

Abstract
Agonist activation of the small GTPase, RhoA, and its effector Rho kinase leads to down-regulation of smooth muscle (SM) myosin light chain phosphatase activity, an increase in myosin light chain (RLC20) phosphorylation and force. Cyclic nucleotides can reverse this process. We report a new mechanism of cAMP-mediated relaxation through Epac, a GTP exchange factor for the small GTPase Rap1 resulting in an increase in Rap1 activity and suppression of RhoA activity. An Epac-selective cAMP analog, 8-pCPT-2′-O-Me-cAMP (“007”), significantly reduced agonist-induced contractile force, RLC20, and myosin light chain phosphatase phosphorylation in both intact and permeabilized vascular, gut, and airway SMs independently of PKA and PKG. The vasodilator PGI2 analog, cicaprost, increased Rap1 activity and decreased RhoA activity in intact SMs. Forskolin, phosphodiesterase inhibitor isobutylmethylxanthine, and isoproterenol also significantly increased Rap1-GTP in rat aortic SM cells. The PKA inhibitor H89 was without effect on the 007-induced increase in Rap1-GTP. Lysophosphatidic acid-induced RhoA activity was reduced by treatment with 007 in WT but not Rap1B null fibroblasts, consistent with Epac signaling through Rap1B to down-regulate RhoA activity. Isoproterenol-induced increase in Rap1 activity was inhibited by silencing Epac1 in rat aortic SM cells. Evidence is presented that cooperative cAMP activation of PKA and Epac contribute to relaxation of SM. Our findings demonstrate a cAMP-mediated signaling mechanism whereby activation of Epac results in a PKA-independent, Rap1-dependent Ca2+ desensitization of force in SM through down-regulation of RhoA activity. Cyclic AMP inhibition of RhoA is mediated through activation of both Epac and PKA.
Keywords: Cyclic Nucleotide Analogs, Cyclic Nucleotides, Rho, Signal Transduction, Smooth Muscle, Ca2+ Desensitization, Epac, Rap1, Contractility
Introduction
Contraction of smooth muscle (SM)3 is a reflection of the ratio of activities of myosin light chain kinase and myosin light chain phosphatase (MLCP). Both are regulated through agonist-induced signaling pathways, which can impact and change the relationship of force versus global [Ca2+]i (for review, see Ref. 1). Agonist or GTPγS activation of permeabilized SM while [Ca2+]i is clamped leads to increased myosin regulatory light chain (RLC20) phosphorylation and force through inhibition of MLCP (2, 5). The term Ca2+ sensitization was coined to describe this phenomenon. One of the major physiologically relevant pathways leading to Ca2+ sensitization is the activation of the small GTPase, RhoA, and its effector Rho kinase (3, 4), which phosphorylates and inhibits myosin phosphatase targeting subunit (MYPT1), resulting in increased RLC20 phosphorylation and force at constant Ca2+ concentration (1, 2, 5–7). On the other hand, intracellular second messengers cAMP and cGMP are important players in the modulation of SM tone under physiological and pathophysiological states. They can reduce the Ca2+ sensitivity of the contractile proteins, resulting in relaxation, termed Ca2+ desensitization (1, 8–11). This proceeds in parallel with processes that reduce [Ca2+]i. Several mechanisms have been proposed to account for cyclic nucleotide-induced relaxation of Ca2+-sensitized force, but their relative importance and temporal and spatial regulation is poorly understood. The known pathways include 1) the phosphorylation and inhibition of RhoA (12–16), 2) activation of MLCP through an interaction between the leucine zipper motifs of protein kinase GIα (PKGIα) and MYPT1 (17) and/or through PKA/PKG phosphorylation of the SM-specific protein telokin (18, 19), and 3) PKA- or PKG-induced reduction of MYPT1 inhibitory phosphorylation at Thr-696/853 (20–23). We now provide evidence that a cAMP-activated, PKA- and PKG-independent, Epac-Rap1 GTPase signaling pathway also contributes to cAMP-induced relaxation of Ca2+-sensitized force in vascular, gut, and airway SM.
Epac, a Rap1 GTP exchange factor directly activated by cAMP, was first reported in two seminal papers (24, 25). This was very surprising as at that time PKA was considered the sole effector of cAMP. In view of the importance of cyclic nucleotide signaling in SM, we set out to investigate whether this Epac1/Rap1 pathway is a tenable mechanism for Ca2+ desensitization of force in SM. Rap1 is a member of the Ras family of small GTPases, which upon exchange of GTP for GDP serves as a molecular switch to couple extracellular signals to a variety of cellular responses. Interestingly, in endothelial cells and neurons, an increase in Rap1-GTP after cAMP activation of Epac through an unknown mechanism leads to a decrease in RhoA-GTP, known to play an important role in these cells regulating permeability and migration, respectively (26, 27). We now show such cross-talk between Rap1 and RhoA signaling pathways in SM. A recent study of intact tracheal SM tissue and cultured tracheal cells based on the use of a Rac inhibitor reported that cAMP activation of Epac lead to relaxation through activation of Rac (28). As Epac GEF (guanine nucleotide exchange factor) activity is selective for Rap1, the pathway leading to Rac activation in their study is unclear. Furthermore, this study did not examine the role of Rap1 activity. We now show that Epac acting through Rap1 is an additional cAMP effector for induction of relaxation in vascular, intestinal, and airway SM. Furthermore, we specifically studied the impact of Epac1/Rap1 signaling on the isolated RhoA-mediated Ca2+sensitization component of SM contractility in SM tissues and Rap1B null cells.
The development of the Epac-specific nucleotide 8-pCPT-2′-O-Me-cAMP (29, 30) has been an important tool for understanding the physiological and pathophysiological roles of Epac. The specificity of this cAMP analog for Epac is thought to arise from distinct structural features in the cAMP domains of Epac and PKA (31, 32). In vivo studies have shown that 8-pCPT-2′-O-Me-cAMP has a >100-fold higher discriminatory affinity for Epac as compared to cAMP (32). In keeping with other publications and for simplicity, we will refer to 8-pCPT-2′-O-Me-cAMP as “007” throughout the manuscript (33). All together, we demonstrate a signaling mechanism whereby isoproterenol, PGI2, or 007 activation of Epac via cAMP results in PKA-independent, Rap1-dependent Ca2+ desensitization of force in SM through down-regulation of RhoA activity. We propose that this is through activation of a Rap1-activated GTPase-activating protein (RhoGAP), to be investigated in future studies. In view of the importance of increased RhoA activity in hypertension, asthma, vasospasm, gut motility, and in the transcriptional regulation of the expression of SM marker proteins (1), we propose that the Epac/Rap1 signaling pathway may offer new therapeutic targets.
EXPERIMENTAL PROCEDURES
Tissue Preparation and Force Measurements
All procedures were carried out according to protocols approved by the Animal Care and Use Committee at the University of Virginia. Methods for dissection, force measurements, α-toxin and β-escin permeabilization, Ca2+ sensitization and desensitization protocols and reagents are detailed in the supplemental Experimental Procedures.
Tissue Screen and Western Blots
Buffers and Western blot analysis are described in the supplemental Experimental Procedures.
Cells Culture and Cell Transfection
Rat aortic (R518), human bronchi, and mouse fundus SM cells were isolated and maintained as described in the supplemental Experimental Procedures. Mouse embryonic fibroblast cells were isolated from Rap1B knock-out mice (34) as described previously (35). Mouse embryonic fibroblasts were cultured in Dulbecco's modified eagle medium supplemented with 10% embryonic stem cell-qualified fetal bovine serum (Invitrogen) and a mixture of penicillin-streptomycin (Invitrogen). Subconfluent, serum-starved SM cells were treated with 1 μm lysophosphatidic acid (LPA) with and without 50 μm 007 or 50 μm 007 alone, isoproterenol, H89, or IBMX for the desired times and used for RhoA or Rap1 activation assays. NIH-3T3 cells were transfected with wild type human pcDNA3-Epac1 and dominant negative pcDNA3-Epac1R279E (a generous gift from Dr. X. Cheng, University of Texas Medical Branch, Galveston, TX) as described in the supplemental Experimental Procedures.
Rap1 and RhoA Activation Assays
The GTP loading status of RhoA and Rap1 in SM cells and tissues were assessed using Rhotekin and RalGDS assays, respectively (36, 37) as detailed in the supplemental Experimental Procedures.
Epac1 Silencing
To knock down endogenous Epac1 protein adeno-associated virus delivery of small hairpin RNA (shRNA) into cultured rat aortic SMCs were used. DNA constructs and viral particles were prepared as described in Kasahara and Aoki (38). Briefly, Epac1-specific insert sequence TTCCTAACCATGAGGAACC (shEpac1) (39) and a non-targeting control sequence, AAGTGGCGCGCTAGGAAGAGA (non-target shRNA), which were separated by loop sequence (TTCAAGAGA) from the reverse complement of the targeting sequences, were synthesized and cloned into the Apa1 and EcoRI sites of the pAdloss shuttle vector. Viral particles were subsequently generated by co-transfecting CRE8 cells with the pAdloss shuttle vector and Ψ5 backbone.
MLCP and RLC20 Phosphorylation
Phosphorylation of MYPT1 Thr-696/853 sites and of RLC20 was determined by SDS-PAGE and Western blotting as detailed in the supplemental Experimental Procedures. After the stimulation protocols, muscle strips were immediately frozen by immersion in acetone, 10% (w/v) trichloroacetic acid and stored at −80 °C followed by an acetone wash, homogenization in sample buffer, and electrophoresis.
Expression of Smooth Muscle Marker Genes
Subconfluent R518 aortic cells were serum-starved for 24 h, treated with 50 μm 007 for 13 h, and lysed in TRIzol reagent (Invitrogen), and RNA was isolated following the manufacturer's instructions. The real time RT-PCR was performed as detailed in the supplemental Experimental Procedures and Table 1. The expression of SM genes was normalized to 18 S rRNA or GAPDH.
Statistics
Mean values and S.E. were obtained from three-eight independent experiments for each condition. Statistical significance of group differences was assessed using Student's t-tests at a p value < 0.05, considered significant. If the data proved highly non-Gaussian, a log transform (log(1 + value)) was used to normalize the data before statistical analysis.
RESULTS
8-pCPT-2′-O-Me-cAMP (007)-induced Relaxation of Force and RLC20 and MLCP Dephosphorylation
In intact SM, maximal forces induced by 0.5 μm phenylephrine in rabbit portal vein (Fig. 1A) or the thromboxane analog, U46619, in rabbit pulmonary artery (data not shown) were relaxed upon the addition of 30 μm 007 by 34 ± 3.5%; p < 0.0001 and 16 ±1.8% (p < 0.0001), respectively. Subsequent addition of forskolin relaxed muscles to base line, 2 ± 2.0%, p < 0.0001. Agonists such as U46619, carbachol, endothelin-1, bradykinin, or phenylephrine were added to α-toxin-permeabilized rabbit pulmonary artery, mouse bronchi, mouse fundus SM, and Ca2+-sensitized force recorded (Fig. 1, B, C, and D). Once force had reached a plateau, 50–100 μm 007 was added, which relaxed the Ca2+ plus agonist-induced force by 73 ± 4.6 and 43 ± 7.0% (n = 7; p < 0.05) in the pulmonary artery and fundus muscle strips, respectively. 50 μm 007 completely relaxed the carbachol, endothelin-1, and bradykinin Ca2+-sensitized force in mouse bronchi muscle strips. Thus, this cyclic nucleotide analog can significantly relax a variety of intact and permeabilized SMs from different species. The 007 IC50 for rabbit pulmonary artery was 35 μm (Fig. 1E). 007-induced relaxation of mouse fundus was accompanied by a significant decrease in RLC20 phosphorylation and MYPT1 Thr-853 phosphorylation (Fig. 1F). RLC20 phosphorylation in pulmonary artery decreased in a concentration-dependent manner to 78 ± 0.9% (p < 0.001) and 45% ± 2.6 (p < 0.001) at 50 and 100 μm 007, respectively (Fig. 1H). This was accompanied by a significant decrease in inhibitory phosphorylation of MLCP regulatory subunit, MYPT1, at the Rho kinase site Thr-853 to 68 ± 4.8% (p < 0.002) and 62 ± 1.0% (p < 0.001) at 50 and 100 μm 007, respectively (Fig. 1H). Phosphorylation of the Thr-696 site on MYPT1 did not change (Fig. 1H). These data suggest that 007-induced Ca2+ independent relaxation is mediated through RLC20 dephosphorylation and disinhibition of MLCP activity. 007 mediated relaxation of U46619-induced Ca2+-sensitized force in pulmonary artery was accompanied by a reduction in the U46619-induced increase in RhoA activity (Fig. 1G). Further changes in RhoA activity were observed in other smooth muscle cells, shown below.
FIGURE 1.

007-induced relaxation, RLC20 dephosphorylation, and disinhibition of MLCP activity as well as a decrease in RhoA activity. A, 007 (30 μm) significantly relaxed phenylephrine (PE)-induced force in intact rabbit portal vein. B, in α-toxin-permeabilized rabbit pulmonary artery precontracted with pCa 6.5, 007 (100 μm) relaxed the thromboxane analog U46619-induced Ca2+-sensitized force. C and D, Ca2+-sensitized force induced by carbachol (Carb), endothelin-1 (ET), and bradykinin (Brad) was completely relaxed by 007 (50 μm) in α-toxin-permeabilized mouse bronchi and mouse fundus in the presence of 2 μm GTP and [Ca2+]i clamped at pCa 6.3 or 6.0. E, shown is a dose-response curve for 007-mediated relaxation of U46619-induced Ca2+-sensitized force in rabbit pulmonary artery. Agonist-induced Ca2+-sensitized force was taken as 100% (mean ± S.E., n = 8). F, Western blot analysis showed a decrease in carbachol-induced RLC20 phosphorylation and MYPT1 phosphorylation at the Rho kinase site Thr-853 after treatment of mouse fundus with 007 (30 μm). G, 007-induced relaxation of U46619-induced Ca2+-sensitized force is associated with a decrease in U46619-induced RhoA activity in rabbit pulmonary artery. H, shown is a summary of normalized RLC20 phosphorylation and MYPT1 phosphorylation at Thr-853 and Thr-696 after treatment of rabbit pulmonary artery with U46619 and 007 (50 or 100 μm). 007 significantly decreased RLC20 phosphorylation and MYPT1 phosphorylation at Thr-853 (mean ± S.E.; #, p < 0.01; *, p < 0.001).
The PGI2 Analog Cicaprost Relaxes SM, Increases Rap1 Activity, and Decreases RhoA Activity
Prostacyclin (PGI2) is the main product of arachidonic acid production with strong hypotensive effects and a vasodilator activity toward all vascular beds (40). Such activity is largely mediated by cAMP production (41, 42). We, therefore, used the physiologically relevant PGI2 analog, cicaprost, and the broad range phosphodiesterase inhibitor, IBMX, and measured effects on Rap1 and RhoA activities. In intact SM, maximal forces induced by EC50 doses of U46619 (0.2 μm) in rabbit pulmonary artery (Fig. 2) or the muscarinic receptor agonist, carbachol (0.15 μm), in rabbit fundus (data not shown) were relaxed upon the addition of 15 μm cicaprost (by 45% in fundus and by 64% in pulmonary artery, respectively). Subsequent addition of 10 μm IBMX further relaxed muscles to near base line (to 80% in fundus). IBMX increased Rap1-GTP (2.7 times) accompanied by a more than 4-fold fall in RhoA activity (Fig. 2, lower panels). Force developed by 0.2 μm U46619 in rabbit pulmonary artery strips coincided with 8-fold increase in active RhoA but no changes in Rap1 activity were observed (Fig. 2, lower panels). The addition of cicaprost significantly increased the level of Rap1-GTP (1.8 times) and decreased RhoA activity almost to base line. An increase in Rap1 activity and decrease in RhoA activity was also measured in intact fundus SM tissue (n = 1). In two experiments there was no detectable change in Rap1, whereas RhoA activity decreased. We have found in general that measurements of changes in Rap1 activity in tissues, but not cultured cells, are more difficult presumably due to loss of GTP during tissue homogenization. All together these findings demonstrate that a physiological stimulation of cAMP by cicaprost in intact SM can increase Rap1 activity and decrease RhoA activity similar to our findings with the Epac selective analog 007.
FIGURE 2.
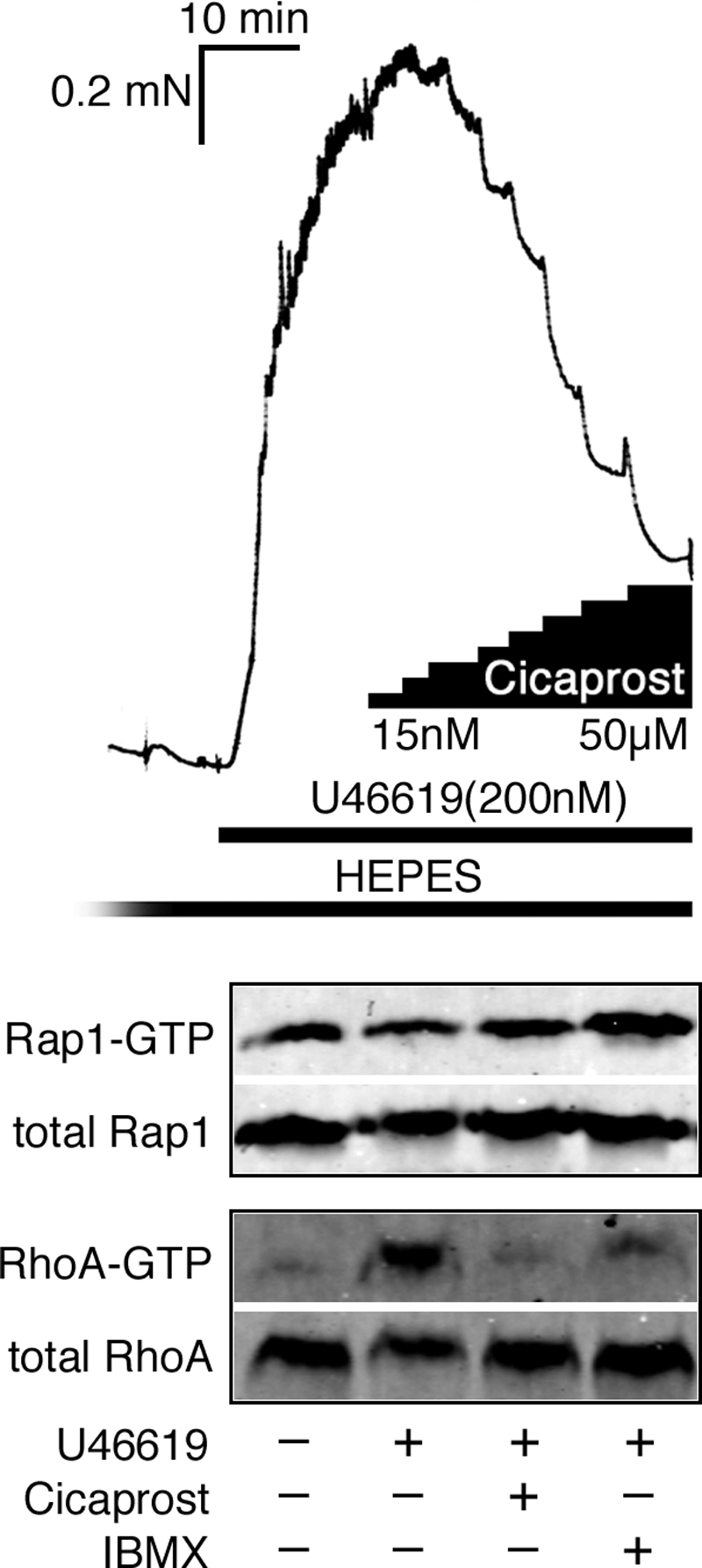
Cicaprost (15 μm) and IBMX (10 μm) significantly relaxed force induced by an EC50 dose of U46619 (0.2 μm) in intact rabbit pulmonary artery. U46619-induced force development coincided with a significant increase in active RhoA, but no changes in Rap1 activity were observed. Subsequent addition of cicaprost and IBMX increased Rap1-GTP and decreased of RhoA activity almost to base line.
Tissue Screens for Epac1, Rap1, and RhoA Proteins
Screening of rabbit tissues for Epac1 showed that protein levels in rabbit pulmonary artery, aorta, and portal vein were comparable with that in heart, which showed the highest protein expression (Fig. 3). Epac1 protein expression in mouse aorta (data not shown) was greater than in ileum, similar to rabbit. Not all gastrointestinal SMs are low in Epac1 expression, as mouse fundus SM Epac1 protein levels were comparable with that of the heart (Fig. 3). Cultured rat aortic SMCs (Fig. 3) expressed Epac1 protein. Rap1 and RhoA expression in aorta, pulmonary artery, and portal vein was ∼2-fold greater than in bladder and ileum when normalized to RLC20 content, taken as a measure of contractile potential (supplemental Fig. S1). Therefore, Epac1, Rap1, and RhoA protein are present in vascular and fundus smooth muscle and to a lesser extent in bladder and ileum of several mammalian species.
FIGURE 3.

Expression profiles of Rap1, RhoA and Epac1 in rabbit, rat, and mouse SM tissues. Western blot analysis of Rap1, RhoA, and Epac1 in various rabbit tissues and cultured rat aortic cells (R518) and Epac1 in various mouse tissues is shown. Note the high Epac content of mouse fundus.
Cyclic Nucleotide Analogues Discriminate between PKA and Epac Activation
U46619 contracted α-toxin-permeabilized pulmonary artery at a constant Ca2+ concentration (Fig. 4A). The non-hydrolysable 8-Br-cAMP relaxed U46619 Ca2+-sensitized force in pulmonary artery by 71 ± 6.1% (data not shown). The PKA-specific analog, 6-Bnz-cAMP, also significantly relaxed these Ca2+-sensitized muscles by 57 ± 2.7% (Fig. 4A). 8-Br-cAMP- was significantly greater than 6-Bnz-cAMP-induced relaxation (p < 0.05, n = 4). The antagonist Rp-cAMP competitively binds to both cAMP binding domains of the PKA regulatory subunit and inhibits dissociation of the catalytic subunit. The Rp-8CPT-cAMP analog of Rp-cAMP inhibited the 6-Bnz-cAMP-induced relaxation by greater than 78% (p < 0.001; Fig. 4A). Rp-cAMP also acts as an inhibitor of Epac guanine nucleotide exchange factor activity toward Rap1 (30). Thus, Rp-8CPT-cAMP inhibited the relaxation induced by 007 as expected (Fig. 4A). The highly specific inhibitor of the PKA catalytic subunit, PKI peptide (pKi = 1.7 nm), inhibited the relaxation induced by 6-Bnz-cAMP of phenylephrine-induced Ca2+-sensitized force in β-escin-permeabilized rabbit portal vein by 67 ± 2.5% (p < 0.002; Figs. 4, B and C) but was without significant effect on the extent of 007-induced relaxation under identical conditions (Figs. 4, B and C). There was a tendency for PKA-specific inhibitory peptide to slow the rate of relaxation (t½) induced by 007, but this did not reach significance (p > 0.06, n = 24). At the same time, PKGI and -II inhibitor (43) Rp-8-pCPT-cGMPS at 25 μm did not affect relaxation induced by 007 in rabbit pulmonary artery, ruling out the possibility that 007 signals either directly or indirectly through PKG (supplemental Fig. S2). Thus, the Epac analog, 007, discriminates between Epac and the cAMP- and cGMP-dependent protein kinases.
FIGURE 4.

cAMP analogs discriminate between PKA and Epac activation in permeabilized smooth muscle. A, U46619 (300 nm) in the presence of 2 μm GTP for 10 min induced Ca2+-sensitized force in α-toxin-permeabilized rabbit pulmonary artery precontracted in pCa 6.7. The PKA-specific activator 6-Bnz-cAMP (1 μm) significantly relaxed U46619-induced Ca2+-sensitized force. Both 6-Bnz-cAMP- and 007 (30 μm)-induced relaxation were significantly inhibited by the RP-8CPT-cAMPS (50 μm) (mean ± S.E., n = 4–26). B and C, the PKA-specific inhibitory peptide (PKI; 10 μm) inhibited the PKA-specific relaxation but not Epac-mediated relaxation of PE-induced Ca2+-sensitized force. In these experiments rabbit portal veins were permeabilized with β-escin to allow PKA-specific inhibitory peptide access to the cytoplasm in the presence of 1 μm calmodulin and subsequently relaxed with 6-Bnz-cAMP (1 μm) or 007 (30 μm). Force was normalized to the pCa plus agonist-induced Ca2+-sensitized force taken as 100% (mean ± S.E., n = 10). The pCa -induced force at the onset of each force trace has been truncated in C. PE, phenylephrine; N.S., not significant.
The PKA independence of 007 stimulation was also supported by the absence of phosphorylation of vasodilator-stimulated phosphoprotein (VASP) at Ser-157. The 007 (50 μm)-induced relaxation of U46619-mediated Ca2+-sensitized force at both 2- and 10-min time points after the addition of 007 was not accompanied by an increase in VASP phosphorylation (Fig. 5A). Positive controls on intact pulmonary artery show VASP phosphorylation was induced by forskolin and the PKA-specific 6-Bnz-cAMP (1 μm) but not by the PKA inhibitor, Rp-8CPT-cAMP (Fig. 5A). Similarly, stimulation of rat aortic SMCs with forskolin (10 μm, 15 min) or 6-Bnz-cAMP (100 μm, 15 min) leads to significant VASP phosphorylation, p < 0.001 and p < 0.03, respectively, whereas 007 (50 μm, 15 min) has no significant effect on VASP phosphorylation compared with control cells (Fig. 5B). The PKA inhibitor H-89 decreased basal, forskolin, and 6-Bnz-cAMP-stimulated levels of VASP phosphorylation (p < 0.001, p < 0.002, and p < 0.03, respectively). VASP phosphorylation values of 007 H89 did not differ significantly from control values with and without H89. Altogether these findings suggest the possibility for a significant contribution of the Epac signaling pathway to cAMP-induced relaxation of Ca2+-sensitized force in smooth muscle. The results under our experimental conditions also support the reported PKA independence of the 007compound (29, 30).
FIGURE 5.

Activation of Epac does not lead to phosphorylation of the PKA specific target VASP. A, both 6-Bnz-cAMP and forskolin increased VASP phosphorylation in intact pulmonary artery. 007, 50 μm (2 min, 10 min), did not increase phosphorylation of VASP at Ser-157 in permeabilized pulmonary arteries precontracted with U46619. Surprisingly, VASP phosphorylation tended to decrease under these conditions. VASP phosphorylation in permeabilized SM before and after U46619 stimulation did not differ. B, both forskolin (FK) and 6-Bnz-cAMP (p < 0.001 and p < 0.03, respectively), but not 007, increased VASP phosphorylation at Ser-157 in rat aortic SMCs. H89 significantly decreased control and forskolin-, 007-, and 6-Bnz-cAMP-induced VASP phosphorylation (p < 0.001; p < 0.002; p < 0.01; and p < 0.03, respectively, n = 4 for each condition. Changes in VASP phosphorylation in the presence of 007 with and without H89 did not differ from control samples. VASP phosphorylation was normalized to total Rap1 (lower panel). Data are shown as a percentage of basal Ser-157-VASP phosphorylation.
Activation of Epac Alters Rap1 and RhoA Activities
Rat aortic SMCs (R518) showed an increase in Rap1-GTP after 007 activation that was not inhibited by the PKA inhibitor H89 (Fig. 6A). Overexpression of wild type Epac1 protein in NIH-3T3 cells did not significantly increase basal Rap1 activity (Fig. 6B). However, stimulation with 007 in the presence of expressed wild type Epac1 protein increased Rap1 activity by 3.5-fold (p < 0.008; Fig. 6B). On the other hand, overexpression of dominant negative Epac1R279E, which has been shown to be defective in cAMP binding (44), significantly decreased basal Rap1 activity by 43 ± 13% compared with control cells (p < 0.03) and prevented 007-induced Rap1 activation (p was not significant). Thus, these experiments demonstrate that activation of Epac increases Rap1 activity and that 007 activates Epac in SM cells and, furthermore, that this is PKA-independent. Significantly, not only 007 but forskolin, IBMX, and isoproterenol treatment of R518 cells also led to a 4.2 ± 1.1, 3.9 ± 1.3, and 4.8 ± 1.9 (p < 0.05)-fold increase in Rap1 activity respectively (Fig. 6C). Importantly, 007 (50 μm) significantly decreased RhoA activity induced by LPA treatment in Rap1B+/+ mouse embryonic fibroblast cells (p < 0.02, n = 3) but not in Rap1B−/− mouse embryonic fibroblast cells (see Fig. 8A). Therefore, the 007-mediated decrease in RhoA activity signals through Rap1B in these fibroblast cells. We next examined whether the ability of 007 to decrease RhoA activity in fibroblasts was also true in different sources of SMCs. Treatment of serum-starved human airway SMCs with 007 significantly increased active Rap1-GTP compared with untreated controls (p < 0.001; Fig. 7, A and C). Stimulation with LPA, a known activator of RhoA, significantly increased RhoA activity in R518, human airway SMCs, and fundus SMCs as expected (Fig. 7, A and B; p < 0.01, p < 0.003, and p < 0.001, respectively). Subsequent addition of the Epac-specific cAMP analog, 007 (50 μm), significantly decreased RhoA-GTP in both the rat aortic (p < 0.007, n = 3) and human airway SMCs (p < 0.04, n = 3). As Epac is expressed in fundus SM and 007 relaxed carbachol-induced Ca2+-sensitized force in this muscle (Fig. 1D), mouse fundus SMCs were also assayed (Fig. 7, A, B, and D). Although in each of the eight paired fundus SMC experiments Rap1 and RhoA activity increased and decreased respectively (Fig. 7, D and B), the magnitude of the changes were highly variable, possibly because of the difficulty in conservation of GTP bound Rap1 and RhoA in the fundus during the necessary homogenization steps. Therefore, statistical analysis was carried out using a log transformation. Fold change Rap1 and RhoA activity of LPA stimulation with and without 007 was 1.8 ± 0.3 versus 4.5 ± 1.5 (p < 0.02) and 5.4 ± 1.4 versus 3.2 ± 1.0 (p < 0.01), respectively. In other assays on tissues, 007 also induced a 47 ± 6.3% (p < 0.03) decrease in RhoA activity in pulmonary arteries analyzed during the contractility assay (Fig. 1B) at 10 min after the addition of 007 (Fig. 1G). This change coincided with the decrease in the Rho kinase target, phospho-Thr-853 of MYPT1, and the fall in RLC20 phosphorylation (Fig. 1H). Next we tested whether LPA-induced RhoA activation is also attenuated by the Gs-coupled receptor agonist, isoproterenol. Treatment of serum-starved R518 cells with LPA (1 μm, 15 min) significantly increased RhoA-GTP (p < 0.02, n = 4) (Fig. 8B), whereas co-stimulation with LPA and isoproterenol (100 μm, 15 min) totally prevented this increase. RhoA activity in cells treated with isoproterenol alone did not differ from the control. Pretreatment of cells with PKA inhibitor, H89 (10 μm, 60 min), significantly increased basal RhoA activity (p < 0.007, n = 4), which did not further increase after LPA stimulation (Fig. 8B) (p was not significant, n = 4). This suggests that cAMP/PKA actively suppresses RhoA activity in the basal state. Co-stimulation with LPA and isoproterenol decreased RhoA activity to the basal level. The inclusion of H89 did not significantly reverse the isoproterenol-induced decrease in RhoA activity.
FIGURE 6.

A and B, 007 increases Rap1 activity independently of PKA. A, 007 (50 μm, 15 min) significantly increased Rap1 activity in rat aortic SMCs in the presence and absence of H89. H89 (5 μm) alone had no significant effect. Rap1-GTP levels were normalized to total Rap1 and are shown as a % of control (mean ± S.E., n = 3); control versus 007: #, p < 0.0001; 007 versus 007 + H89: *, p < 0.001. B, 007 activates wild type Epac1 but not a mutant dominant negative Epac1. Overexpression of wild type Epac1 in NIH-3T3 significantly increased Rap1 activity upon 007 (50 μm, 15 min) stimulation but had no effect on basal Rap1 activity, whereas overexpression of dominant-negative Epac1 totally blocked Rap1 activation after 007 treatment as well as decreased basal Rap1 activity (mean ± S.E., n = 3–5; *, p < 0.008; #, p < 0.001 versus Epac1-WT plus 007; +, p < 0.03 versus non-stimulated control). C, forskolin, IBMX and isoproterenol (iso) increase Rap1 activity in aortic cells. Serum-starved rat aortic SMCs were treated with 10 μm forskolin (FK), 1 mm IBMX, or 100 μm isoproterenol for 15 min and subsequently assayed for Rap1 activation. p < 0.05, n = 4–5.
FIGURE 8.

A, 007 decreases LPA-induced RhoA activity in WT but not Rap1B null mouse embryonic fibroblasts. Serum-starved cells were treated with 1 μm LPA ± 50 μm 007 for 15 min. In Rap1B+/+ cells, 007 significantly decreased RhoA-GTP levels (*, p < 0.02, n = 3) but not in the Rap1B−/− cells. The inset shows the absence of Rap1B protein in Rap1B−/− cells. B, isoproterenol (iso, 100 μm) decreases LPA-induced RhoA activity in a PKA-independent manner. LPA stimulation (1 μm, 15 min) of serum-starved rat aortic SMCs significantly increased RhoA activity that was decreased to control levels by co-treatment with isoproterenol. Pretreatment of cells with H89 (1 μm, 1 h) significantly increased basal RhoA activity that was not further increased by LPA stimulation. H89 did not inhibit the isoproterenol-induced decrease in RhoA activity (mean ± S.E.; *, p < 0.006; #, p < 0.02, n = 4). C, D, and E, Epac1 shRNA decreases both Epac1 mRNA and protein levels and totally abolished isoproterenol-induced increases in Rap1 activity. Rat aortic SMCs were infected with either Epac1 shRNA- or non-targeting shRNA-carrying virus and incubated for 36 h followed by serum-free medium for another 16 h. Shown are mRNA (C) and Epac1 (D) mRNA and protein levels, respectively (*, p < 0.05; #, p < 0.0005, n = 3). E, isoproterenol (100 μm, 15 min) significantly increased Rap1 activity in non-targeting shRNA infected cells but not in Epac1 shRNA-infected cells (*, p < 0.0005, n = 4). F, isoproterenol decreased LPA-induced RhoA activity in an Epac1-independent manner. LPA stimulation (1 μm, 15 min) of serum-starved rat aortic SMCs significantly increased RhoA activity that was subsequently decreased to basal levels by co-treatment with isoproterenol (100 μm). A 50% knockdown of Epac1 significantly increased basal RhoA-GTP that did not change after LPA addition. However, knockdown of Epac1 did not prevent the isoproterenol-induced decrease in RhoA activity (*, p < 0.02; #, p < 0.04, n = 4). G, simultaneous inhibition of both Epac1 and PKA pathways significantly inhibits the isoproterenol-induced decrease of RhoA-GTP levels in rat aortic SMCs. Note that basal RhoA activity is significantly increased by treatment with both H89 and Epac1 shRNA (*, p < 0.0005, n = 4). Isoproterenol treatment = 15 min.
FIGURE 7.

007 activates Rap1 and decreases RhoA activity in SMCs. A, a Western blot shows 007-induced increased Rap1 and decreased RhoA activity for serum-starved rat aortic, human airway, and mouse fundus SMCs. B, LPA (1 μm) significantly increased RhoA activity in all three cell types (p < 0.01, p < 0.003, and p < 0.001, respectively) that were reduced by treatment with 007 (50 μm) (p < 0.007 n = 3, p < 0.04 n = 3, p < 0.01, respectively). A, C, and D, 007 significantly increased Rap1 activity in rat aortic (n = 3), human airway (n = 3), and fundus (n = 8) SMCs compared with control cells (p < 0.03 (*), p < 0.001(#), and p < 0.02 (+) respectively). B, statistical significance for each comparison designated by brackets was p < 0.05.
To further investigate a role of Epac1 pathway in down-regulation of RhoA signaling, we have silenced Epac1 in R518 cells using adeno-associated virus delivery of shRNA. Forty-eight hours after infecting cells with shEpac1 virus, Epac1 mRNA and protein significantly decreased by 69 ± 7% and 54 ± 4%, respectively, as compared with cells infected with non-targeting control shRNA virus (Fig. 8, C and D). We first examined whether this decrease is sufficient to attenuate Epac1-mediated Rap1 activation. Stimulation with isoproterenol (100 μm, 15 min) significantly increased Rap1-GTP levels in non-targeting control cells (p < 0.0005, n = 4), whereas in Epac1 knocked-down cells the Rap1-GTP levels did not change (Fig. 8E). Thus, the isoproterenol-induced increase in Rap1 activity is through activation of Epac1 (the difference in magnitude of changes in Rap1 activation between this experiment and Fig. 6C is a result of using a different lot of RalGDS beads). Next we examined the effect of Epac1 silencing on RhoA activation. Epac1 shRNA-treated cells showed a significantly increased basal level of RhoA-GTP (p < 0.02, n = 4) that did not increase further after LPA stimulation (10 μm, 15 min) (Fig. 8F). This suggests that Epac1 is active and suppresses RhoA activity in the basal state. Co-stimulation of cells with LPA and isoproterenol significantly prevented RhoA activation in cells treated with the non-targeting shRNA (p < 0.02, n = 4) (Fig. 8F). However, surprisingly, knockdown of Epac1 did not significantly reverse the isoproterenol-induced decrease in RhoA activity. These findings are consistent with either Epac or PKA being capable of mediating the isoproterenol-induced decrease in RhoA activity. Therefore, we next used H89 treatment of the SMCs, in which Epac1 was knocked down by 50%. We found under these conditions a highly significant partial reversal of the isoproterenol-induced decrease in RhoA activity was achieved (Fig. 8G). Altogether, based on these experiments, we propose that β-adrenergic stimulation of cAMP leads to signaling through both Epac1 and PKA to reduce RhoA activity and induce relaxation.
Further evidence that activation of Epac in SMCs reduces RhoA activity is shown in supplemental Fig. S3. We have previously shown that in SMCs, RhoA-dependent regulation of the actin cytoskeleton selectively regulates SMC differentiation marker gene expression by modulating SRF-dependent transcription (45). 007 (50 μm) significantly decreased the transcription of the SM-specific differentiation marker genes SM22, SM myosin heavy chain, and SM actin by 73, 49, and 35% respectively (p < 0.05 for expression of all three genes). All together, these data suggest that 007 activation of Epac in SM can lead to a significant decrease in RhoA activity.
DISCUSSION
Our findings demonstrate the presence of a β-adrenergic- and PGI2-activated cAMP-mediated activation of Epac. Cyclic AMP activation of Epac results in a PKA-independent, Rap1-dependent Ca2+-desensitization and relaxation of force in SM through down-regulation of RhoA activity. The significance of our findings lies in the importance of cyclic nucleotides as physiologic mediators of basal tone and relaxation of SM and in the importance of SM tone in hypertension, asthma, cerebral, and coronary vasospasm, and gut motility disorders.
Briefly, we find that the potent β-adrenergic vasodilators cicaprost and isoproterenol and the Epac-specific analog 007 increased Rap1 and decreased RhoA activities in SM tissues and cultured cells, resulting in Ca2+ desensitization. Evidence for Ca2+ desensitization is based on the findings that 007 and cicaprost 1) induced relaxation of agonist-induced force in intact tissue and, in the case of 007, in permeabilized vascular, gut, and airway SM, where [Ca2+]i is held constant, 2) reduced RhoA activity, and 3) decreased RLC20 phosphorylation and MLCP phosphorylation at the Rho kinase site Thr-853. That 007 signals through RhoA was also supported by our finding that transcription of SM α-actin and SM22, known to be regulated by RhoA, was decreased by activation of Epac.
Isoproterenol and 007 signal through Epac/Rap1, as silencing of Epac1 inhibited isoproterenol-induced activation of Rap1. Also, 007 inhibition of LPA-stimulated RhoA activity is abolished in Rap1B null cells. Overexpression of Epac1 or a dominant negative Epac1 mutant increased or decreased Rap1 activity, respectively. Furthermore, isoproterenol, forskolin, and IBMX significantly increased Rap1-GTP in rat aortic SM cells. Thus, 007 and isoproterenol signal through Rap1, and Rap1 is upstream of the fall in RhoA activity.
Here we show Epac and Rap1 proteins are expressed in a number of SM tissues and cells from various species. Epac expression in the vasculature and fundus was severalfold greater than in the ileum and bladder, perhaps contributing to their different functional behaviors. For example, vaso-intestinal peptide (VIP)-induced cAMP generation plays an important role in neuron-induced relaxation of fundus SM.
Our findings are consistent with 007 working predominantly through Epac/Rap1 rather than through PKA/PKG. Epac specificity arises from a conserved Glu found in PKA that is replaced by Gln in Epac1 and differences between the binding sites including roominess, hydrophobicity/polarity, and side chain flexibility. Although 007 has been shown to be highly selective for Epac (29, 30), there are currently no Epac inhibitors available. Therefore, it is presently difficult to accurately assess the relative contributions of PKA and Epac in response to activation of cAMP in various SMs. Our evidence that 007 does not activate PKA is based on our finding that the potent-specific PKA inhibitor peptide (PKI) inhibited the 6-Bnz-cAMP but not 007-induced relaxation of phenylephrine-induced Ca2+-sensitized force. In addition, phosphorylation at Ser-157, the PKA specific site of VASP, was increased by 6-Bnz cAMP and forskolin but not by 007 in permeabilized Ca2+-sensitized pulmonary artery and rat aortic SMCs. 007 does not increase cAMP through inhibition of phosphodiesterases or increase cGMP by inhibiting cGMP-phosphodiesterases and contributing to relaxation, as H89 did not alter VASP phosphorylation in 007-treated SM cells. Likewise the PKG inhibitor Rp-8-pCPT-cGMP did not inhibit 007-induced relaxation of Ca2+-sensitized force (supplemental Fig. S2). Our finding that the PKA inhibitor H89 does not inhibit 007-induced activation of Rap1 in rat aortic SMCs also supports the specificity of 007 for Epac.
In Rp-cAMPS, a sulfur replaces one of the free oxygens of the phosphate group that has been shown to be critical for the activation of both PKA and Epac (30), consistent with our finding that it inhibits both. Although there is agreement concerning Rp-cAMPS antagonistic effects on PKA activation, the effects on Epac are controversial, and this is discussed in the supplemental discussion). Recently, 8-pCPT-conjugated cAMP analogs have been reported to have thromboxane receptor antagonist activity (46). This could contribute to the 007-induced relaxation of U46619 force but should not contribute to relaxation of phenylephrine-, endothelin1-, carbachol-, or bradykinin-induced Ca2+-sensitized force nor to the inhibition of expression of SM actin, SM myosin heavy chain, and SM22, known to be regulated by RhoA activity (Figs. 1 and supplemental Fig. S3) (45). Furthermore, the study reporting the thromboxane receptor antagonist activity did not find an increase Rap1-GTP upon 007 treatment of platelets, unlike the increase measured in our rat aortic, human airway, and fundus SMCs.
An earlier study reported 007-induced relaxation of intact rat aorta precontracted with noradrenalin or phenylephrine but concluded that the effects could be mediated via endothelial cells or SMCs (47). Our data are the first to report that activation of Epac leads to relaxation of SM Ca2+-sensitized force through activation of Rap1 accompanied by a fall in RhoA activity and a decrease in both MYPT1 phosphorylation at Thr-853 and RLC20 phosphorylation.
Rap1 has been widely studied as a suppressor of oncogenic transformation, but more recently many more functions of Rap1 have become apparent (48–50). Interestingly, and in support of our findings, an increase in Rap1-GTP after cAMP activation leads to a decrease in RhoA-GTP in endothelial cells and in neurons (26, 27), suggesting that the Rap1-induced regulation of barrier function and neurite outgrowth is also because of down-regulation of RhoA. The mechanism leading to the Rap1-activated decrease in RhoA activity was not identified in the endothelial cell study (26), but in neurons a Rap1-activated RhoGAP, Ra-RhoGAP, was identified as a downstream target of Rap1 capable of inactivating RhoA and promoting neurite outgrowth (27). Therefore, we suggest that a Rap1-activated RhoGAP such as Ra-RhoGAP or ARAP3, shown to be directly stimulated by Rap1 (51, 52), may down-regulate RhoA activity in SM (Fig. 9). The identification of the relevant Rap1-activated RhoGAP(s) in SM is currently under investigation and is outside of the scope of the present study. It is also possible that Epac or Rap1-GTP could, through some other mechanism, lead to a decrease in RhoA activity (i.e. through an increase in RhoGDI activity or through an increase in degradation of RhoA). Increased degradation of RhoA is, however, unlikely to occur in the short 10–15-min time period of 007 or isoproterenol or cicaprost treatments used in our experiments. Furthermore, total RhoA in whole homogenates did not differ from control samples (Fig. 7).
FIGURE 9.

Scheme illustrating cAMP induced relaxation of RhoA-mediated Ca2+-sensitized force in smooth muscle via two signaling pathways, PKA and Epac. PKA induces SM relaxation through decreasing [Ca2+]i by inhibitory phosphorylation of RhoA at Ser-188 (12–16) by phosphorylation of telokin (18, 19), which activates MLCP activity or by binding to and activating MYPT1 by an unknown mechanism. cAMP activation of Epac leads to GTP exchange onto Rap1, which possibly by activation of a Rap1-activated RhoGAP results in a decrease in RhoA activity and relaxation through disinhibition of MLCP and a fall in RLC20 phosphorylation and Ca2+-sensitized force. GEF, guanine nucleotide exchange factor. AC, adenyl cyclase.
An intriguing recent study, based on use of a Rac1 inhibitor in intact tracheal SM proposes that 007 suppression of RhoA is due to 007 activation of Rac1, although the mechanism of activation is not defined (28, 53). They propose that 007 skews the balance of RhoA/Rac1 activation toward Rac1, a concept previously suggested to contribute to EC barrier regulation (53, 54). The role of Rac1 through activation of p21-activated kinases in SM is controversial. p21-activated kinases can phosphorylate and inhibit myosin light chain kinase (55), resulting in relaxation, and can also induce Ca2+-independent contraction through phosphorylating caldesmon and desmin and uncoupling force from RLC20 phosphorylation (56). Thus, in the intact tracheal preparations used in their study, signaling through p21-activated kinases or Epac activation of Ca2+-dependent ATP or K channels (57, 58) could also contribute to the 007-induced relaxation, as suggested by these authors. Their study did not investigate the role of Rap1, a focus of our study. Indeed, we found that 007-induced suppression of RhoA activity was abolished in Rap1B null cells, and Rap1 activation by isoproterenol was inhibited by shRNA down-regulation of Epac1 expression (Fig. 8A). Therefore, if 007 signals through Rac1, it must be downstream of Rap1B. Rap1 activity was also increased by forskolin, IBMX, and isoproterenol in aortic cells, and the PKA inhibitor, H89, did not inhibit 007-induced Rap1 activation, indicating that cAMP via a PKA-independent mechanism increases Rap1 activity. Furthermore, 007 increased Rap1 activity in aortic cells expressing WT Epac1 but not Epac1R279E (Fig. 6B), confirming that the 007 nucleotide activates Epac1 as expected.
Altogether our findings are consistent with isoproterenol and 007 working predominantly through Epac/Rap1 rather than through PKA or a nonspecific pathway. It is, however, important to note that both PKA and Epac signaling pathways respond to physiological low μm concentrations of cAMP. In support of isoproterenol-induced activation of both pathways, we found that whereas either H89 or silencing Epac1 alone did not inhibit the isoproterenol-induced decrease in RhoA activity (Fig. 8, B and F) treatment with both partially reversed the inhibition of RhoA activity. The lack of a complete reversal may reflect the incomplete silencing of Epac1 (Fig. 8, C and D) or incomplete inhibition of PKA by H89. Alternatively, other unknown isoproterenol-activated pathways may down-regulate RhoA activity. Interestingly, both Epac1 and PKA were found to be active in control SMCs, as RhoA activity increases ∼1.9-fold upon treatment with an shRNA targeting Epac1 expression or H89 alone or in combination (Fig. 8, B, F, and G). Therefore, Epac and PKA signaling may be mutually reinforcing to drive relaxation and maintain basal tone. Interestingly, an endogenous regulator of blood pressure, urocortin, mediated vasodilation via activation of cAMP but was reported to be only partially inhibited by PKA inhibitors, raising the possibility for an additional role of Epac in hypertension (20). In cardiac myocytes, the cAMP-PKA and cAMP-Epac response are both associated with muscle-specific protein kinase A-anchoring protein and cooperate to control cAMP levels (59). Both Epac and PKA also contribute to endothelial cell barrier function (26, 60–62). We propose that a similar cooperative cAMP activation of PKA and Epac contributes to the relaxation of SM (Fig. 9). Furthermore, based on findings that cAMP signaling is highly compartmentalized, resulting in different dynamics and responses to local cAMP and substrate concentrations (29, 63, 64), Epac1 may have additional specialized functions in SM. For example, Epac has been shown to accelerate SMC and fibroblast migration, whereas stimulation with a PKA-specific nucleotide inhibited migration (65, 66). Thus, Epac could have distinct roles in development or in angiogenesis and vasculogenesis. In addition, PKA and Epac may play distinct roles in neointima formation during ductus arteriosus closure (67) or PKB activation (68).
In summary, we demonstrate that β-adrenergic and PGI2 stimulation of the cAMP-activated GTP exchange factor for Rap1, Epac, provides a novel PKA-independent mechanism capable of mediating relaxation of RhoA-mediated Ca2+-sensitized force through disinhibition of MLCP activity in vascular, fundus, and airway smooth muscle.
Supplementary Material
Acknowledgments
We are most grateful to Dr. H. Kasahara, University of Florida for help in adenovirus generation. We thank Dr. J. L. Bos, University Medical Center, Utrecht for the RalGDS plasmid and Dr. X. Cheng, University of Texas, Galveston, TX for providing the Epac1 WT and DN constructs. We are grateful to Howard Phipps for help with preparation of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants R01GM086457-02 and 1R01DK088905-01 (to A. V. S.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Table 1 and Figs. 1–3.
- SM
- smooth muscle
- SMC
- SM cell
- GTPγS
- guanosine 5′-3-O-(thio)triphosphate
- MLCP
- myosin light chain phosphatase
- IBMX
- isobutylmethylxanthine
- LPA
- lysophosphatidic acid
- RLC20
- regulatory light chain
- VASP
- vasodilator-stimulated phosphoprotein
- Rap1-GTP
- Rp-cAMP, adenosine-3′,5′-cyclic monophosphorothioate, Rp isomer
- 6-Bnz-cAMP
- N6-benzoyladenosine-3′,5′-cyclic monophosphate
- PKA
- protein kinase A.
REFERENCES
- 1. Somlyo A. P., Somlyo A. V. (2003) Physiol. Rev. 83, 1325–1358 [DOI] [PubMed] [Google Scholar]
- 2. Somlyo A. P., Kitazawa T., Himpens B., Matthijs G., Horiuti K., Kobayashi S., Goldman Y. E., A. V. S. (1989) Adv. Prot. Phosphatases 5, 181–195 [Google Scholar]
- 3. Matsui T., Amano M., Yamamoto T., Chihara K., Nakafuku M., Ito M., Nakano T., Okawa K., Iwamatsu A., Kaibuchi K. (1996) EMBO J. 15, 2208–2216 [PMC free article] [PubMed] [Google Scholar]
- 4. Uehata M., Ishizaki T., Satoh H., Ono T., Kawahara T., Morishita T., Tamakawa H., Yamagami K., Inui J., Maekawa M., Narumiya S. (1997) Nature 389, 990–994 [DOI] [PubMed] [Google Scholar]
- 5. Kitazawa T., Masuo M., Somlyo A. P. (1991) Proc. Natl. Acad. Sci. U.S.A. 88, 9307–9310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Puetz S., Lubomirov L. T., Pfitzer G. (2009) Physiology 24, 342–356 [DOI] [PubMed] [Google Scholar]
- 7. Swärd K., Dreja K., Susnjar M., Hellstrand P., Hartshorne D. J., Walsh M. P. (2000) J. Physiol. 522, 33–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Carvajal J. A., Germain A. M., Huidobro-Toro J. P., Weiner C. P. (2000) J. Cell. Physiol. 184, 409–420 [DOI] [PubMed] [Google Scholar]
- 9. Himpens B., Matthijs G., Somlyo A. P. (1989) J. Physiol. 413, 489–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Walsh M. P., Kargacin G. J., Kendrick-Jones J., Lincoln T. M. (1995) Can. J. Physiol. Pharmacol. 73, 565–573 [DOI] [PubMed] [Google Scholar]
- 11. Weisbrod R. M., Griswold M. C., Yaghoubi M., Komalavilas P., Lincoln T. M., Cohen R. A. (1998) Br. J. Pharmacol. 125, 1695–1707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ellerbroek S. M., Wennerberg K., Burridge K. (2003) J. Biol. Chem. 278, 19023–19031 [DOI] [PubMed] [Google Scholar]
- 13. Gudi T., Chen J. C., Casteel D. E., Seasholtz T. M., Boss G. R., Pilz R. B. (2002) J. Biol. Chem. 277, 37382–37393 [DOI] [PubMed] [Google Scholar]
- 14. Lang P., Gesbert F., Delespine-Carmagnat M., Stancou R., Pouchelet M., Bertoglio J. (1996) EMBO J. 15, 510–519 [PMC free article] [PubMed] [Google Scholar]
- 15. Sauzeau V., Le Jeune H., Cario-Toumaniantz C., Smolenski A., Lohmann S. M., Bertoglio J., Chardin P., Pacaud P., Loirand G. (2000) J. Biol. Chem. 275, 21722–21729 [DOI] [PubMed] [Google Scholar]
- 16. Sawada N., Itoh H., Yamashita J., Doi K., Inoue M., Masatsugu K., Fukunaga Y., Sakaguchi S., Sone M., Yamahara K., Yurugi T., Nakao K. (2001) Biochem. Biophys. Res. Commun. 280, 798–805 [DOI] [PubMed] [Google Scholar]
- 17. Surks H. K., Mochizuki N., Kasai Y., Georgescu S. P., Tang K. M., Ito M., Lincoln T. M., Mendelsohn M. E. (1999) Science 286, 1583–1587 [DOI] [PubMed] [Google Scholar]
- 18. Wu X., Haystead T. A., Nakamoto R. K., Somlyo A. V., Somlyo A. P. (1998) J. Biol. Chem. 273, 11362–11369 [DOI] [PubMed] [Google Scholar]
- 19. Wu X., Somlyo A. V., Somlyo A. P. (1996) Biochem. Biophys. Res. Commun. 220, 658–663 [DOI] [PubMed] [Google Scholar]
- 20. Lubomirov L. T., Reimann K., Metzler D., Hasse V., Stehle R., Ito M., Hartshorne D. J., Gagov H., Pfitzer G., Schubert R. (2006) Circ. Res. 98, 1159–1167 [DOI] [PubMed] [Google Scholar]
- 21. Nakamura K., Koga Y., Sakai H., Homma K., Ikebe M. (2007) Circ. Res. 101, 712–722 [DOI] [PubMed] [Google Scholar]
- 22. Neppl R. L., Lubomirov L. T., Momotani K., Pfitzer G., Eto M., Somlyo A. V. (2009) J. Biol. Chem. 284, 6348–6360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wooldridge A. A., MacDonald J. A., Erdodi F., Ma C., Borman M. A., Hartshorne D. J., Haystead T. A. (2004) J. Biol. Chem. 279, 34496–34504 [DOI] [PubMed] [Google Scholar]
- 24. de Rooij J., Zwartkruis F. J., Verheijen M. H., Cool R. H., Nijman S. M., Wittinghofer A., Bos J. L. (1998) Nature 396, 474–477 [DOI] [PubMed] [Google Scholar]
- 25. Kawasaki H., Springett G. M., Mochizuki N., Toki S., Nakaya M., Matsuda M., Housman D. E., Graybiel A. M. (1998) Science 282, 2275–2279 [DOI] [PubMed] [Google Scholar]
- 26. Cullere X., Shaw S. K., Andersson L., Hirahashi J., Luscinskas F. W., Mayadas T. N. (2005) Blood 105, 1950–1955 [DOI] [PubMed] [Google Scholar]
- 27. Yamada T., Sakisaka T., Hisata S., Baba T., Takai Y. (2005) J. Biol. Chem. 280, 33026–33034 [DOI] [PubMed] [Google Scholar]
- 28. Roscioni S. S., Maarsingh H., Elzinga C. R., Schuur J., Menzen M., Halayko A. J., Meurs H., Schmidt M. (2010) J. Cell. Mol. Med., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Christensen A. E., Selheim F., de Rooij J., Dremier S., Schwede F., Dao K. K., Martinez A., Maenhaut C., Bos J. L., Genieser H. G., Døskeland S. O. (2003) J. Biol. Chem. 278, 35394–35402 [DOI] [PubMed] [Google Scholar]
- 30. Rehmann H., Schwede F., Døskeland S. O., Wittinghofer A., Bos J. L. (2003) J. Biol. Chem. 278, 38548–38556 [DOI] [PubMed] [Google Scholar]
- 31. Dao K. K., Teigen K., Kopperud R., Hodneland E., Schwede F., Christensen A. E., Martinez A., Døskeland S. O. (2006) J. Biol. Chem. 281, 21500–21511 [DOI] [PubMed] [Google Scholar]
- 32. Enserink J. M., Christensen A. E., de Rooij J., van Triest M., Schwede F., Genieser H. G., Døskeland S. O., Blank J. L., Bos J. L. (2002) Nat. Cell Biol. 4, 901–906 [DOI] [PubMed] [Google Scholar]
- 33. Gloerich M., Bos J. L. (2010) Annu. Rev. Pharmacol. Toxicol. 50, 355–375 [DOI] [PubMed] [Google Scholar]
- 34. Chrzanowska-Wodnicka M., Smyth S. S., Schoenwaelder S. M., Fischer T. H., White G. C., 2nd (2005) J. Clin. Invest. 115, 680–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Conner D. A. (2001) Curr. Protoc. Mol. Biol., Chapter 23, Unit 23.22 [Google Scholar]
- 36. Franke B., Akkerman J. W., Bos J. L. (1997) EMBO J. 16, 252–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ren X. D., Schwartz M. A. (2000) Methods Enzymol. 325, 264–272 [DOI] [PubMed] [Google Scholar]
- 38. Kasahara H., Aoki H. (2005) Methods Mol. Med. 112, 155–172 [DOI] [PubMed] [Google Scholar]
- 39. Shi G. X., Rehmann H., Andres D. A. (2006) Mol. Cell. Biol. 26, 9136–9147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Moncada S. (1983) Stroke 14, 157–168 [DOI] [PubMed] [Google Scholar]
- 41. Clapp L. H., Finney P., Turcato S., Tran S., Rubin L. J., Tinker A. (2002) Am. J. Respir. Cell Mol. Biol. 26, 194–201 [DOI] [PubMed] [Google Scholar]
- 42. Sobolewski A., Jourdan K. B., Upton P. D., Long L., Morrell N. W. (2004) Am. J. Physiol. Lung Cell Mol. Physiol. 287, L352–L359 [DOI] [PubMed] [Google Scholar]
- 43. Poppe H., Rybalkin S. D., Rehmann H., Hinds T. R., Tang X. B., Christensen A. E., Schwede F., Genieser H. G., Bos J. L., Doskeland S. O., Beavo J. A., Butt E. (2008) Nat. Methods 5, 277–278 [DOI] [PubMed] [Google Scholar]
- 44. Qiao J., Mei F. C., Popov V. L., Vergara L. A., Cheng X. (2002) J. Biol. Chem. 277, 26581–26586 [DOI] [PubMed] [Google Scholar]
- 45. Mack C. P., Somlyo A. V., Hautmann M., Somlyo A. P., Owens G. K. (2001) J. Biol. Chem. 276, 341–347 [DOI] [PubMed] [Google Scholar]
- 46. Sand C., Grandoch M., Börgermann C., Oude Weernink P. A., Mahlke Y., Schwindenhammer B., Weber A. A., Fischer J. W., Jakobs K. H., Schmidt M. (2010) Thromb. Haemost. 103, 662–678 [DOI] [PubMed] [Google Scholar]
- 47. Sukhanova I. F., Kozhevnikova L. M., Popov E. G., Podmareva O. N., Avdonin P. V. (2006) Dokl Biol. Sci. 411, 441–444 [DOI] [PubMed] [Google Scholar]
- 48. Bos J. L., de Rooij J., Reedquist K. A. (2001) Nat. Rev. Mol. Cell Biol. 2, 369–377 [DOI] [PubMed] [Google Scholar]
- 49. Roscioni S. S., Elzinga C. R., Schmidt M. (2008) Naunyn Schmiedebergs Arch. Pharmacol. 377, 345–357 [DOI] [PubMed] [Google Scholar]
- 50. Boettner B., Van Aelst L. (2009) Curr. Opin Cell Biol. 21, 684–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Krugmann S., Anderson K. E., Ridley S. H., Risso N., McGregor A., Coadwell J., Davidson K., Eguinoa A., Ellson C. D., Lipp P., Manifava M., Ktistakis N., Painter G., Thuring J. W., Cooper M. A., Lim Z. Y., Holmes A. B., Dove S. K., Michell R. H., Grewal A., Nazarian A., Erdjument-Bromage H., Tempst P., Stephens L. R., Hawkins P. T. (2002) Mol. Cell 9, 95–108 [DOI] [PubMed] [Google Scholar]
- 52. Krugmann S., Williams R., Stephens L., Hawkins P. T. (2004) Curr. Biol. 14, 1380–1384 [DOI] [PubMed] [Google Scholar]
- 53. Birukova A. A., Zagranichnaya T., Alekseeva E., Bokoch G. M., Birukov K. G. (2008) J. Cell. Physiol. 215, 715–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Birukova A. A., Zagranichnaya T., Fu P., Alekseeva E., Chen W., Jacobson J. R., Birukov K. G. (2007) Exp. Cell Res. 313, 2504–2520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sanders L. C., Matsumura F., Bokoch G. M., de Lanerolle P. (1999) Science 283, 2083–2085 [DOI] [PubMed] [Google Scholar]
- 56. Van Eyk J. E., Arrell D. K., Foster D. B., Strauss J. D., Heinonen T. Y., Furmaniak-Kazmierczak E., Côté G. P., Mak A. S. (1998) J. Biol. Chem. 273, 23433–23439 [DOI] [PubMed] [Google Scholar]
- 57. Purves G. I., Kamishima T., Davies L. M., Quayle J. M., Dart C. (2009) J. Physiol. 587, 3639–3650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ster J., De Bock F., Guérineau N. C., Janossy A., Barrère-Lemaire S., Bos J. L., Bockaert J., Fagni L. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 2519–2524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Dodge-Kafka K. L., Soughayer J., Pare G. C., Carlisle Michel J. J., Langeberg L. K., Kapiloff M. S., Scott J. D. (2005) Nature 437, 574–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Fukuhara S., Sakurai A., Sano H., Yamagishi A., Somekawa S., Takakura N., Saito Y., Kangawa K., Mochizuki N. (2005) Mol. Cell. Biol. 25, 136–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kooistra M. R., Corada M., Dejana E., Bos J. L. (2005) FEBS Lett. 579, 4966–4972 [DOI] [PubMed] [Google Scholar]
- 62. Wittchen E. S., Worthylake R. A., Kelly P., Casey P. J., Quilliam L. A., Burridge K. (2005) J. Biol. Chem. 280, 11675–11682 [DOI] [PubMed] [Google Scholar]
- 63. DiPilato L. M., Cheng X., Zhang J. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 16513–16518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Houslay M. D., Baillie G. S., Maurice D. H. (2007) Circ. Res. 100, 950–966 [DOI] [PubMed] [Google Scholar]
- 65. Yokoyama U., Minamisawa S., Quan H., Akaike T., Jin M., Otsu K., Ulucan C., Wang X., Baljinnyam E., Takaoka M., Sata M., Ishikawa Y. (2008) Am. J. Physiol. Heart Circ. Physiol. 295, H1547–H1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yokoyama U., Patel H. H., Lai N. C., Aroonsakool N., Roth D. M., Insel P. A. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 6386–6391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Yokoyama U., Minamisawa S., Quan H., Akaike T., Suzuki S., Jin M., Jiao Q., Watanabe M., Otsu K., Iwasaki S., Nishimaki S., Sato M., Ishikawa Y. (2008) J. Biol. Chem. 283, 28702–28709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Mei F. C., Qiao J., Tsygankova O. M., Meinkoth J. L., Quilliam L. A., Cheng X. (2002) J. Biol. Chem. 277, 11497–11504 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.