quantitative way, which has not been performed by previous methylation analysis. QDMR can be used to analyse the methylation profiles from most of the current methylation mapping techniques as summarized in Supplementary Table S12.
With the emergence of cost-effective high-throughput sequencing techniques (for example, single-molecule sequencing and nanopore sequencing), it may become less expensive to profile the methylation status in various tissues and other states (9,22). The identification of DMRs from those high-throughput data may be the foundation of further functional genomics analysis. In addition to the identification of T-DMRs, QDMR could be applied to identify C-DMRs, D-DMRs, R-DMRs, Intra-DMRs, Inter-DMRs and DMRs in other biological processes. The quantification of methylation difference and identification of DMRs in multifarious temporal and spatial methylomes should provide comprehensive survey of genome-wide epigenetic functions.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Funding for open access charge: National Natural Science Foundation of China (61075023 and 30971645); Natural Science Foundation of Heilongjiang Province (C201012).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
Y.Z., H.L. and J.L. contributed equally to this work and are regarded as co-first authors. The authors would like to thank three anonymous referees for their important suggestions, Dr Yaoping Lei and Dr Diansong Zhou for providing constructive comments.
REFERENCES
- 1.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 2.Rakyan VK, Down TA, Thorne NP, Flicek P, Kulesha E, Graf S, Tomazou EM, Backdahl L, Johnson N, Herberth M, et al. An integrated resource for genome-wide identification and analysis of human tissue-specific differentially methylated regions (tDMRs) Genome Res. 2008;18:1518–1529. doi: 10.1101/gr.077479.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 2009;41:178–186. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science. 2001;293:1089–1093. doi: 10.1126/science.1063443. [DOI] [PubMed] [Google Scholar]
- 5.Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, Zhang X, Bernstein BE, Nusbaum C, Jaffe DB, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454:766–770. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Doi A, Park IH, Wen B, Murakami P, Aryee MJ, Irizarry R, Herb B, Ladd-Acosta C, Rho J, Loewer S, et al. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat. Genet. 2009;41:1350–1353. doi: 10.1038/ng.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bjornsson HT, Sigurdsson MI, Fallin MD, Irizarry RA, Aspelund T, Cui H, Yu W, Rongione MA, Ekstrom TJ, Harris TB, et al. Intra-individual change over time in DNA methylation with familial clustering. JAMA. 2008;299:2877–2883. doi: 10.1001/jama.299.24.2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bock C, Walter J, Paulsen M, Lengauer T. Inter-individual variation of DNA methylation and its implications for large-scale epigenome mapping. Nucleic Acids Res. 2008;36:e55. doi: 10.1093/nar/gkn122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lister R, Ecker JR. Finding the fifth base: genome-wide sequencing of cytosine methylation. Genome Res. 2009;19:959–966. doi: 10.1101/gr.083451.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shen CK, Maniatis T. Tissue-specific DNA methylation in a cluster of rabbit beta-like globin genes. Proc. Natl Acad. Sci. USA. 1980;77:6634–6638. doi: 10.1073/pnas.77.11.6634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kawai J, Hirotsune S, Hirose K, Fushiki S, Watanabe S, Hayashizaki Y. Methylation profiles of genomic DNA of mouse developmental brain detected by restriction landmark genomic scanning (RLGS) method. Nucleic Acids Res. 1993;21:5604–5608. doi: 10.1093/nar/21.24.5604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frommer M, McDonald LE, Millar DS, Collis CM, Watt F, Grigg GW, Molloy PL, Paul CL. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl Acad. Sci. USA. 1992;89:1827–1831. doi: 10.1073/pnas.89.5.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eckhardt F, Lewin J, Cortese R, Rakyan VK, Attwood J, Burger M, Burton J, Cox TV, Davies R, Down TA, et al. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat. Genet. 2006;38:1378–1385. doi: 10.1038/ng1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weber M, Davies JJ, Wittig D, Oakeley EJ, Haase M, Lam WL, Schubeler D. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat. Genet. 2005;37:853–862. doi: 10.1038/ng1598. [DOI] [PubMed] [Google Scholar]
- 15.Weber M, Hellmann I, Stadler MB, Ramos L, Paabo S, Rebhan M, Schubeler D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 2007;39:457–466. doi: 10.1038/ng1990. [DOI] [PubMed] [Google Scholar]
- 16.Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meissner A, Gnirke A, Bell GW, Ramsahoye B, Lander ES, Jaenisch R. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 2005;33:5868–5877. doi: 10.1093/nar/gki901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Down TA, Rakyan VK, Turner DJ, Flicek P, Li H, Kulesha E, Graf S, Johnson N, Herrero J, Tomazou EM, et al. A Bayesian deconvolution strategy for immunoprecipitation-based DNA methylome analysis. Nat. Biotechnol. 2008;26:779–785. doi: 10.1038/nbt1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Serre D, Lee BH, Ting AH. MBD-isolated Genome sequencing provides a high-throughput and comprehensive survey of DNA methylation in the human genome. Nucleic Acids Res. 2010;38:391–399. doi: 10.1093/nar/gkp992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ball MP, Li JB, Gao Y, Lee JH, LeProust EM, Park IH, Xie B, Daley GQ, Church GM. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat. Biotechnol. 2009;27:361–368. doi: 10.1038/nbt.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Edwards JR, O’Donnell AH, Rollins RA, Peckham HE, Lee C, Milekic MH, Chanrion B, Fu Y, Su T, Hibshoosh H, et al. Chromatin and sequence features that define the fine and gross structure of genomic methylation patterns. Genome Res. 2010;20:972–980. doi: 10.1101/gr.101535.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Laird PW. Principles and challenges of genome-wide DNA methylation analysis. Nat. Rev. Genet. 2010;11:191–203. doi: 10.1038/nrg2732. [DOI] [PubMed] [Google Scholar]
- 23.Bock C, Lengauer T. Computational epigenetics. Bioinformatics. 2008;24:1–10. doi: 10.1093/bioinformatics/btm546. [DOI] [PubMed] [Google Scholar]
- 24.Bibikova M, Chudin E, Wu B, Zhou L, Garcia EW, Liu Y, Shin S, Plaia TW, Auerbach JM, Arking DE, et al. Human embryonic stem cells have a unique epigenetic signature. Genome Res. 2006;16:1075–1083. doi: 10.1101/gr.5319906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Byun HM, Siegmund KD, Pan F, Weisenberger DJ, Kanel G, Laird PW, Yang AS. Epigenetic profiling of somatic tissues from human autopsy specimens identifies tissue- and individual-specific DNA methylation patterns. Hum. Mol. Genet. 2009;18:4808–4817. doi: 10.1093/hmg/ddp445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rakyan VK, Hildmann T, Novik KL, Lewin J, Tost J, Cox AV, Andrews TD, Howe KL, Otto T, Olek A, et al. DNA methylation profiling of the human major histocompatibility complex: a pilot study for the human epigenome project. PLoS Biol. 2004;2:e405. doi: 10.1371/journal.pbio.0020405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fan S, Zhang X. CpG island methylation pattern in different human tissues and its correlation with gene expression. Biochem. Biophys. Res. Commun. 2009;383:421–425. doi: 10.1016/j.bbrc.2009.04.023. [DOI] [PubMed] [Google Scholar]
- 28.Shannon CE. The mathematical theory of communication. MD Comput. 1997;14:306–317. [PubMed] [Google Scholar]
- 29.Fuhrman S, Cunningham MJ, Wen X, Zweiger G, Seilhamer JJ, Somogyi R. The application of shannon entropy in the identification of putative drug targets. Biosystems. 2000;55:5–14. doi: 10.1016/s0303-2647(99)00077-5. [DOI] [PubMed] [Google Scholar]
- 30.Schug J, Schuller WP, Kappen C, Salbaum JM, Bucan M, Stoeckert CJ., Jr Promoter features related to tissue specificity as measured by Shannon entropy. Genome Biol. 2005;6:R33. doi: 10.1186/gb-2005-6-4-r33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karolchik D, Hinrichs AS, Furey TS, Roskin KM, Sugnet CW, Haussler D, Kent WJ. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004;32:D493–D496. doi: 10.1093/nar/gkh103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Su J, Zhang Y, Lv J, Liu H, Tang X, Wang F, Qi Y, Feng Y, Li X. CpG_MI: a novel approach for identifying functional CpG islands in mammalian genomes. Nucleic Acids Res. 2010;38:e6. doi: 10.1093/nar/gkp882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reimand J, Kull M, Peterson H, Hansen J, Vilo J. g:Profiler–a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Res. 2007;35:W193–W200. doi: 10.1093/nar/gkm226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 35.Barrett T, Troup DB, Wilhite SE, Ledoux P, Rudnev D, Evangelista C, Kim IF, Soboleva A, Tomashevsky M, Marshall KA, et al. NCBI GEO: archive for high-throughput functional genomic data. Nucleic Acids Res. 2009;37:D885–D890. doi: 10.1093/nar/gkn764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D, Zhang J, Soden R, Hayakawa M, Kreiman G, et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl Acad. Sci. USA. 2004;101:6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rhead B, Karolchik D, Kuhn RM, Hinrichs AS, Zweig AS, Fujita PA, Diekhans M, Smith KE, Rosenbloom KR, Raney BJ, et al. The UCSC Genome Browser database: update 2010. Nucleic Acids Res. 2010;38:D613–D619. doi: 10.1093/nar/gkp939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kadota K, Ye J, Nakai Y, Terada T, Shimizu K. ROKU: a novel method for identification of tissue-specific genes. BMC Bioinformatics. 2006;7:294. doi: 10.1186/1471-2105-7-294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Y, Lv J, Liu H, Zhu J, Su J, Wu Q, Qi Y, Wang F, Li X. HHMD: the human histone modification database. Nucleic Acids Res. 2010;38:D149–D154. doi: 10.1093/nar/gkp968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, Cui K, Roh TY, Peng W, Zhang MQ, et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008;40:897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hubbell E, Liu WM, Mei R. Robust estimators for expression analysis. Bioinformatics. 2002;18:1585–1592. doi: 10.1093/bioinformatics/18.12.1585. [DOI] [PubMed] [Google Scholar]
- 42.Yagi S, Hirabayashi K, Sato S, Li W, Takahashi Y, Hirakawa T, Wu G, Hattori N, Ohgane J, Tanaka S, et al. DNA methylation profile of tissue-dependent and differentially methylated regions (T-DMRs) in mouse promoter regions demonstrating tissue-specific gene expression. Genome Res. 2008;18:1969–1978. doi: 10.1101/gr.074070.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Laurent L, Wong E, Li G, Huynh T, Tsirigos A, Ong CT, Low HM, Kin Sung KW, Rigoutsos I, Loring J, et al. Dynamic changes in the human methylome during differentiation. Genome Res. 2010;20:320–331. doi: 10.1101/gr.101907.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ernst C, Deleva V, Deng X, Sequeira A, Pomarenski A, Klempan T, Ernst N, Quirion R, Gratton A, Szyf M, et al. Alternative splicing, methylation state, and expression profile of tropomyosin-related kinase B in the frontal cortex of suicide completers. Arch. Gen. Psychiatr. 2009;66:22–32. doi: 10.1001/archpsyc.66.1.22. [DOI] [PubMed] [Google Scholar]
- 45.Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T. Identification of tissue-specific microRNAs from mouse. Curr. Biol. 2002;12:735–739. doi: 10.1016/s0960-9822(02)00809-6. [DOI] [PubMed] [Google Scholar]
- 46.Alvarez-Garcia I, Miska EA. MicroRNA functions in animal development and human disease. Development. 2005;132:4653–4662. doi: 10.1242/dev.02073. [DOI] [PubMed] [Google Scholar]
- 47.Khraiwesh B, Arif MA, Seumel GI, Ossowski S, Weigel D, Reski R, Frank W. Transcriptional control of gene expression by microRNAs. Cell. 2010;140:111–122. doi: 10.1016/j.cell.2009.12.023. [DOI] [PubMed] [Google Scholar]
- 48.Schoenborn JR, Dorschner MO, Sekimata M, Santer DM, Shnyreva M, Fitzpatrick DR, Stamatoyannopoulos JA, Wilson CB. Comprehensive epigenetic profiling identifies multiple distal regulatory elements directing transcription of the gene encoding interferon-gamma. Nat. Immunol. 2007;8:732–742. doi: 10.1038/ni1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hark AT, Schoenherr CJ, Katz DJ, Ingram RS, Levorse JM, Tilghman SM. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature. 2000;405:486–489. doi: 10.1038/35013106. [DOI] [PubMed] [Google Scholar]
- 50.Klochkov D, Rincon-Arano H, Ioudinkova ES, Valadez-Graham V, Gavrilov A, Recillas-Targa F, Razin SV. A CTCF-dependent silencer located in the differentially methylated area may regulate expression of a housekeeping gene overlapping a tissue-specific gene domain. Mol. Cell. Biol. 2006;26:1589–1597. doi: 10.1128/MCB.26.5.1589-1597.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Takeichi M. The cadherins: cell-cell adhesion molecules controlling animal morphogenesis. Development. 1988;102:639–655. doi: 10.1242/dev.102.4.639. [DOI] [PubMed] [Google Scholar]
- 52.Schaefer CB, Ooi SK, Bestor TH, Bourc’his D. Epigenetic decisions in mammalian germ cells. Science. 2007;316:398–399. doi: 10.1126/science.1137544. [DOI] [PubMed] [Google Scholar]
- 53.Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–112. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wei G, Wei L, Zhu J, Zang C, Hu-Li J, Yao Z, Cui K, Kanno Y, Roh TY, Watford WT, et al. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity. 2009;30:155–167. doi: 10.1016/j.immuni.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li E. Chromatin modification and epigenetic reprogramming in mammalian development. Nat. Rev. Genet. 2002;3:662–673. doi: 10.1038/nrg887. [DOI] [PubMed] [Google Scholar]
- 57.Movassagh M, Choy MK, Goddard M, Bennett MR, Down TA, Foo RS. Differential DNA methylation correlates with differential expression of angiogenic factors in human heart failure. PLoS One. 2010;5:e8564. doi: 10.1371/journal.pone.0008564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim HR, Hwang KA, Kim KC, Kang I. Down-regulation of IL-7Ralpha expression in human T cells via DNA methylation. J. Immunol. 2007;178:5473–5479. doi: 10.4049/jimmunol.178.9.5473. [DOI] [PubMed] [Google Scholar]
- 59.Bird AP. CpG-rich islands and the function of DNA methylation. Nature. 1986;321:209–213. doi: 10.1038/321209a0. [DOI] [PubMed] [Google Scholar]
- 60.Garin E, Lemieux M, Coulombe Y, Robinson GW, Jeannotte L. Stromal Hoxa5 function controls the growth and differentiation of mammary alveolar epithelium. Dev. Dyn. 2006;235:1858–1871. doi: 10.1002/dvdy.20822. [DOI] [PubMed] [Google Scholar]
- 61.Strathdee G, Sim A, Soutar R, Holyoake TL, Brown R. HOXA5 is targeted by cell-type-specific CpG island methylation in normal cells and during the development of acute myeloid leukaemia. Carcinogenesis. 2007;28:299–309. doi: 10.1093/carcin/bgl133. [DOI] [PubMed] [Google Scholar]
- 62.Watson RE, Curtin GM, Hellmann GM, Doolittle DJ, Goodman JI. Increased DNA methylation in the HoxA5 promoter region correlates with decreased expression of the gene during tumor promotion. Mol. Carcinog. 2004;41:54–66. doi: 10.1002/mc.20043. [DOI] [PubMed] [Google Scholar]
- 63.Barrera LO, Li Z, Smith AD, Arden KC, Cavenee WK, Zhang MQ, Green RD, Ren B. Genome-wide mapping and analysis of active promoters in mouse embryonic stem cells and adult organs. Genome Res. 2008;18:46–59. doi: 10.1101/gr.6654808. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.