


Abstract
We study the number of causal variants and associated regions identified by top SNPs in rankings given by the popular 1 df chi-squared statistic, support vector machine (SVM) and the random forest (RF) on simulated and real data. If we apply the SVM and RF to the top 2r chi-square-ranked SNPs, where r is the number of SNPs with P-values within the Bonferroni correction, we find that both improve the ranks of causal variants and associated regions and achieve higher power on simulated data. These improvements, however, as well as stability of the SVM and RF rankings, progressively decrease as the cutoff increases to 5r and 10r. As applications we compare the ranks of previously replicated SNPs in real data, associated regions in type 1 diabetes, as provided by the Type 1 Diabetes Consortium, and disease risk prediction accuracies as given by top ranked SNPs by the three methods. Software and webserver are available at http://svmsnps.njit.edu.
INTRODUCTION
Genome-wide association studies aim to identify genetic variants associated with disease, drug response and various phenotypes (1). The standard method of ranking SNPs from genome-wide association studies is the one or two degree of freedom chi-squared test (2).
Previous studies have examined the performance of the chi-squared statistic in ranking SNPs (3), proposed techniques to improve the rankings under two-stage designs (4), and to correct for overestimated significance values and apply the false discovery rate control method thereafter (5). Other approaches instead of chi-square have also been proposed for ranking SNPs. These include the trend test (6), Bayes factors (1), random forests (RFs) (7–9), support vector machine (SVM; 10), L1 penalized logistic regression (11,12) and a hidden Markov model method (13).
Chi-square-based rankings have been found similar to other univariate tests such as Bayes factors and likelihood ratios (1,3). Our experiments with information theoretic methods (14) and the MAX-rank trend test (6) also show strong similarity to chi-square-based rankings on simulated data.
In this article, we study the number of causal variants and associated regions identified by top SNPs in rankings given by the popular 1 df chi-squared statistic and two popular multivariate feature selection methods: the SVM (15) and the RF method (16). Both have been studied extensively for the problem of gene selection from microarray data (17–20). While the SVM and RF have previously been applied to genome-wide association studies (7–10), here we explicitly study their performance in ranking causal SNPs and those from associated regions and their performance as a function of the input as explained below.
We apply each of the two methods to the top kr chi-square-ranked SNPs where r is the number of SNPs with P-values at most 0.05 divided by the total number of SNPs. This corrected P-value threshold is also known as the Bonferroni correction (21) for multiple hypothesis and is a common cutoff in genome-wide association studies (22,23). We show that the SVM(2r) method, which is basically the SVM method applied to the top 2r chi-square-ranked SNPs, and RF(2r) contain more causal variants and those from associated regions compared to chi-square when we examine the top-ranked SNPs at the Bonferroni threshold. The SVM performs the best followed by RF and chi-square. However, at the 5r and 10r thresholds the improvement is less, if at all, in both methods. We also show that SVM(2r) has the highest power followed by RF(2r) and chi-square, but this progressively decreases at the 5r and 10r threshold.
As applications on real data, we show that both SVM(2r) and RF(2r) improve the ranks of previously replicated SNPs on Wellcome Trust Case Control Consortium (WTCCC) (1) genome-wide studies and identify more known associated type 1 diabetes regions—as given by the Type 1 Diabetes Consortium (24)—than chi-square at the Bonferroni cutoff. We also show that top ranked SVM(2r) SNPs achieve the highest AUC for type 1 diabetes, arthritis and simulated data disease risk prediction in testing across independent cohorts and in cross-validation studies.
In the rest of the article, we provide brief descriptions of the SVM and RF, followed by the real and simulated data used in our study. We then present detailed experimental results including an empirical power study to compare the three methods followed by results on disease risk prediction. Software and data to reproduce the results in this article along with a webserver are available at http://svmsnps.njit.edu.
METHODS
Here, we describe the SVM and RF methods along with their implementations used in this study. The input to each method is a case control study with the top kr chi-square-ranked SNPs where r is the number of SNPs with P-values within the Bonferroni correction (0.05 divided by total number of SNPs m) and k is an integer ≥1. Before applying each method, we encode each genotype in the real data as the number of copies of major allele. We use our own implementation of the one degree of freedom chi-squared statistic in C which we provide freely.
Support vector machine
The SVM is the optimally separating hyperplane between two sets of points each belonging to a different class. The sign of the SVM discriminant wT x + w0 determines the class of input x and the distance to the hyperplane is given by 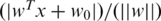 (25). The SVM can be represented by the vector w and scalar w0 that minimizes
(25). The SVM can be represented by the vector w and scalar w0 that minimizes 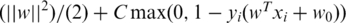 where xi is the genotype vector of the i-th individual, yi is an integer specifying case (+1) or control (−1), max(0, 1 − yi(wT xi + w0)) is the hinge loss function, and C is the loss-complexity tradeoff parameter. The solution w and w0 is obtained by applying Lagrange multipliers to obtain the dual problem which is a quadratic program and can be solved by standard methods see (25,26 for details). In the SVM, optimization criterion the term max(0, 1 − yi (wT xi + w0)) measures how well the discriminant w, which is basically our model, fits the training data and ||w||2 measures the complexity of the model. We set the parameter C to its default value of
where xi is the genotype vector of the i-th individual, yi is an integer specifying case (+1) or control (−1), max(0, 1 − yi(wT xi + w0)) is the hinge loss function, and C is the loss-complexity tradeoff parameter. The solution w and w0 is obtained by applying Lagrange multipliers to obtain the dual problem which is a quadratic program and can be solved by standard methods see (25,26 for details). In the SVM, optimization criterion the term max(0, 1 − yi (wT xi + w0)) measures how well the discriminant w, which is basically our model, fits the training data and ||w||2 measures the complexity of the model. We set the parameter C to its default value of 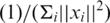 , where i loops over all subjects in the training set, as provided in the SVM-light software package. We obtain a SNP ranking from the SVM discriminant w as described below.
, where i loops over all subjects in the training set, as provided in the SVM-light software package. We obtain a SNP ranking from the SVM discriminant w as described below.
Obtaining a SNP ranking from the SVM discriminant vector w
We can obtain an ordering of the SNPs using the absolute value of the entries of w. The input to the discriminant is the set of top mr chi-square ranked SNPs s1, s2, … smr. The i-th entry of w represents the weight of the i-th SNP in the input. Let w = (w1, … , wmr) and |w| = (|w1|, … , |wmr|). Now consider the entries of |w| in sorted descending order. We denote this ordering by the vector p such that 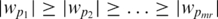 . Using p we obtain an ordering on the input SNP identifiers
. Using p we obtain an ordering on the input SNP identifiers  which gives us the SNP ranking.
which gives us the SNP ranking.
Implementation
We use the popular and freely available SVM-light SVM implementation (27). We run it with the linear kernel and all other parameters set to their default values.
Random forest
A classification tree is built by the recursive partitioning method (25). At each step the feature, which is a SNP in this study, with the highest impurity, usually measured by entropy or the Gini index, is selected and then split into k children where k is the number of values the SNP can take. This process is repeated until all nodes are pure, meaning that all sets of decisions leading to that node result in the same class. In a RF (16), several classification trees are created each by drawing n subjects with replacement from the original data, where n is the total number of case and control subjects. The SNPs are then ranked by a classification based variable importance index that considers interaction between the SNPs (28).
Implementation
We use the freely available willows software package (28) for generating random forests and obtaining variable importance indices. We set the number of trees to 10 000 and use the default values for other parameters as provided by the program.
Datasets
Real data
We use real data from two sources: the Wellcome Trust Case Control Consortium (WTCCC) and The Genetics of Kidneys in Diabetes (GoKinD). The WTCCC provides two sets of controls and one set of cases each for type 1 diabetes, rheumatoid arthritis, Crohn's disease and type 2 diabetes (1) and GoKinD provides a type 1 diabetes case set (29,30). The WTCCC also provides case subjects for bipolar disorder, hypertension and coronary artery disease. However, we omit them from this study for two reasons. First, much fewer replicated SNPs are catalogued for them in comparison to the other four. In Ref. (31), which is from where we obtain these SNPs, there are just three listed for bipolar, one for coronary artery disease and none for hypertension. Second, none of the listed SNPs or those in linkage disequilibrium with them are captured by twice or even five times the number of top chi-square-ranked SNPs within the Bonferroni correction for bipolar disorder, whereas for coronary artery disease the single previously replicated SNP is already ranked as the top one by chi-square.
We follow the standard protocol of removing SNPs with >1% missing entries and those that deviate from Hardy–Weinberg equilibrium with P-values below 5 × 10−7 (1). See Supplementary Table S6 for more details and the number of subjects and SNPs in each dataset.
Simulated data
The GWAsimulator program (32) produces case and control genome-wide SNP genotypes under a logistic regression disease model. It takes as input a control file that specifies various parameters such as relative risk and sample size and phased genotype data, and simulates SNP genotypes with the same linkage disequilibrium structure as the input genotype data. It outputs data in a numerical format as the number of copies of the causal allele. We use the HapMap CEU phased genotypes provided with the software package as input. These genotypes were produced by the Illumina HumanHap300 SNP chip. The program generates one causal SNP on a specified position of a chromosome and then simulates remaining SNPs according to a moving window algorithm (33).
We simulated data across several different parameters from the followings sets of control files. In each case though each causal SNP follows a multiplicative model. This means that if λ is the relative risk for one copy of the risk allele than λ2 is the risk for two copies of that allele. Except for the power study case we generate one simulated study per control file.
General performance on different relative risks: 50 control files each for relative risk 1.25, 1.5 and 2. Each control file contains 15 randomly selected SNPs as causal, one per chromosomes 1 through 15 each with a specified relative risk. The disease prevalence is set to .01, and case and control sample sizes each to 1000. We simulate a 1000 SNPs on either side of each causal one which adds up to a total of approximately 30 000 SNPs per dataset.
Performance as a function of sample size: 50 control files of relative risk 1.25 and two additional case and control sizes of 2000 and 4000. Remaining parameters same as above.
Performance on low causal allele frequencies: 10 control files each for relative risk 1.25 and 1.5 and two case and control sizes of 2000 and 4000, and causal allele frequencies of at most 5%. Other parameters as above in the general performance setting.
Power study: First five control files for relative risks 1.25, 1.5 and 2. We simulated 50 studies for each control file thereby giving a total of 250 simulated studies for each relative risk setting. Remaining parameters same as the general performance setting.
Disease risk prediction: 50 control files each for relative risk 1.25, 1.5 and 2 and same settings as the general performance case except that 100 case and 100 control subjects are generated instead of 1000 each.
We provide all simulated studies, input control files and HapMap CEU phased genotypes to the GWAsimulator program at http://www.cs.njit.edu/usman/SVMSNPs.
RESULTS
We are interested in measuring the number of causal variants and associated regions identified by top SNPs in a given ranking. In simulated data, we define an associated region as the set of SNPs in linkage disequilibrium (34) with the causal one. In other words, the squared correlation coefficient is at least 0.05, which is a standard threshold for defining associated regions (12,35). In order to simulate a scenario where causal variants are not necessarily genotyped we make a copy of each simulated study without the causal SNPs and then compute chi-square, SVM, and random RFs. We also compute rankings on the original studies with the causal SNPs.
It is straightforward to measure the number of causal variants in the number of top-ranked SNPs given by a method. To measure the number of associated regions we count the number of unique regions covered by top ranked SNPs. For example, consider a ranking of five SNPs: s1, s2, s3, s4, s5. Suppose that SNPs s1 and s3 belong to region r1, SNPs s4 and s5 belong to region r2, and the s2 does not belong to any known region. In this ranking, we have two regions covered by the five SNPs.
We first study the effect of the P-value threshold on the two methods including both methods applied to all SNPs in GWAS, effect of sample sizes at relative risk 1.25 and performance on data with low causal allele frequencies. We then compare the power of the three methods, their running times on simulated and real data, and stability of SNP rankings given by the different methods. Finally, we study ranks of previously replicated SNPs on real data, associated regions in type 1 diabetes, and disease risk prediction accuracies of logistic regression as given by top ranked SNPs by the methods.
Effect of P-value threshold on the support vector machine and random forest
Let r be the number of SNPs with P-values within the Bonferroni threshold. We reorder the top 2r, 5r and 10r chi-square-ranked SNPs with the SVM and RF separately. At the relative risk 1.25 setting r is 0 for some datasets and so we exclude them from the analysis.
In Table 1, we show the mean number of causal variants identified by the SVM and RF when applied to the top 2r, 5r and 10r chi-square-ranked SNPs as input as well as the entire GWAS. We examine the top r ranked SNPs in each method. The larger input to the SVM and RF contains many more false positives and this clearly deteriorates their SNP rankings. Similarly, the larger P-value also deteriorates the number of associated regions detected by the two methods as shown in Table 1.
Table 1.
Mean number of causal variants and associated regions in top r SNPs given by each method at the three different relative risks
| RR | χ2 | SV(2r) | SV(5r) | SV(10r) | RF(2r) | RF(5r) | RF(10r) |
|---|---|---|---|---|---|---|---|
| Mean number of causal variants | |||||||
| 1.25 | 1.2 | 1.2 | 0.9 | 0.5 | 1.2 | 1.1 | 1 |
| 1.5 | 8.9 | 10.8 | 7.4 | 6.6 | 9.7 | 9.3 | 9.4 |
| 2 | 14 | 14.6 | 13.6 | 13.1 | 14.1 | 14.1 | 13.7 |
| Mean number of associated regions | |||||||
| 1.25 | 1 | 0.8 | 0.8 | 0.8 | 0.9 | 0.8 | 0.8 |
| 1.5 | 4.5 | 5.9 | 3.5 | 2.6 | 5.3 | 4.9 | 4.8 |
| 2 | 10.6 | 11.9 | 9.6 | 9.4 | 11 | 10.9 | 10.8 |
A comparison of the SVM and RF shows that while SVM(2r) is the best performing method, RF(5r) and RF(10r) are better than the SVM counterparts. This holds true for detecting causal variants and associated regions at all three relative risks.
The improvement given by SVM(2r) and RF(2r) is small at relative risk 1.25 but increases as we move to relative risk 1.5 and 2. However, the signal in these studies depend upon sample size among other things. If we increase the total sample size to 4000 and 8000 with each containing half cases and half controls then r, which is the number of SNPs with P-values within Bonferroni, increases and so does the improvement given by SVM(2r) as shown in Table 2 below. In Supplementary Figures S2–S4, we report mean number of causal variants and associated regions for different thresholds of top ranked SNPs instead of the just the top r ranked SNPs. There too we find similar patterns reported here.
Table 2.
Mean number of causal variants detected in top r ranked SNPs given by each method on different sample sizes at relative risk 1.25
| Sample size | Mean r | χ2 | SVM(2r) | RF(2r) |
|---|---|---|---|---|
| 2000 | 2.1 | 1.2 | 1.2 | 1.2 |
| 4000 | 9.2 | 4.8 | 5.7 | 4.4 |
| 8000 | 31.7 | 11.0 | 12.1 | 8.5 |
The simulated data above has random causal allele frequencies. In Table 3, we compare the methods on relative risk 1.25 and 1.5 but with low causal allele frequencies of at most 5% and with 4000 and 8000 subjects each containing half cases and half controls. We find both SVM(2r) and RF(2r) to perform better than chi-square at these settings.
Table 3.
Mean number of causal variants detected in top r ranked SNPs by each method on data with causal allele frequencies at most 5% and two different sample sizes and relative risks
| Sample size and relative risk | χ2 | SVM(2r) | RF(2r) |
|---|---|---|---|
| 4000, 1.25 | 0.5 | 1 | 1 |
| 8000, 1.25 | 1.7 | 2.5 | 2.5 |
| 4000, 1.5 | 4.8 | 6.3 | 6.1 |
| 8000, 1.5 | 12.7 | 13.2 | 13.3 |
We also studied the SVM and RF applied to the entire GWAS and found that they perform worse than chi-square and SVM(2r) and RF(2r) (Table 4). Note that we use the SVM with an automatic setting of the value of C which controls the tradeoff between error on training data and model complexity. To be fair to the SVM we ran it on all the simulated studies of relative risk 1.5 with fixed values of C from the set {10−6, 10−5, 10−4, 10−3, 10−2, 10−1}. At C = 10−3 the SVM identifies the same mean number of causal variants as chi-square which is 8.9 and at the remaining values it is lower.
Table 4.
Mean number of causal variants and associated regions in SVM and RF applied to all SNPs in the GWAS
| RR | Causal variants |
Associated regions |
||||
|---|---|---|---|---|---|---|
| χ2 | SVM | RF | χ2 | SVM | RF | |
| 1.25 | 1.2 | 1.3 | 1 | 1 | 0.8 | 0.9 |
| 1.5 | 8.9 | 8.1 | 8.7 | 4.8 | 4.3 | 4.8 |
| 2 | 14 | 12.4 | 14 | 10.6 | 9.4 | 10.7 |
Power study
We now compare the empirical power of the chi-square, SVM and RF to rank causal variants from simulated data of relative risk 1.5. We define the empirical power of a method to be the percentage of simulated datasets where the top r ranked SNPs given by the method, where r is the number of SNPs with P-values within Bonferroni correction, contain k causal variants. In Figure 1, we plot this value for k ranging from 1 to 15 which is the total number of causal SNPs in the simulated data. We see that SVM(2r) has the highest power followed by RF(2r).
Figure 1.

Empirical power to detect k causal variants in simulated data of relative risk 1.5.
In Supplementary Figures S5, we compare the empirical power of the three methods on simulated data of relative risk 1.25 and 2. At the 1.25 setting chi-square has highest power for detecting one causal variant, RF(2r) and SVM(2r) both have the highest power for detecting two and three causal variants, and after that all three methods have same power. At relative risk of 2 all three methods have the same power up to value of k = 12. After that SVM(2r) has highest power.
Running time comparisons
In Supplementary Tables S1–S3, we show running times on real data and the simulated one with different relative risks, sample sizes and causal allele frequency. This running time includes the time for chi-square since both methods require a chi-square ranking to start with. These were measured on AMD Opteron model 2218 machines each with 2.6 GHz speed and 8 GB RAM. Our results show that running time of all methods depends unsurprisingly on the sample size. However, the running time of SVM(2r) and RF(2r) also depends on the value of r which in turn depends on the relative risk and causal allele frequency. We also see that both RF(2r) and SVM(2r) are much faster than their 10r counterparts and the running time for SVM(2r) is comparable to that of chi-square.
Stability of rankings
In line with recent studies that examine stability of ranked gene and SNP lists, we do the same for the SVM and RF methods on our simulated data (36,37). Following these studies we create a jacknifed dataset by randomly removing 10% of the subjects from a given simulated study. In this manner, we create 100 jacknifed studies and compute chi-square, SVM and RF rankings with the four P-value thresholds of r, 2r, 5r and 10r on each one. As before r denotes the number of SNPs with P-values within the Bonferroni correction. We perform this on process on simulated studies one through five. For each of the three methods we then compute the correlation coefficient between the ranks of the top r SNPs captured by chi-square on the original datasets with their mean rank in the jacknifed studies.
We study two variants of the random forest method. In RF(100), we set the number of trees in the forest to 100 and in RF(10 000) we set this to 10 000. Note that the latter setting is the one we used in the experiments throughout this paper. In Table 5, we see that the correlation is high at the r threshold for all methods but progressively decreases as the P-value threshold increases. We also find that the random forest with 10 000 trees has much better stability than with just 100 trees even though the former has a higher running time.
Table 5.
Correlation coefficient between original SNP ranks and mean SNP ranks across 100 jacknifed datasets of relative risk 1.5
| r | 2r | 5r | 10r | |
|---|---|---|---|---|
| χ2 | 0.99 | 0.99 | 0.97 | 0.94 |
| SVM | 0.99 | 0.98 | 0.97 | 0.96 |
| RF(100) | 0.87 | 0.84 | 0.65 | 0.59 |
| RF(10000) | 0.99 | 0.98 | 0.95 | 0.91 |
Calle et al. (36) report a low correlation when they study the stability of the RF applied to all SNPs in a real study. In agreement with their results, we find that the correlation of RF(100) and RF(10 000) are both very low when applied to the entire GWAS. In Supplementary Tables S4 and S5, we show the stability at relative risk 1.25 and 2. There too we find high stability at the r and 2r thresholds and RF(10 000) doing much better than RF(100).
Applications on real data
We demonstrate some applications of our work by studying ranks of previously replicated SNPs, associated regions in type 1 diabetes and prediction of disease risk as given by top-ranked SNPs by the three methods.
Ranks of previously replicated SNPs in WTCCC studies
The Bonferroni corrections r for the WTCCC type 1 diabetes, arthritis, Crohn's disease and type 2 diabetes are 452, 176, 63 and 14, respectively. We compute SVM and RF rankings with the three thresholds and show results in Supplementary Tables S7 and S8. In type 1 diabetes and arthritis, we see a clear improvement in rank by SVM(2r) and a less pronounced one by RF(2r). As the threshold increases from 2r to 5r and 10r the ranking given by SVM and RF deteriorates. In Crohn's disease and type 2 diabetes the rankings are comparable. Note that the value of r for these two diseases is also much smaller than the ones for the other two.
Type 1 diabetes-associated regions
As in the simulated data, we define an associated region for each replicated as the set of all SNPs with squared correlation coefficient at least 0.05 with the replicated SNP. We also examine associated regions defined by the Type 1 Diabetes Consortium (24) and list SNPs and boundaries of both sets of regions in Section 8 of the Supplementary Data. We consider a region as detected if the top r SNPs of a ranking contains at least one SNP from the region. The Bonferroni corrected P-value threshold is about 10−7 which yields 452 SNPs. If we double this to 904 SNPs the P-value threshold increases to about 0.002 and includes many more regions not detected by chi-square. Table 6 shows that the SVM(2r) and RF(2r) can lift the ranks of many SNPs from these undetected regions to above 452.
Table 6.
Number of type 1 diabetes associated regions given by top r SNPs of chi-square, RF(2r) and SVM(2r)
| Regions defined by replicated SNPs in Ref. (31) | T1D Base regions | |
|---|---|---|
| χ2 | 5 | 5 |
| RF(2r) | 8 | 13 |
| SVM(2r) | 9 | 15 |
Prediction of type 1 diabetes risk on independent studies
The previous few sections have demonstrated that the SVM(2r) and RF(2r) can lift the ranks of causal SNPs and those from associated regions compared to chi-square on simulated data. We expect that top ranked SNPs given by the SVM should be enough for predicting disease risk accurately since they are mostly causal and cover several associated regions. To test this hypothesis, we measure the ROC area under curve of a logistic regression based} composite odds ratio score (31,38,39) for predicting type 1 diabetes risk as a function of top-ranked SNPs given by the three methods including chi-square. See the Supplementary Data for details of the composite odds ratio score (Section 1), cross-validation results on the WTCCC arthritis study (Supplementary Figures S9) and risk prediction on simulated data with this risk estimator (Supplementary Figures S10 and S11).
We compute SNPs rankings on the WTCCC study and then classify subjects in the GoKinD study plus WTCCC coronary artery disease samples as controls using top ranked SNPs from the three different methods. We also repeat these steps by computing SNP rankings on the GoKinD study and predicting on the WTCCC one. In Figure 2, we show the composite odds ratio AUC as a function of top ranked SNPs in the three rankings. SVM(r) achieves the highest AUC of 0.83 with 21 SNPs followed by random forest and chi-square AUCs of 0.81 each with 29 and 17 SNPs, respectively. See Supplementary Figure S8 for graphs comparing AUCs of the SVM score for predicting disease risk (40).
Figure 2.

ROC area under curve of the composite odds ratio score on the GoKinD type 1 diabetes study as a function of top-ranked SNPs obtained from the WTCCC study by the three different methods.
We make similar observations if we compute the rankings on the GoKinD study and predict risk on the WTCCC study. Figure 2 and its reverse counterpart in Supplementary Figure S7 show that many initial thresholds of top SVM-ranked SNPs are consistent in their prediction AUC. With chi-square the AUC is highest for a few top ranked SNPs after which it begins to fall quickly. In fact, this also happens for arthritis as shown in Supplementary Figure S9. The rapid drop in type 1 diabetes and arthritis prediction with chi-square-ranked SNPs is also observed by Evans et al. (31) who use a composite odds ratio score similar to ours.
DISCUSSION
The work presented here sheds light into the performance of the SVM and the RF method for ranking SNPs in genome-wide association studies. As the P-value threshold increases the ranking of causal SNPs and those from associated regions deteriorates by each method suggesting that non-causal SNPs and those not from associated regions affect the performance of these two discriminative multivariate methods.
In unpublished work, we make similar observations with three other multivariate feature selection methods: L2 norm regularized logistic regression (41), the weighted maximum margin criterion (42) and ridge regression (25). We use the Bundle software package (41) for regularized logistic regression and our own implementations of the latter two methods. After cross-validating parameters in each method, we find that at the 2r threshold level all methods improve upon chi-square to different degrees but the improvement decreases at higher thresholds.
The strategy of removing features in high-dimensional data using a simple statistic before applying a more sophisticated method has been studied previously but not exactly in the manner that we do and not on genome-wide studies. Take the winners of the NIPS 2003 feature selection contest. They used a simple univariate statistic to obtain a smaller input size before applying a more sophisticated neural network plus tree-based multivariate procedure for final feature selection (43). Fan and Lv (44) provide theoretical and empirical arguments for the same idea: removing many features with a simple statistic before applying a sophisticated multivariate one for final selection. Finally, Chen and Lin (45) show that removing features with a simple univariate ‘F-score’ statistic improves classification performance of the SVM on all but one of the datasets used in the NIPS 2003 contest. This is not the same as SVM-based feature selection but is relevant to our work because it says something about SVM discriminant computed with full versus a culled set of features.
The SVM has previously been shown to rank non-causal variables higher than causal ones empirically (46) and theoretically (47). In both studies, culling the input dataset by a univariate filter was not considered. In light of our results here and the studies cited above it is possible that the SVM may yield better results in those studies if the input is first culled.
When the SVM is applied to the entire GWAS we find its ranking of causal variants and associated regions to be similar to chi-square and better than SVM(5r) and SVM(10r). This can be explained by the automatic setting of the SVM loss-complexity tradeoff parameter 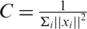 , where i loops over all subjects in the training set. When the entire GWAS is considered each ||xi||2 is large which makes C a very small number particularly in comparison to the value obtained in SVM(5r) and SVM(10r). This affects the discriminant w which is actually given by w = ∑i αi yi xi where each αi ≤ C (25), yi is +1 if xi is case and −1 otherwise. With a very small value of C all of αi are small and the same thus effectively reducing the discriminant value of the j-th SNP to be
, where i loops over all subjects in the training set. When the entire GWAS is considered each ||xi||2 is large which makes C a very small number particularly in comparison to the value obtained in SVM(5r) and SVM(10r). This affects the discriminant w which is actually given by w = ∑i αi yi xi where each αi ≤ C (25), yi is +1 if xi is case and −1 otherwise. With a very small value of C all of αi are small and the same thus effectively reducing the discriminant value of the j-th SNP to be 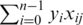 where n is the total number of subjects in the study, yi as defined above, and xij is the j-th encoded SNP of the i-th subject. We verify this manually on simulated study number zero and find the SVM ranking to be similar to the one obtained using the above formula.
where n is the total number of subjects in the study, yi as defined above, and xij is the j-th encoded SNP of the i-th subject. We verify this manually on simulated study number zero and find the SVM ranking to be similar to the one obtained using the above formula.
It is important to note that our work with the SVM presented here does not cross-validate the tuning parameter C which controls tradeoff between error on the training data and the classifier complexity. As mentioned earlier, we use a default value of C provided by the SVM-light software. We did perform the same experiments by cross-validating C and found no difference in the performance of the support vector machine at the 2r threshold. At the larger 5r and 10r thresholds the SVM performs better with the cross-validated C than the automatic one. The improvement, however, is not large enough to justify an expensive cross-validation procedure which is why we omit the procedure from this study altogether.
The less pronounced differences between the multivariate methods and chi-square on the WTCCC Crohn's disease and type 2 diabetes studies as well as on simulated data of relative risk 1.25 suggest that the advantage from multivariate methods over univariate in genome-wide studies may be gained only on studies where the value of r, which is the number of SNPs with P-values within Bonferroni, is non-trivial. This becomes clear if we compare the values of r across the three different relative risks at fixed sample sizes of 2000 and across the different sample sizes at relative risk 1.25.
Our risk prediction results show limited improvement with SVM-ranked SNPs compared to chi-square and RF ones. However, there are several aspects of this improvement that are noteworthy: (i) we see it consistently on many simulated datasets, (ii) it becomes larger at higher relative risks, and (iii) the AUC peaks earlier with a few top SVM ranked SNPs when compared to the chi-square ranked ones. In Supplementary Figures S10 and S11, we provide risk prediction results on simulated data that support the above observations. There we see that the improvement given by a few top SVM-ranked SNPs is highest at relative risk 2 and progressively decreases at lower relative risks. This suggests that there is a potential to gain higher risk prediction accuracy with SVM ranked SNPs if there is sufficient signal in a GWAS. This may very well be the case with GWAS that have larger sample sizes and more SNPs than current ones. It is part of our ongoing research to test these methods on such GWAS.
Although we have not explored this in detail here, the SVM(2r) and RF(2r) methods both have the potential to detect interacting SNPs. A straightforward, yet computationally expensive, solution would be to first recode all pairs of SNPs into new numerical values between 0 and 8 instead of encoding each SNP 0, 1 or 2. Then we would apply SVM(2r) and RF(2r) in the same way as done in this article and examine the top r ranked variables for interacting SNPs.
We also rank all SNPs in the GWAS by the SVM and apply chi-square to the top 100 ranked ones to determine if it would improve upon the support vector machine ranking. Supplementary Figures S6 shows that this offers no improvement over chi-square applied to the entire GWAS.
Finally, it is straightforward to incorporate non-genetic variables such as age, sex and principal components for population substructure into the SVM(kr) and RF(kr) methods. They would simply be additional columns in the culled data matrix that is given as input.
CONCLUSION
We find the support vector machine to rank causal SNPs and those from associated regions higher than random forest and chi-square if applied to the top 2r chi-square-ranked SNPs, where r is the number of SNPs with p-values within Bonferroni, and the value of r is sufficiently large.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
The CIPRES cluster supported by the National Science Foundation (EF0331654) and the Kong cluster at NJIT; Wellcome Trust under award 076113. Funding for open access charge: U.S. National Science Foundation and U.S. National Institutes of Health.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
This study makes use of data generated by the Wellcome Trust Case-Control Consortium. A full list of the investigators who contributed to the generation of the data is available from www.wtccc.org.uk.
REFERENCES
- 1. doi: 10.1038/nature05911. Welcome Trust Case Control Consortium. (2007) Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature, 447, 661–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jewell NP. Statistics for Epidemiology. New York, USA: Chapman & Hall; 2003. [Google Scholar]
- 3.Stromberg U, Bjork J, Vineis P, Broberg K, Zeggini E. Ranking of genome-wide association scan signals by different measures. Int. J. Epidemiol. 2009;38:1364–1373. doi: 10.1093/ije/dyp285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li J. Prioritize and select SNPs for association studies with multi-stage designs. J. Computat. Biol. 2007;15:241–257. doi: 10.1089/cmb.2007.0090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li C, Li M, Lange EM, Watanabe RM. Prioritized subset analysis: improving power in genome-wide association studies. Hum. Heredity. 2008;65:129–141. doi: 10.1159/000109730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li Q, Yu K, Li Z, Zheng G. Max-rank: a simple and robust genome-wide scan for case-control association studies. Hum. Genet. 2008;123:617–623. doi: 10.1007/s00439-008-0514-8. [DOI] [PubMed] [Google Scholar]
- 7.Schwarz DF, Knig IR, Ziegler A. On safari to random jungle: a fast implementation of random forests for high-dimensional data. Bioinformatics. 2010;26:1752–1758. doi: 10.1093/bioinformatics/btq257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meng Y, Yu Y, Cupples LA, Farrer L, Lunetta K. Performance of random forest when SNPs are in linkage disequilibrium. BMC Bioinformatics. 2009;10:78. doi: 10.1186/1471-2105-10-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mao W, Mao J. In Proceedings of International Conference on Technology and Applications in Biomedicine. USA: IEEE; 2008. The application of random forest in genetic case-control studies; pp. 370–373. [Google Scholar]
- 10.Ban H-J, Heo JY, Oh K-S, Park K-J. Identification of type 2 diabetes-associated combination of snps using support vector machine. BMC Genetics. 2010;11:26. doi: 10.1186/1471-2156-11-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu TT, Chen YF, Hastie T, Sobel E, Lange K. Genome-wide association analysis by lasso penalized logistic regression. Bioinformatics. 2009;25:714–721. doi: 10.1093/bioinformatics/btp041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoggart CJ, Whittaker JC, Iorio MD, Balding DJ. Simultaneous analysis of all snps in genome-wide and re-sequencing association studies. PLoS Genet. 2008;4:e1000130. doi: 10.1371/journal.pgen.1000130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wei Z, Sun W, Wang K, Hakonarson H. Multiple testing in genome-wide association studies via hidden markov models. Bioinformatics. 2009;25:2802–2808. doi: 10.1093/bioinformatics/btp476. [DOI] [PubMed] [Google Scholar]
- 14.Chanda P, Sucheston L, Zhang A, Brazeau D, Freudenheim JL, Ambrosone CB, Ramanathan M. Ambience: a novel approach and efficient algorithm for identifying informative genetic and environmental interactions associated with complex phenotypes. Genetics. 2008;180:1191–210. doi: 10.1534/genetics.108.088542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vapnik V. The Nature of Statistical Learning Theory. Cambridge, Massachusetts, USA: Springer; 1998. [Google Scholar]
- 16.Breiman L. Random forests. Mach. Learning. 2001;45:532. [Google Scholar]
- 17.Guyon I, Weston J, Barnhill S, Vapnik V. Gene selection for cancer classification using support vector machines. Mach. Learning. 2002;46:389–422. [Google Scholar]
- 18.Niijima S, Kuhara S. Recursive gene selection based on maximum margin criterion: a comparison with svm-rfe. BMC Bioinformatics. 2006;7:543. doi: 10.1186/1471-2105-7-543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Diaz-Uriarte R, Alvarez de Andres S. Gene selection and classification of microarray data using random forest. BMC Bioinformatics. 2006;7:3. doi: 10.1186/1471-2105-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Statnikov A, Wang L, Constantin Aliferis A comprehensive comparison of random forests and support vector machines for microarray-based cancer classification. BMC Bioinformatics. 2008;9:319. doi: 10.1186/1471-2105-9-319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hochberg Y. A sharper Bonferroni procedure for multiple tests of significance. Biometrika. 1988;75:800–802. [Google Scholar]
- 22. doi: 10.1176/appi.ajp.2008.08091354. Psychiatric GWAS Consortium Coordinating Committee. (2009) Genomewide association studies: history, rationale, and prospects for psychiatric disorders. Am. J. Psychiat., 166, 540–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pearson TA, Manolio TA. How to interpret a genome-wide association study. 2008;299:1335–1344. doi: 10.1001/jama.299.11.1335. [DOI] [PubMed] [Google Scholar]
- 24.Hulbert EM, Smink LJ, Adlem EC, Allen JE, Burdick DB, Burren OS, Cavnor CC, Dolman GE, Flamez D, Friery KF, et al. T1DBase: integration and presentation of complex data for type 1 diabetes research. Nucleic Acids Res. 2007;35:D742–D746. doi: 10.1093/nar/gkl933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alpaydin E. Machine Learning. Cambridge, MA, USA: MIT Press; 2004. [Google Scholar]
- 26.Schölkopf B, Smola AJ. Learning with Kernels: Support Vector Machines, Regularization, Optimization, and Beyond. Cambridge, MA, USA: MIT Press; 2001. [Google Scholar]
- 27.Joachims T. Making large-scale svm learning practical. In: Schölkopf B, Burges C, Smola A, editors. Advances in Kernel Methods - Support Vector Learning. Cambridge, MA, USA: MIT Press; 1999. [Google Scholar]
- 28.Zhang H, Wang M, Chen X. Willows: a memory efficient tree and forest construction package. BMC Bioinformatics. 2009;10:130. doi: 10.1186/1471-2105-10-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mueller PW, Rogus JJ, Cleary PA, Zhao Y, Smiles AM, Steffes MW, Bucksa J, Gibson TB, Cordovado SK, Krolewski AS, et al. Genetics of kidneys in diabetes (GoKinD) study: a genetics collection available for identifying genetic susceptibility factors for diabetic nephropathy in Type 1 diabetes. J. Am. Soc. Nephrol. 2006;17:1782–1790. doi: 10.1681/ASN.2005080822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. The Gain Collaborative Research Group. (2007) New models of collaboration in genome-wide association studies: the genetic association information network. Nature, 39, 1045–1051. [DOI] [PubMed] [Google Scholar]
- 31.Evans DM, Visscher PM, Wray NR. Harnessing the information contained within genome-wide association studies to improve individual prediction of complex disease risk. Hum. Mol. Genet. 2009;18:3525–3531. doi: 10.1093/hmg/ddp295. [DOI] [PubMed] [Google Scholar]
- 32.Li C, Li M. GWAsimulator: a rapid whole-genome simulation program. Bioinformatics. 2008;24:140–142. doi: 10.1093/bioinformatics/btm549. [DOI] [PubMed] [Google Scholar]
- 33.Durrant C, Zondervan KT, Cardon LR, Hunt S, Deloukas P, Morris AP. Linkage disequilibrium mapping via cladistic analysis of single-nucleotide polymorphism haplotypes. Am. J. Hum. Genet. 2004;75:35–43. doi: 10.1086/422174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gillespie JH. Population Genetics: A Concise Guide. Baltimore, MD, USA: The Johns Hopkins University Press; 2004. [Google Scholar]
- 35.Smith CP, Nielsen DM, Suchindran S. Does strong linkage disequilibrium guarantee redundant association results? Genet. Epidemiol. 2008;32:546–552. doi: 10.1002/gepi.20328. [DOI] [PubMed] [Google Scholar]
- 36.Calle ML, Urrea V. Letter to the Editor: stability of random forest importance measures. Brief. Bioinformatics. 2010 doi: 10.1093/bib/bbq011. [DOI] [PubMed] [Google Scholar]
- 37.Boulesteix A-L, Slawski M. Stability and aggregation of ranked gene lists. Brief. Bioinformatics. 2009;10:556–568. doi: 10.1093/bib/bbp034. [DOI] [PubMed] [Google Scholar]
- 38.Wray NR, Goddard ME, Visscher PM. Prediction of individual genetic risk to disease from genome-wide association studies. Genome Res. 2007;17:1520–1528. doi: 10.1101/gr.6665407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gail MH. Discriminatory accuracy from single-nucleotide polymorphisms in models to predict breast cancer risk. N. Engl. J. Med. 2008;100:1037–1041. doi: 10.1093/jnci/djn180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wei Z, Wang K, Qu H-Q, Zhang H, Bradfield J, Kim C, Frackleton E, Hou C, Glessner JT, Chiavacci R, et al. From disease association to risk assessment: an optimistic view from genome-wide association studies on type 1 diabetes. PLoS Genet. 2009;5:e1000678. doi: 10.1371/journal.pgen.1000678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Teo CH, Smola A, Vishwanathan SVN, Le QV. In KDD '07: Proceedings of the 13th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. New York, NY, USA: ACM; 2007. A scalable modular convex solver for regularized risk minimization; pp. 727–736. [Google Scholar]
- 42.Zheng W, Zou C, Zhao L. Weighted maximum margin discriminant analysis with kernels. Neurocomputing. 2005;67:357–362. [Google Scholar]
- 43.Guyon I, Gunn S, Ben-Hur A, Dror G. Result analysis of the nips 2003 feature selection challenge. In: Saul LK, Weiss Y, Bottou L, editors. Advances in Neural Information Processing Systems 17. Cambridge, MA: MIT Press; 2005. pp. 545–552. [Google Scholar]
- 44.Fan J, Lv J. Sure independence screening for ultrahigh dimensional feature space. J. Roy. Stat. Soc. Ser. B. 2008;70:849–911. doi: 10.1111/j.1467-9868.2008.00674.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen Y-W, Lin C-J. Combining svms with various feature selection strategies. In: Guyon I, Nikravesh M, Gunn S, Zadeh L, editors. Feature Extraction. 2006. Vol. 207 of Studies in Fuzziness and Soft Computing, Springer Berlin/Heidelberg, Germany, pp. 315–324. [Google Scholar]
- 46.Statnikov A, Hardin D, Aliferis CF. In Proceedings of Neural Information Processing Systems (NIPS) Workshop on Causality and Feature Selection. Cambridge, MA, USA: The MIT Press; 2006. Using svm weight-based methods to identify causally relevant and non-causally relevant variables. [Google Scholar]
- 47.Hardin D, Tsamardinos I, Aliferis CF. In ICML '04: Proceedings of the Twenty-First International Conference on Machine Learning. New York, NY, USA: ACM; 2004. A theoretical characterization of linear svm-based feature selection; p. 48. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.