

Abstract
C4a-hydroperoxyflavin is found commonly in the reactions of flavin-dependent monooxygenases, in which it plays a key role as an intermediate that incorporates an oxygen atom into substrates. Only recently has evidence for its involvement in the reactions of flavoprotein oxidases been reported. Previous studies of pyranose 2-oxidase (P2O), an enzyme catalyzing the oxidation of pyranoses using oxygen as an electron acceptor to generate oxidized sugars and hydrogen peroxide (H2O2), have shown that C4a-hydroperoxyflavin forms in P2O reactions before it eliminates H2O2 as a product (Sucharitakul, J., Prongjit, M., Haltrich, D., and Chaiyen, P. (2008) Biochemistry 47, 8485–8490). In this report, the solvent kinetic isotope effects (SKIE) on the reaction of reduced P2O with oxygen were investigated using transient kinetics. Our results showed that D2O has a negligible effect on the formation of C4a-hydroperoxyflavin. The ensuing step of H2O2 elimination from C4a-hydroperoxyflavin was shown to be modulated by an SKIE of 2.8 ± 0.2, and a proton inventory analysis of this step indicates a linear plot. These data suggest that a single-proton transfer process causes SKIE at the H2O2 elimination step. Double and single mixing stopped-flow experiments performed in H2O buffer revealed that reduced flavin specifically labeled with deuterium at the flavin N5 position generated kinetic isotope effects similar to those found with experiments performed with the enzyme pre-equilibrated in D2O buffer. This suggests that the proton at the flavin N5 position is responsible for the SKIE and is the proton-in-flight that is transferred during the transition state. The mechanism of H2O2 elimination from C4a-hydroperoxyflavin is consistent with a single proton transfer from the flavin N5 to the peroxide leaving group, possibly via the formation of an intramolecular hydrogen bridge.
Keywords: Enzyme Catalysis, Enzyme Kinetics, Enzyme Mechanisms, FAD, Flavin, Flavoproteins, Isotope Effects, Kinetics, Oxidase
Introduction
Hydrogen peroxide (H2O2), a molecule known to induce cellular oxidative stress, can be generated from reactions of flavin-dependent oxidases or monooxygenases (1, 2). Flavoprotein oxidases catalyze two-electron transfer from organic substrates to oxygen, generating oxidized substrates and H2O2 as products (3). A flavin cofactor (flavin mononucleotide or FAD) plays a central role in this redox process by receiving the two electrons from organic substrates during the first (reductive) half-reaction and transferring them to oxygen during the second (oxidative) half-reaction. H2O2 can also be generated by flavin-dependent monooxygenases, which catalyze the monooxygenation of organic compounds using reactive C4a-hydroperoxyflavin intermediates as monooxygenating reagents (1, 2, 4, 5). In the absence of organic substrates, the C4a-hydroperoxyflavin intermediate eliminates H2O2 instead of performing monooxygenation (4–11). However, the mechanism by which H2O2 is eliminated from free or enzyme-bound C4a-hydroperoxyflavin remains unknown.
Although the formation of C4a-hydroperoxyflavin commonly occurs in flavin-dependent monooxygenase reactions, its existence in flavoprotein oxidases reactions under natural turnover conditions has only been observed with pyranose 2-oxidase from wood-rotting fungi Trametes multicolor (12–14). Based on investigations by Bruice and co-workers (15–17), the initial step in the reaction of reduced flavin and oxygen occurs through the formation of a radical pair of flavin semiquinone and superoxide (see Fig. 1) via a single electron transfer. For monooxygenases and pyranose oxidase, it is clear that this radical pair collapses to form C4a-hydroperoxyflavin prior to monooxygenation or H2O2 elimination (see path 1 in Fig. 1). The lack of a C4a-hydroperoxyflavin in other flavoprotein oxidase reactions could be explained by two possible reaction routes (18). The first route involves a direct second electron transfer from flavin semiquinone to superoxide to generate H2O2 as a product, bypassing the formation of C4a-hydroperoxyflavin (see path 2 in Fig. 1). The second route involves the formation of a C4a-hydroperoxyflavin intermediate, but the H2O2 elimination step occurs so rapidly that the kinetic detection of C4a-hydroperoxyflavin is prevented (1, 18). However, these two possibilities have not been distinguished because the mechanism of H2O2 elimination from C4a-hydroperoxyflavin in the free or an enzyme-bound form is not known. Previous studies of solvent kinetic isotope effects (SKIE) with flavoprotein oxidases (3, 19–26) have mostly focused on the mechanism of flavin reduction or oxygen activation, and the enzymes used in these studies do not stabilize C4a-hydroperoxyflavin as an intermediate. Therefore, the unique property of P2O2 in that the enzyme reacts with oxygen and stabilizes the C4a-hydroperoxyflavin intermediate makes the P2O system an ideal model for investigating SKIE on the H2O2 elimination step in a flavoprotein oxidase and for understanding the general mechanism underlying H2O2 elimination from enzyme-bound C4a-hydroperoxyflavins.
FIGURE 1.

The reaction of reduced flavin and oxygen. Path 1 is generally observed for flavin-dependent monooxygenases and P2O, whereas path 2 is observed for most flavoprotein oxidases.
P2O is a flavoprotein oxidase containing an FAD covalently linked to the N3 atom of His167. This enzyme catalyzes the oxidation of a wide variety of aldopyranoses at the sugar C2 position (27) and uses oxygen as an electron acceptor to generate H2O2. The x-ray structures of wild-type and mutant enzymes in free and sugar-bound forms have shown that the enzyme exists in open and closed conformations, which are relevant for sugar oxidation (reductive half-reaction) and the oxidation of reduced enzyme (oxidative half-reaction), respectively (28–32). The results from pre-steady state kinetics investigations using 2-d-d-glucose and other d-glucose isotopes as substrates have shown that a hydride equivalent is transferred regiospecifically from the d-glucose C2 position to the flavin N5 position during the reductive half-reaction (13, 14). In the ensuing oxidative half-reaction, the reduced enzyme reacts with oxygen to form a C4a-hydroperoxyflavin intermediate, which eliminates H2O2 (path 1 in Fig. 1) (12–14). The formation of C4a-hydroperoxy-FAD in P2O is known to require optimized interactions, especially around the FAD N5/C4a locus because mutations of Thr169 to Ser/Ala/Asn/Gly resulted in the abolishment of the intermediate formation (31).
In this report, we elucidated the mechanism underlying the elimination of H2O2 from C4a-hydroperoxy-FAD in P2O using the results of kinetic isotope effects and transient kinetics. The data clearly show that the transfer of the flavin N5 proton to the peroxide leaving group is the key step controlling the process of H2O2 elimination from C4a-hydroperoxyflavin.
EXPERIMENTAL PROCEDURES
Reagents
d-glucose (99.5% purity) and horseradish peroxidase were purchased from Sigma-Aldrich Chemie GMG. Deuterated glucose (2-d-d-glucose), sodium deuteroxide (99% purity), and deuterium oxide (99.9% purity) were purchased from the Cambridge Isotope Laboratory. ABTS was purchased from Sigma-Aldrich. Wild-type P2O was cloned and expressed without a His6 tag, to avoid the interfering properties of the tag, and prepared as described previously (12). Concentrations of the following compounds were determined using the known absorption coefficients at pH 7.0: ϵ403 = 1 × 105 m−1 cm−1 for peroxidase and ϵ458 = 1.13 × 104 m−1 cm−1 for the wild-type enzyme (12). The flavin absorption coefficient assumes one FAD bound per subunit.
Spectroscopic Studies
UV-visible absorbance spectra were recorded using a Hewlett-Packard diode array spectrophotometer (HP8453), a Shimadzu 2501PC spectrophotometer or a Cary 300Bio double-beam spectrophotometer. All spectrophotometers were equipped with temperature-controlled cell compartments. Enzyme activities were determined by continuous assays using coupled reactions of horseradish peroxidase and its substrate, ABTS, as described previously (33). Initial rates were calculated from the increase of absorbance at 420 nm resulting from the oxidation of ABTS by H2O2 using the molar absorption coefficient of 4.2 × 104 m−1cm−1 (per one molar of d-glucose consumed).
Rapid Reaction Experiments
Reactions were conducted in 100 mm sodium phosphate buffer (pH 7.0) at 4 °C, unless otherwise specified. Measurements were obtained using a TgK Scientific Model SF-61DX stopped-flow spectrophotometer in either single-mixing or double-mixing mode, as described previously (14). The optical path length of the observation cell was 1 cm. The stopped-flow apparatus was made anaerobic by flushing the flow system with an anaerobic buffer solution containing 10 mm sodium dithionite in 50 mm sodium phosphate buffer, pH 7.0. The buffer for the sodium dithionite solution was made anaerobic by equilibration with oxygen-free nitrogen (ultra high purity) that had been passed through an Oxyclear oxygen removal column (Labclear). The anaerobic buffer was allowed to stand in the flow system overnight. The flow unit was then rinsed with the anaerobic buffer prior to performing the experiments. Apparent rate constants (kobs) were calculated from the kinetic traces using exponential fits and the software packages Kinetic Studio (TgK Scientific, Salisbury, UK) and Program A.3
To study the reactions of the reduced enzyme with oxygen in H2O buffer, an anaerobic enzyme solution was equilibrated in an anaerobic glove box (Belle Technology, Weymouth, UK) to maintain the concentration of oxygen at levels below 3 ppm, and the enzyme was then reduced with a solution of 10 mm d-glucose in 100 mm sodium phosphate buffer, pH 7.0. While adding the solution of d-glucose, enzyme spectra were recorded using a spectrophotometer inside the glove box to ensure complete reduction. The reduced enzyme solution was placed in a glass tonometer and loaded onto the stopped-flow spectrophotometer.
To study solvent kinetic isotope effects on flavin oxidation, all solutions were exchanged with sodium phosphate buffer made with deuterium oxide as described previously (13). In brief, solid sodium phosphate powder of 1.38 g was dissolved in ∼30 ml of 99.9% purity deuterium oxide, and the resulting solution was equilibrated for 13–15 h (overnight) inside the anaerobic glove box. The equilibrated solution was then evaporated at 60° C for 3 h using a rotary evaporator to obtain H2O-free sodium phosphate powder. The resulting powder was added to deuterium oxide, and the same process was repeated to ensure that the buffer contained at least 99.9% deuterium oxide (v/v). The dried powder was then redissolved in ∼95 ml of 99.9% purity deuterium oxide. The buffer pD was adjusted by adding sodium deuteroxide solution into the solution while monitoring pH using a pH meter (pD = pH measured + 0.4) (34). The volume of the resulting buffer was adjusted to 100 ml with D2O. D-glucose (0.018 g) was dissolved in ∼30 ml of 99.9% purity deuterium oxide, and the solutions were dried twice as described above. To prepare a 10 mm D-glucose stock solution, the dried substrate powder was dissolved in 10 ml of 100 mm sodium phosphate buffer in deuterium oxide. The enzyme in D2O buffer was reduced by D-glucose, and the reduced enzyme solution was left overnight (∼18 h) prior to the stopped-flow experiment. This was to assure that the enzyme was well equilibrated in D2O, and all exchangeable sites of the reduced enzyme were incorporated with deuterium.
The oxidation of reduced P2O by molecular oxygen was monitored using the stopped-flow spectrophotometer by following the absorbance at 395 nm for detection of C4a-hydroperoxyflavin and 458 nm for oxidized flavin. Solutions with various concentrations of oxygen were made by bubbling certified oxygen/nitrogen gas mixtures (v/v) of 20%, 50%, 100%, and 100% on ice through syringes for 8 min. After mixing, this procedure resulted in oxygen concentrations of 0.13, 0.31, 0.61, and 0.96 mm, respectively. Equilibration of a buffer on ice with a 100% oxygen/nitrogen gas mixture enables the buffer solution to contain 1.92 mm oxygen before mixing.
P2O was kept at −80 °C in 100 mm MOPS buffer, pH 7.0, prior to its use in solvent kinetic isotope experiments. The concentration of the thawed enzyme solution was increased by reducing the solution volume to ∼200 μl using a 15-ml Amicon concentrator device with a membrane cut-off size of 10 kDa. The concentrated enzyme was loaded onto a PD-10 column (GE Healthcare) that had been equilibrated with either 100 mm sodium phosphate buffer in D2O, pD 7.0, or 100 mm sodium phosphate in H2O, pH 7.0. D2O or H2O buffer was added to elute the enzyme from the column. The resulting solution of P2O in D2O or H2O buffer (∼2.5 ml) was retrieved, and D2O or H2O buffer was added to obtain the desired volume and enzyme concentration (∼0.6 of absorbance or ∼53 μm).
Rate constants were obtained from plots of kobs versus the concentration of oxygen using Marquardt-Levenberg nonlinear fitting algorithms included in KaleidaGraph (version 4.0; Synergy Software). Simulations were performed by numerical methods using Runge-Kutta algorithms implemented in Berkeley Madonna 8.3 and a time step of 1 × 10−4 s. The model and methods used for simulations of the P2O oxidative half-reaction have been described previously (12–14).
Proton Inventory
Two hundred microliters of the solution of concentrated enzyme were loaded onto a PD-10 column equilibrated with either 100 mm sodium phosphate buffer in D2O, pD 7.0, or 100 mm sodium phosphate in H2O, pH 7.0. D2O, or H2O buffer was added to the column to elute the enzyme. H2O or D2O buffer was added to the equilibrated enzyme solution to obtain a final volume of 9 ml with absorbance at 458 nm ∼ 0.6. The enzyme solutions were equilibrated inside the anaerobic glove box for 30 min to remove oxygen. A solution of 10 mm d-glucose in 100 mm sodium phosphate buffer, pH 7.0, or 10 mm 2-d-d-glucose in 100 mm sodium phosphate buffer in D2O, pD 7.0, was added to the enzyme solution to generate the reduced enzyme in H2O or D2O buffer, as described in the previous section. Reduced enzyme solutions in various mixtures of H2O and D2O buffers were placed inside tonometers under anaerobic conditions. The atom fractions of D/H (n) were prepared and calculated based on the volume ratios of D2O and H2O in a total volume of 3 ml according to methods described previously: n = 0 (100% (v/v) H2O), n = 0.25 (2.25 ml of H2O + 0.75 ml of D2O), n = 0.50 (1.5 ml of H2O + 1.5 ml of D2O), n = 0.74 (0.75 ml of H2O + 2.25 ml of D2O), n = 1 (100% (v/v) D2O) (34).
All tonometers containing different mole fractions of deuterium were left overnight (∼18 h) at 4 °C to ensure complete equilibration between the enzyme and deuteroxide buffer. Buffers used in substrate syringes (total volume of 5 ml) were prepared with the same volume ratios of D2O and H2O as in the enzyme tonometers to ensure that the atom fractions of D/H were equivalent in both sides of the stopped-flow syringes. The resulting buffers were bubbled with certified oxygen/nitrogen gas mixtures to achieve the desired oxygen concentrations and left overnight (∼18 h) in tightly closed screw cap tubes inside an anaerobic glove box.
RESULTS
Reaction of Reduced P2O with Oxygen in D2O
The reactions of reduced P2O with oxygen in this report were conducted in 100 mm sodium phosphate, pH(D) 7.0, rather than 50 mm as in previous reports (12, 14) because increased ionic strength improves the solubility of reduced enzymes in both H2O and D2O buffers (13).
The oxidation of the reduced enzyme (26 μm) by 0.96 mm oxygen (the concentrations after mixing) in H2O (filled circle traces in Fig. 2A) and D2O (solid traces in Fig. 2A) were monitored at 395 and 458 nm. The kinetic traces in Fig. 2 show that the reactions were biphasic in both D2O and H2O buffers. The first phase (0.002–0.04 s) was characterized by an increase of absorbance at 395 nm due to the formation of a C4a-hydroperoxyflavin intermediate (Fig. 2A) (12). The second phase (0.04–0.86 s) consisted of an absorbance decrease at 395 nm and an absorbance increase at 458 nm because of the elimination of H2O2 to form the oxidized enzyme (Figs. 2A and 3). The observed rate constants (kobs1) for the first phase of the reactions in H2O buffer were dependent on the oxygen concentration and exhibit a slope of 5.6 ± 0.2 × 104 m−1 s−1 (k1app in Table 1) and an intercept of 23 ± 1 s−1 (× symbols in Fig. 4A), similar to the values reported previously (12). A plot of the observed rate constants of the second phase versus the oxygen concentrations shows the apparent rate constant of the H2O2 elimination from C4a-hydroperoxyflavin (k2app) of 24 ± 2 s−1 (× symbols in Fig. 4B, Table 1). The plots of the first and second observed phases in H2O buffer (Fig. 4, A and B) were analyzed according to Equations 1 and 2, respectively (12). The derivations of Equations 1 and 2 have been demonstrated in Ref. 12.
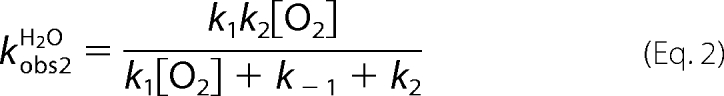 |
According to Equation 1, the intercept of the plot in Fig. 4A (× symbols) is equal to k−1 + k2. Therefore, kinetic simulations using a two-step consecutive model (Fig. 3), as described previously (12), were used to estimate the k−1 and k2 values and to confirm the validity of the kinetic constants calculated from the graphic method described. Simulations using the parameters listed in Table 1 yielded kinetic traces that agree well with the experimental data (Table 1, data not shown). The analysis based on the kinetic simulations indicates a bimolecular rate constant for the formation of C4a-hydroperoxyflavin (k1sim) of 6.5 ± 0.3 × 104 m−1s−1, a reverse rate constant (k−1sim) of 2.0 ± 0.1 s−1, and a rate constant for H2O2 elimination from the intermediate (k2sim) of 18 ± 1 s−1 in H2O (Table 1 and Fig. 3).
FIGURE 2.

The reactions of reduced P2O with oxygen in H2O and D2O buffers. A, filled circle traces are the kinetics monitored from mixing reduced P2O (26 μm) in H2O buffer (100 mm sodium phosphate, pH 7.0) with the same buffer containing 0.96 mm oxygen under a stopped-flow spectrophotometer at 4 °C. Solid line traces are the kinetics monitored from mixing reduced P2O (26 μm) in D2O buffer (100 mm sodium phosphate, pD 7.0), with the same D2O buffer under a similar condition as the reaction in H2O. All of the concentrations described are after mixing. The kinetics of the absorbance change at 395 nm shows two phases, with the absorbance increase resulting from the formation of C4a-hydroperoxyflavin and the absorbance decrease due to the H2O2 elimination from the intermediate; the kinetics of the flavin oxidation monitored at 458 nm coincided with the absorbance decrease of the intermediate at 395 nm. B, the solid lines show the reactions shown in A, but they were carried out in D2O buffer at oxygen concentrations of 0.13, 0.31, 0.61, and 0.96 mm (shown as the lower to upper traces). The dotted lines are kinetic simulations using the rate constants listed in Table 1 and the model in Fig. 3, as described in Ref. 12.
FIGURE 3.

The reaction mechanism of the reduced P2O with oxygen.
TABLE 1.
The effects of the solvent isotope on the kinetics of the P2O oxidative half-reaction
The reactions were performed in 100 mm sodium phosphate pH(D) 7.0, at 4 °C in a stopped-flow spectrophotometer.
| Parameters | H2O buffer | D2O buffer |
|---|---|---|
| k1app (m−1 s−1) | 5.6 ± 0.2 × 104 | 5.9 ± 0.3 × 104 |
| k1sim (m−1 s−1) | 6.5 ± 0.3 × 104 | 6.5 ± 0.3 × 104 |
| k−1sim (s−1) | 2 ± 0.1 | 2 ± 0.1 |
| k2app (s−1) | 24 ± 2 | 6.9 ± 0.5 |
| k2sim (s−1) | 18 ± 1 | 6.4 ± 0.3 |
FIGURE 4.

The kinetic analysis of the reactions of reduced P2O with oxygen in D2O and H2O buffers. The observed rate constants from the kinetic traces in Fig. 2 versus the oxygen concentrations (0.13, 0.31, 0.61, and 0.96 mm) are shown. A shows a plot of the observed rate constants for the first phase (kobs1) in H2O (× symbols) and in D2O (open circles). The kobs1H2O values from the low to high oxygen concentrations are 25.9 ± 0.9 s−1, 29.5 ± 0.3 s−1, 44.8 ± 0.1 s−1, and 62.2 ± 0.7 s−1, respectively, whereas the kobs1D2O values are 10.6 ± 0.04 s−1, 19.2 ± 0.24 s−1, 35.5 ± 0.16 s−1, and 63.4 ± 0.48 s−1, respectively. B shows plots of the observed rate constants of the second phase (kobs2) from the reaction in H2O (× symbols) and D2O (open circles). The kobs2H2O values according to the low to high oxygen concentrations are 5.99 ± 0.02 s−1, 12.0 ± 0.07 s−1, 15.9 ± 0.03 s−1, and 17.1 ± 0.09 s−1, respectively, whereas the kobs2D2O values are 4.81 ± 0.02 s−1, 6.43 ± 0.08 s−1, 6.63 ± 0.03 s−1, and 6.08 ± 0.05 s−1, respectively. A vertical line at each data point indicates the S.D. of the measurement.
For the reactions in D2O buffer, the enzyme oxidation observed using various oxygen concentrations (0.13, 0.31, 0.61, and 0.96 mm from the lower to upper traces in Fig. 2B) yielded kobs1 for the formation of C4a-hydroperoxyflavin as shown in Fig. 4A (open circles). A plot of the kobs1 versus the oxygen concentration shows a slope of 5.9 ± 0.3 × 104 m−1 s−1 and an intercept of ∼ 8.5 s−1 (open circles in Fig. 4A and Table 1). A plot of the observed rate constants of the second phase versus the oxygen concentration yielded an apparent rate constant for the H2O2 elimination from C4a-hydroperoxyflavin (k2app) of 6.9 ± 0.5 s−1.
Equations 1 and 2 and kinetic simulations using the model in Fig. 3, similar to those used for the analysis of the H2O reaction described above, were used for the analysis of the data of the D2O reaction. The analysis yielded a bimolecular rate constant (k1sim) for the formation of C4a-hydroperoxyflavin of 6.5 ± 0.3 × 104 m−1 s−1 and a reversible rate constant of 2 ± 0.1 s−1 (k−1sim), which are similar to the rate constants of the reaction in H2O buffer (Table 1). The rate constant for H2O2 elimination from C4a-hydroperoxyflavin (k2sim) in D2O was 6.4 ± 0.3 s−1 (Table 1), showing an SKIE of 2.8 (18 s−1 versus 6.4 s−1). Kinetic traces obtained from the simulations using these kinetic parameters agree well with the experimental data (dotted-line traces versus solid line traces, Fig. 2B).
The results above indicate that there is a negligible SKIE on k1 (formation of the C4a-hydroperoxyflavin intermediate) because D2Ok1sim = 1 (6.5 s−1/6.5 s−1) (Table 1), whereas D2O significantly diminishes the value of k2 (Fig. 4B and Table 1). We determined that the SKIE of D2Ok2sim was 2.8 (18 s−1/6.4 s−1) (Table 1). This SKIE value is not an artifact of the ∼0.49 unit increase of the pKa associated with a reaction when a pH-dependent reaction is performed in D2O (34) because the observed rate constants for H2O2 elimination from C4a-hydroperoxyflavin in the pH range of 6.5–7.5 are pH-independent and deviate within ±0.5 s−1 (data not shown).
Proton Inventory Analysis
Reactions of reduced P2O with oxygen at various atom fractions of D/H were carried out to analyze the number of proton sites involved in the elimination of H2O2 from the C4a-hydroperoxyflavin. The proton inventory was analyzed according to the Gross-Butler Equation (34, 35) (Equation 3),
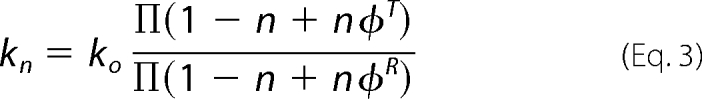 |
where n is the atom fraction of D2O in the solvent mixture, kn is the observed rate constant in a D2O/H2O mixture, k0 is the observed rate constant in H2O, and φT and φR are fractionation factors for proton sites at the transition state and the initial state, respectively.
Solutions of the reduced enzyme in buffers with various atom fractions of D/H were mixed with the same buffers containing 0.96 mm oxygen (the concentration after mixing). Kinetic traces monitored at 395 nm are shown in Fig. 5A, in which the traces from the lowest to the highest correspond to the n values of 0, 0.25, 0.50, 0.74, and 1.00, respectively. The reactions in the medium with n = 0 (100% v/v H2O, the bottom trace) and n = 1 (100% (v/v) D2O, the top trace), and in the mixtures of D2O/H2O mixtures, each showed biphasic kinetics; the first phase corresponded to the formation of the C4a-hydroperoxyflavin intermediate, and the second phase corresponded to the H2O2 elimination from the C4a-hydroperoxyflavin, which was similar to the results in Fig. 2 and Table 1. For example, the reaction with n = 0.74 (the second kinetic trace from the top, Fig. 5A) showed that the first phase (0.002–0.04 s) was the intermediate formation with an observed rate constant of 70 s−1, and the second phase (0.04–0.5 s) was the H2O2 elimination from C4a-hydroperoxyflavin with an observed rate constant of 8.2 s−1. A slow third phase (∼2.4 s−1), which accounted for ∼10% of the total absorbance change at 458 and 395 nm, was observed in the reaction with n = 0.25. This phase was a small decrease in the absorbance at 395 nm and a small increase in the absorbance at 458 nm. The origin of this slow phase is unknown, though it might be due to the slow equilibration of the newly generated oxidized enzyme, which resulted from the H2O2 elimination, with the outside solvent of a D2O/H2O mixture. Although this small additional kinetic phase may suggest some hidden complexity of the reaction in the D2O/H2O mixture, it did not affect the rate constants of the preceding two major phases used for the evaluation of SKIE and does not disagree with the kinetic model in Fig. 3.
FIGURE 5.

The proton inventory analysis for the observed constants of the H2O2 elimination from C4a-hydroperoxyflavin. Solutions of the reduced enzyme (26 μm) were mixed with buffer solutions containing 0.96 mm oxygen in a stopped-flow spectrophotometer at 4 °C. Both solutions of the reduced enzyme and the oxygenated buffers were prepared in various atom fractions of D/H, and the solutions with the same mole fractions were mixed under the stopped-flow experiments (see “Experimental Procedures”). A, the reactions were monitored at 395 nm for measuring the kinetics of the formation and H2O2 elimination of the C4a-hydroperoxyflavin intermediate at various atom fractions of D/H (n). Mixtures of 100 mm sodium phosphate, pH 7.0, in H2O and 100 mm sodium phosphate, pD 7.0, in D2O were employed to attain n = 0, 0.25, 0.50, 0.74, and 1.00, and their results are shown according to the lower to upper traces. All of the concentrations described are after mixing. B, a plot of the observed rate constants (kn) for the H2O2 elimination from C4a-hydroperoxyflavin is shown; the second phase with a decrease in absorbance at 395 nm in A versus the atom fractions of D/H (n) shows a linear relationship. The observed rate constants (kn) according to the low to high values of D/H (n) are 17.1 ± 0.09 s−1, 14.0 ± 0.17 s−1, 11.2 ± 0.23 s−1, 8.21 ± 0.04 s−1, and 6.12 ± 0.04 s−1, respectively. The data are consistent with a model involving one proton-in-flight during the transition state. A vertical line at each data point indicates the S.D. of the measurement.
The observed rate constants of the second phase (kobs2) were used for proton inventory analysis to identify the number of protons associated with the elimination of H2O2 from C4a-hydroperoxyflavin. Plotting the kobs2 versus the atom fraction of D (n) (Fig. 5B) reveals a linear relationship, suggesting that only a single proton site contributes to the SKIE on k2. Therefore, the plot was analyzed according to Equation 4, a simplified form of Equation 3 in which only one proton bridge is involved during the transition state.
The analysis yielded a fractionation factor φT of 0.34 ± 0.02 and an SKIE (D2Okobs2) of 2.9. Because the rate constant for the H2O2 elimination in H2O is dependent on the oxygen concentration (12), the proton inventory was also performed at a lower oxygen concentration (0.61 mm after mixing) to verify whether the same result is observed at lower oxygen concentrations. The results obtained using an oxygen concentration of 0.61 mm (φT = 0.32 ± 0.03 and SKIE (D2Okobs2) = 3.1) were quite similar to those observed using 0.96 mm oxygen. These results confirm that both the fractionation factor and SKIE are independent of the oxygen concentration and are derived from intrinsic properties associated with H2O2 elimination from C4a-hydroperoxyflavin. Although the plot in Fig. 5B is linear and fits with a one-proton transfer model, another independent piece of evidence was sought to confirm this conclusion. Therefore, experiments (presented in Figs. 6 and 7) were carried out to identify the nature and the identity of the proton involved with the H2O2 elimination process.
FIGURE 6.

The nature of a proton site causing the solvent kinetic isotope effect. The dotted-line trace was obtained from mixing reduced enzyme (22 μm), which was prepared in H2O buffer (100 mm sodium phosphate pH 7.0), with 0.96 mm oxygen in D2O buffer (100 mm sodium phosphate, pD 7.0). All of the concentrations described are after mixing. The solid line trace is from mixing reduced enzyme (22 μm), which was prepared in H2O buffer (100 mm sodium phosphate, pH 7.0), with 0.96 mm oxygen in H2O buffer (100 mm sodium phosphate, pH 7.0). The reaction was monitored as the absorbance change at 395 nm in a stopped-flow spectrophotometer at 4 °C. The results show that the kinetics of the H2O2 elimination (the second phase) of both traces are similar and only dependent on the solution in which P2O was prepared and independent of the buffer in the substrate syringe. For a reference, the kinetic trace of the reaction of the reduced enzyme, which was prepared in D2O buffer, mixed with oxygen (0.96 mm) in D2O buffer (data from Fig. 2) is shown as the open circle trace. The open circle trace was multiplied by a factor of 0.85 to bring the absorbance signal to a similar range as the other traces.
FIGURE 7.

The kinetic isotope effects of the reduced enzyme specifically labeled with deuterium at the flavin N5 position in H2O buffer. A, oxidation of the reduced enzyme labeled with N5-D was investigated using double-mixing stopped-flow spectrophotometry. During the first mixing, a solution of the oxidized enzyme (78 μm before mixing) was mixed with an equal volume of 100 μm 2-d-d-glucose (before mixing) in H2O buffer under anaerobic conditions. The mixture was continued for 80 s after the first mixing, before the reduced enzyme was mixed with 100 mm sodium phosphate buffer containing 0.96 mm oxygen (final concentration) (filled circle line). Only H2O buffers were employed in this experiment. The reaction was monitored at 395 nm to determine the rate constant for the H2O2 elimination from C4a-hydroperoxyflavin. The control experiment, where a solution of 100 μm d-glucose was used instead of 100 μm 2-d-d-glucose, was carried out in a similar fashion and is shown as a solid line trace. The results show that the kinetics of the trace with filled circles (N5-D reduced flavin) was similar to that of the reduced enzyme in D2O buffer reacting with 0.96 mm oxygen in D2O buffer (data in Fig. 2), whereas the kinetics of the double-mixing reaction employing d-glucose (solid line) was similar to the reaction carried out in H2O buffer (dotted line trace, data from Fig. 2). The dotted line trace was multiplied by a factor of 0.7 and offset by + 0.005 absorbance units to bring the absorbance signal to a similar range as the traces from the double-mixing experiment. B, oxidation of the reduced enzyme labeled with N5-D was carried out using a single-mixing stopped-flow spectrophotometer. Equal volumes of solutions of the oxidized enzyme 88 μm in H2O buffer (before mixing) in syringe A and 100 μm 2-d-d-glucose in H2O buffer (before mixing) in syringe B were manually pushed and mixed in syringe D under anaerobic conditions. The reaction in syringe D was allowed to proceed for 80–100 s to obtain the completely reduced P2O labeled with N5-D before the single-mixing stopped-flow experiment took place by mixing syringe D solution with an aerobic buffer in syringe C. The final reaction contained 22 μm of the reduced enzyme specifically labeled with N5-D and 0.96 mm oxygen in 100 mm sodium phosphate in H2O, pH 7.0 (open circle trace). The open circle trace is similar to the kinetic trace from the reduced enzyme in D2O buffer reacting with 0.96 mm oxygen in D2O buffer (Fig. 2). For a reference, the trace from Fig. 2B was multiplied by a factor of 0.66, offset by + 0.044 absorbance units and is shown as a dotted line. The solid line trace was obtained from the control reaction, in which a solution of 100 μm d-glucose was used instead of 100 μm 2-d-d-glucose, and the reaction was carried out in a similar fashion to that for the open circle trace. The results of the single-mixing stopped-flow in Fig. 7B are similar to the results of the double-mixing stopped-flow experiment in Fig. 7A. The data in this figure indicate that the solvent kinetic isotope effect on the H2O2 elimination from C4a-hydroperoxyflavin is mainly contributed by the N5-H bond breaking.
Nature of Proton Site Causing SKIE
The results from the previous section suggest that only one proton site is likely to be responsible for the SKIE on the H2O2 elimination step. The experiments in this section were carried out to determine whether the proton-in-flight can be rapidly exchanged with the outside solvent. A reduced enzyme solution (∼26 μm in H2O) was mixed with 100 mm sodium phosphate buffer (in either D2O or H2O) equilibrated with an oxygen concentration of 0.96 mm (after mixing). The reactions were monitored at 395 nm to detect the formation and decay of the intermediate. The dotted-line trace (Fig. 6) represents the reaction of reduced enzyme prepared in H2O buffer that was mixed with the oxygenated D2O buffer. This kinetic trace is similar to that of the control reaction, which was mixed with the oxygenated H2O buffer (solid trace in Fig. 6). For a reference, the trace of the reaction carried out in D2O (from Fig. 2B) is shown as the open circle trace (Fig. 6). These results indicate that the reaction kinetics are primarily dependent on the buffer in which the enzyme was prepared, not the buffer added during the stopped-flow mixing. These data also suggest that the exchange rate of the proton-in-flight takes longer than the period monitored in the stopped-flow experiment (∼1 s).
In another experiment, the exchange rate of the proton responsible for the SKIE on the H2O2 elimination step was investigated using double-mixing spectrophotometry. A solution of the reduced enzyme (78 μm before mixing) in 100 mm sodium phosphate in D2O, pD = 7.0, was mixed with the same buffer in H2O under anaerobic conditions during the first mixing step of the double-mixing stopped-flow experiments. The second mixing step added an aerobic buffer (100 mm sodium phosphate, pH 7.0 equilibrated with 0.96 mm O2) to the solution resulting from the first mixing step. The effects of various age times between the first and second mixings (0.01 s, 0.1, 1, 100, 200, and 300 s) were also examined. A longer age time should allow the exchange process to be more complete, that is, more of the deuterium in the reduced P2O should be replaced by protium. Reactions were monitored at 395 nm to measure the kinetics of C4a-hydroperoxyflavin formation and H2O2 elimination, and at 458 nm to detect flavin oxidation. The observed rate constants for the H2O2 elimination from C4a-hydroperoxyflavin at 0.96 mm O2 after mixing (∼6.1 s−1 in D2O versus ∼17 s−1 in H2O, Fig. 4) were used to indicate whether the proton-in-flight causing the SKIE was exchangeable within the age times examined. The results indicated that the rate constant for the H2O2 elimination did not exceed 6.1 s−1 (data not shown) for any time point examined. This result suggests that the t½ of the exchange rate was >300 s or the rate constant of the exchange was equivalent to or <2.3 × 10−3 s−1. These data also indicate that the exchange of the dissociable proton causing the SKIE on the H2O2 elimination step was negligible within a period of 300 s.
Origin of Proton-in-flight Causing SKIE on H2O2 Elimination from C4a-hydroperoxyflavin
To identify the location of the proton-in-flight that controls the process of H2O2 elimination from C4a-hydroperoxyflavin, a reduced enzyme in H2O buffer that was specifically labeled with deuterium at the flavin N5 was prepared using two stopped-flow mixing methods and tested to determine whether the labeled enzyme generated a similar KIE as the reaction in D2O (Fig. 2). The first method used double-mixing stopped-flow spectrophotometry. A solution of oxidized P2O in H2O buffer (78 μm before mixing) was mixed with an equal volume of a solution of 100 μm 2-d-d-glucose in H2O buffer (before mixing) under anaerobic conditions in the first mixing of the double-mixing stopped-flow experiment to produce a reduced enzyme specifically labeled with deuterium at the flavin N5 by a hydride transfer from 2-d-d-glucose (13, 14). This mixing process was continued for 80 s to allow for complete flavin reduction by 2-d-d-glucose prior to the second mixing with an aerobic buffer (a final oxygen concentration of 0.96 mm). This experiment allowed the N5-D-reduced flavin to react with oxygen. All buffers employed in this experiment were prepared in H2O (100 mm sodium phosphate buffer, pH 7.0). Therefore, under these conditions, any kinetic isotope effect detected should be due to the N5-D of the reduced P2O.
Kinetic traces detected at 395 nm showed that the kinetic isotope effect resulting from the N5-D of the reduced P2O (filled circle trace, Fig. 7A) was similar to the effect detected when the reaction was carried out in all D2O buffers at the same oxygen concentration (0.96 mm, Fig. 2A) because both traces yielded a similar kobs2 for the H2O2 elimination step. C4a-hydroperoxyflavin resulting from the reaction of reduced P2O labeled with N5-D in H2O buffer eliminated H2O2 at a rate constant of 7.6 s−1 (t½ ∼ 0.09 s) (filled circle trace in Fig. 7A), which is similar to the rate constant of 6.08 s−1 (t½ ∼ 0.11 s) observed for the reaction in D2O buffer (data in Figs. 2 and 4). A control experiment in which the oxidized enzyme was reduced by a solution of 100 μm d-glucose (before mixing) using the same double-mixing stopped-flow spectrophotometry setup resulted in an observed rate constant of 19 s−1 (solid line trace in Fig. 7A), which is similar to the rate constant observed using H2O buffer (17.1 ± 0.09 s−1, data from Fig. 4 were overlaid for a reference as the dotted line). The slight difference between the observed rate constant of 7.6 s−1 for the H2O2 elimination of N5-D C4a-hydroperoxyflavin (filled circle trace in Fig. 7A) versus the value of 6.08 ± 0.05 s−1 observed when the reaction was pre-equilibrated and carried out in all D2O buffers may be due to the small loss of N5-D caused by an exchange with the outside H2O buffer. The results in Fig. 7A clearly suggest that the bond breakage of N5-D during the transition state gives rise to the observed KIE of 2.5 (19 s−1/7.6 s−1) and that this step is the rate-limiting factor for the H2O2 elimination from C4a-hydroperoxyflavin.
In another experiment, the KIE resulting from the N5-D of the reduced P2O was measured using the stopped-flow single-mixing mode. We used the internal flow paths of the stopped-flow machine to generate the N5-D-labeled reduced P2O under anaerobic conditions. A solution of oxidized P2O (88 μm, before mixing) in H2O buffer was loaded into syringe A of the stopped-flow machine (TgK Scientific) and then was mixed with an equal volume of a solution of 100 μm 2-d-d-glucose in H2O buffer, which was loaded into syringe B. Both solutions were manually mixed under anaerobic conditions by pushing the solutions of syringes A and B into syringe D to produce the reduced P2O specifically labeled with deuterium at the flavin N5 (13, 14). This process was allowed to proceed for 80–100 s to achieve complete reduction before the solution in syringe D was mixed with aerobic buffer in syringe C using the single-mixing mode of the stopped-flow machine and monitored at 395 and 458 nm. After mixing and under the stopped-flow measurement, the reaction contained 22 μm of the reduced P2O labeled with N5-D and 0.96 mm O2 in 100 mm sodium phosphate buffer, pH 7.0. The kinetics of the reaction is shown as the open circle trace in Fig. 7B, which indicates a rate constant for H2O2 elimination from the intermediate of 7.2 s−1. The kinetic trace of the reduced P2O that was prepared by reduction with 2-d-d-glucose in H2O buffer (open circle trace) is similar to the reaction in D2O buffer (Fig. 2, overlaid dotted line trace in Fig. 7B). When the control reaction for which the same mixing setup was carried out as in the open circle trace in Fig. 7B, but with d-glucose as a reductant, the observed rate constant of the H2O2 elimination was 17.1 s−1 (solid line in Fig. 7B). The results in Fig. 7B indicate that the reduced P2O labeled with N5-D shows a KIE of 2.4 (17.1 s−1/7.2 s−1) for the H2O2 elimination step, which is similar to the observed KIE of 2.5 from the result in Fig. 7A. The two different mixing setup experiments confirm that the reaction of reduced P2O specifically labeled at N5-D with oxygen in H2O buffer gives rise to an observed KIE that is similar to the observed SKIE of the reduced P2O pre-equilibrated in D2O buffer reacting with 0.96 mm oxygen (SKIE of 2.8 from 17.1 s−1/6.08 s−1 (data from Fig. 4)). These results suggest that the observed SKIE is mainly a result of the flavin N5-H(D) breakage and that this step mainly controls the H2O2 elimination from C4a-hydroperoxyflavin.
DISCUSSION
This work has provided the first evidence of SKIE on H2O2 elimination from C4a-hydroperoxyflavin. The results indicated that for P2O from T. multicolor, an SKIE (D2Ok2) of 2.8 ± 0.2 was found for the H2O2 elimination step and that the N5 proton of reduced flavin is the proton-in-flight that causes this SKIE. Another group has examined SKIE using bacterial luciferase reactions but did not detect SKIE for the formation and decay of C4a-hydroperoxyflavin mononucleotide (36).
Our results (Figs. 2 and 4 and Table 1) show that D2O had a negligible effect on the bimolecular rate constant for C4a-hydroperoxyflavin formation (k1 in Fig. 3) and the intermediate decay (k−1). The currently accepted mechanism of the reaction between reduced flavin and oxygen (Fig. 1) predicts that the first step of the reaction involves a one-electron transfer to form a radical pair of flavin semiquinone and superoxide radical, which rapidly collapses to form C4a-hydroperoxyflavin (1, 2, 15–18). Although a net transfer of one proton is required for the formation of C4a-hydroperoxyflavin (path 1 in Fig. 1), our results do not identify any solvent kinetic isotope effect on this step. These data indicate that the protonation process to form C4a-hydroperoxyflavin is rapid and is not the rate-limiting step in the formation of the intermediate. We propose that the residue His548, which is located near the flavin ring, may donate a proton for this rapid protonation (29, 30). Because this residue is proposed to be a catalytic base that deprotonates a proton from d-glucose C2-OH during the reductive half-reaction (13), it may be possible for the resulting protonated His548 to donate a proton for the C4a-hydroperoxyflavin formation during the oxidative half-reaction.
The proton inventory analysis revealed that the plot of the rate constants for H2O2 elimination from C4a-hydroperoxyflavin at various D2O fractions (kn) versus n is linear (Fig. 5B), suggesting that a one-proton transfer process is a major factor controlling the rate of this step (35). This result suggests that only one proton-in-flight was involved during the transition state. The proton inventory analysis also yielded an SKIE of = 2.9 ± 0.2 and a fractionation factor for the transition state (φT) of 0.34 ± 0.02 (Fig. 5B), implying that deuterium binds to the exchangeable site less tightly than it binds average bulk water molecules (34). A value of φT in the range of 0.3–0.6 is typically found with proton transfers among oxygen, nitrogen, or sulfur atoms (34, 35, 37, 38). Our results in Fig. 7 have confirmed that a single proton bridge contributes to the SKIE.
The results in Figs. 5–7 clearly show that the proton bridge responsible for the SKIE in the H2O2 elimination step is the flavin N5 proton and that the environment surrounding this site is rather enclosed. The first mixing in the stopped-flow experiments (Fig. 7) specifically labeled the flavin N5 position with deuterium by transferring a hydride equivalent from 2-d-d-glucose. Although these experiments were performed in an H2O medium, the kinetics of the reaction were similar to those observed in a D2O medium, in which the enzyme was pre-equilibrated ∼18 h in the D2O buffer (Fig. 2). The observed KIE of the H2O2 elimination step (at 0.96 mm O2) for the N5-D-labeled flavin performed in the H2O buffer was 2.5 (19 s−1/7.6 s−1) from the experiment shown in Fig. 7A that used a double-mixing stopped-flow set up to generate the N5-D-labeled flavin, and 2.4 (17.1 s−1/7.2 s−1) from the experiment that manually mixed the oxidized enzyme with 2-d-glucose and used the single-mixing mode of the stopped-flow machine to follow the reaction (Fig. 7B); these results were similar to an observed SKIE of 2.8 (17.1 s−1/6.08 s−1) (data in Fig. 4) for the reaction performed in D2O at 0.96 mm O2. Therefore, the bond breaking of the flavin N5-H is the key step controlling the overall process of H2O2 elimination from C4a-hydroperoxyflavin. The exchange rate between deuterium and protium at the flavin N5 site was shown to be quite slow by the fact that the deuterium of the N5-D-labeled flavin remained intact in H2O medium before the second stopped-flow mixing (∼80–100 s, Fig. 7). Based on the stopped-flow double-mixing experiment with different buffers, the exchange rate constant at the flavin N5 position was estimated to be <2.3 × 10−3 s−1 (data not shown). Previous work has shown that the exchange rate constant of the N5 proton of free reduced flavin mononucleotide at pH ∼ 7 is ∼242 s−1, which is equivalent to a t1/2 of ∼ 0.0029 s at 25 °C (39). These results indicate that the active site environment surrounding the N5 proton in P2O must be enclosed to impede the exchange rate with the outside solvent. During the oxidative half-reaction of P2O, the substrate loop is thought to be in a closed conformation, which is thought to increase the hydrophobicity of the active site environment (12, 29, 30, 32).
Based on the current data, the reaction mechanism underlying the H2O2 elimination from C4a-hydroperoxyflavin is proposed to involve an intramolecular H-bridge that facilitates the H2O2 elimination (as shown in Fig. 8). The data reported here clearly show that the N5 proton is responsible for SKIE, and it is the proton-in-flight during the transition state. The mechanism of H2O2 elimination from C4a-hydroperoxyflavin is likely to involve a single proton bridge transfer from the flavin N5 position to a peroxide leaving group (Fig. 8). Although an x-ray structure of the C4a-hydroperoxyflavin transient intermediate of P2O is not available, a potential configuration at the C4a position of the intermediate can be postulated based on the structure of the C4a-flavin oxygen adduct of choline oxidase, a flavoprotein oxidase in the same superfamily (glucose-methanol-choline oxidoreductases) as P2O (40). According to the choline oxidase adduct structure, the C4a carbon of the flavin adduct assumes an sp3 configuration, similar to that which would be expected if the flavin adduct were a free compound (40). Based on this information, a three-dimensional representation of C4a-hydroperoxyflavin was generated using ChemDraw three-dimensional Pro to approximate the distance between the flavin N5 proton and the proximal oxygen atom of the intermediate. Because the configuration of the proximal oxygen is fixed in the structure of C4a-hydroperoxyflavin, measurements based on this theoretical structure estimate that the distance between the flavin N5 proton and the proximal oxygen is ∼2.2 Å (data not shown). This distance is too far to allow the flavin N5 proton and proximal oxygen to engage in direct H-bonding interactions to facilitate a one-step proton transfer. Therefore, we propose that the flavin N5-H may initially form an H-bond with the distal oxygen (Fig. 8). When the reaction proceeds, the bond between the flavin C4a and the proximal oxygen becomes more extended because the flavin ring becomes more planar when the flavin N5 and C4a assume their new hybridization and the iminium double bond starts to form. This should decrease the distance between the flavin N5-H and the proximal oxygen and permit the H-bond interactions (Fig. 8) that facilitate the protonation of the proximal oxygen to generate a stable H2O2 leaving group (Fig. 8).
FIGURE 8.

The proposed reaction mechanism for the H2O2 elimination from C4a-hydroperoxyflavin. Dotted lines represent hydrogen bonds that may form between the flavin N5 proton and the distal and proximal oxygen during the transition state.
The intramolecular H-bridge facilitating the H2O2 elimination mechanism proposed in Fig. 8 is also supported by our previous results showing that mutations of Thr169 (a residue with its side chain [Oγ] close enough to interact with the flavin N5) to Ser, Ala, or Gly, abolish the formation of the C4a-hydroperoxyflavin intermediate (Fig. 9A) (31). For the wild-type enzyme, the H-bond interaction between the flavin N5 and the Oγ of the Thr169 side chain may divert the interaction of the intramolecular H-bridge that facilitates H2O2 elimination (Fig. 9B). In Thr169 mutants, the optimum H-bonding interaction between the Oγ of Thr169 and the flavin N5 is removed, resulting in an environment with lower dielectric constants, which promotes the intra-molecular H-bridge formation and thus facilitates H2O2 elimination. Therefore, the abolishment of C4a-hydroperoxyflavin that is observed in Thr169 mutants may be due to an increased decay of the intermediate (31), that is, an increased rate constant of H2O2 elimination. The model in Fig. 8 is also supported by the recent investigation of a flavin-containing monooxygenase showing that an N78S mutant, in which the site near the C4a/N5 locus is widened and in which the H-bonding interaction between the flavin N5 and NADP+ is altered, voids its ability to form a C4a-hydroperoxyflavin intermediate (41).
FIGURE 9.

The P2O active site structure. A, the active site of P2O in the closed conformation indicates that the Oγ of Thr169 can make an H-bond interaction with the flavin N5 (29, 30). B, this H-bonding interaction in the wild-type enzyme may divert the intramolecular H-bridge proton transfer, which assists in the H2O2 elimination from C4a-hydroperoxyflavin. Therefore, C4a-hydroperoxyflavin is observed in the wild-type enzyme but not in the Thr169 variants (31).
The intramolecular H-bridge proton transfer proposed in Fig. 8 would be supported if the pKa values of the flavin N5-H and the peroxide leaving group were in the same range. The pKa of H2O2 is known to be ∼11.8 (42), whereas a hydroperoxide of a free flavin derivative is ∼9.2 (16). When bound to the enzyme, the pKa of flavin-C4a-hydroperoxide may be significantly varied. Indeed, it has been reported to be 8.4 for the reaction of cyclohexanone monooxygenase (43) or >10.0 for the oxygenase component of p-hydroxyphenylacetate 3-hydroxylase (44). The pKa of the N5-H of C4a-hydroperoxyflavin has not been measured experimentally but has been estimated to be <17 (45). According to NMR studies, the pKa of the N5-H in free reduced flavin mononucleotide is high (≥20) (42) because it is the second deprotonation of the flavin ring. Merenyi et al. (45) argued that the pKa of the flavin N5-H in the presence of a C4a-hydroperoxy substituent would likely be decreased to below ∼17 because the flavin ring is neutral. In addition, the change of nitrogen hybridization from the amine (pKa < 17) in the C4a-hydroperoxyflavin (a reactant) to the imine with sp2 hybridization in the oxidized flavin (a product) should help lower the pKa of the flavin N5-H to significantly <17. The pKa of C4a = N5-H in the oxidized flavin was estimated to be about −8.3 (16). Recently, we have shown that the pKa associated with the H2O2 elimination from C4a-hydroperoxyflavin in the reaction of the oxygenase component of p-hydroxyphenylacetate hydroxylase is >9.4 (44). Taken together, our data suggest that the pKa values of the flavin N5-H and the C4a-flavin hydroperoxide may not differ significantly and may permit proton transfer via an intra-molecular H-bridge as proposed in Fig. 8.
In conclusion, our results clearly show for the first time that the H2O2 elimination from C4a-hydroperoxyflavin in a flavoprotein oxidase reaction is controlled by a proton transfer from the flavin N5 to the peroxide leaving group. Our data indicate that the mechanism might involve the formation of an intramolecular H-bridge that facilitates the H2O2 elimination process. The findings in this report also provide a framework to explain the mechanism that might be involved in the elimination of H2O2 from the C4a-hydroperoxyflavin in other flavin-dependent oxidases and monooxygenases.
Acknowledgments
We are grateful to Bruce A. Palfey (University of Michigan, Ann Arbor) and Richard L. Schowen (University of Kansas) for useful discussions about the SKIE results. We thank Tienthong Thongpanchang (Mahidol University) for discussion regarding the mechanism of H2O2 elimination and Kittisak Thotsaporn (Chulalongkorn University) for the graphical illustration in Fig. 9. We thank Thikumporn Luanloet for providing us with the enzyme used in the experiment depicted in Fig. 7B and Ted King (TgK Scientific) for help with the experiment shown in Fig. 7B. We thank Barrie Entsch (University of New England, New South Wales, Australia) for critical reading of the manuscript. We thank Methinee Prongjit for sharing unpublished data.
This work was supported by The Thailand Research Fund Grants BRG5480001 (to P. C.) and MRG5380240 (to J. S.) and grants from the Faculty of Science, Mahidol University (to P. C.), from the National Research University Project of Commission on Higher Education and the Ratchadaphiseksomphot Endowment Fund (HR1166I), Chulalongkorn University (to J. S.), and from National Science and Technology Development Agency (to T. W.). This work was presented at the Gordon Research Conference 2010 (July 19–23) at Waterville Valley Resort, NH, Enzymes, Co-enzymes & Metabolic Pathways.
Program A was written at the University of Michigan by Rong Chang, Jung-yen Chiu, Joel Dinverno, and David Penfield Ballou.
- P2O
- pyranose 2-oxidase
- FMN
- flavin mononucleotide
- KIE
- kinetic isotope effect
- SKIE
- solvent kinetic isotope effect(s)
- ABTS
- 2,2′-azino-bis(3)-ethybenzenethiazoline-6-sulfonic acid diammonium salt
- D/H
- D2O/H2O
- N5-D
- the deuterium incorporated flavin N5
- N5-H
- the protium incorporated flavin N5.
REFERENCES
- 1. Massey V. (1994) J. Biol. Chem. 269, 22459–22462 [PubMed] [Google Scholar]
- 2. Palfey B. A., Massey V. (1998) Comprehensive Biological Catalysis: Flavin-dependent Enzymes, pp. 83–153, Academic Press, CA [Google Scholar]
- 3. Fitzpatrick P. F. (2001) Acc. Chem. Res. 34, 299–307 [DOI] [PubMed] [Google Scholar]
- 4. Ballou D. P., Entsch B., Cole L. J. (2005) Biochem. Biophys. Res. Commun. 338, 590–598 [DOI] [PubMed] [Google Scholar]
- 5. van Berkel W. J., Kamerbeek N. M., Fraaije M. W. (2006) J. Biotechnol. 124, 670–689 [DOI] [PubMed] [Google Scholar]
- 6. Sucharitakul J., Chaiyen P., Entsch B., Ballou D. P. (2006) J. Biol. Chem. 281, 17044–17053 [DOI] [PubMed] [Google Scholar]
- 7. Torres Pazmiño D. E., Winkler M., Glieder A., Fraaije M. W. (2010) J. Biotechnol. 146, 9–24 [DOI] [PubMed] [Google Scholar]
- 8. Palfey B. A., Ballou. D. P., Massey V. (1995) Active Oxygen in Biochemistry: Oxygen Activation by Flavins and Pterins, pp. 37–83, Chapman & Hall, Glasgow, Scotland, UK [Google Scholar]
- 9. Palfey B. A., McDonald C. A. (2010) Arch. Biochem. Biophys. 493, 26–36 [DOI] [PubMed] [Google Scholar]
- 10. Chaiyen P. (2010) Arch. Biochem. Biophys. 493, 62–70 [DOI] [PubMed] [Google Scholar]
- 11. Entsch B., Cole L. J., Ballou D. P. (2005) Arch. Biochem. Biophys. 433, 297–311 [DOI] [PubMed] [Google Scholar]
- 12. Sucharitakul J., Prongjit M., Haltrich D., Chaiyen P. (2008) Biochemistry 47, 8485–8490 [DOI] [PubMed] [Google Scholar]
- 13. Sucharitakul J., Wongnate T., Chaiyen P. (2010) Biochemistry 49, 3753–3765 [DOI] [PubMed] [Google Scholar]
- 14. Prongjit M., Sucharitakul J., Wongnate T., Haltrich D., Chaiyen P. (2009) Biochemistry 48, 4170–4180 [DOI] [PubMed] [Google Scholar]
- 15. Bruice T. C. (1984) Isr. J. Chem. 24, 54–61 [Google Scholar]
- 16. Eberlein G., Bruice T. C. (1983) J. Am. Chem. Soc. 105, 6685–6697 [Google Scholar]
- 17. Eberlein G., Bruice T. C. (1982) J. Am. Chem. Soc. 104, 1449–1452 [Google Scholar]
- 18. Mattevi A. (2006) Trends Biochem. Sci. 31, 276–283 [DOI] [PubMed] [Google Scholar]
- 19. Sobrado P., Fitzpatrick P. F. (2003) Biochemistry 42, 15208–15214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pompon D., Iwatsubo M., Lederer F. (1980) Eur. J. Biochem. 104, 479–488 [DOI] [PubMed] [Google Scholar]
- 21. Gadda G. (2008) Biochemistry 47, 13745–13753 [DOI] [PubMed] [Google Scholar]
- 22. Rungsrisuriyachai K., Gadda G. (2010) Biochemistry 49, 2483–2490 [DOI] [PubMed] [Google Scholar]
- 23. Su Q., Klinman J. P. (1999) Biochemistry 38, 8572–8581 [DOI] [PubMed] [Google Scholar]
- 24. Klinman J. P. (2007) Acc. Chem. Res. 40, 325–333 [DOI] [PubMed] [Google Scholar]
- 25. Henderson Pozzi M., Fitzpatrick P. F. (2010) Arch. Biochem. Biophys. 498, 83–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pennati A., Gadda G. (2011) Biochemistry 50, 1–3 [DOI] [PubMed] [Google Scholar]
- 27. Leitner C., Haltrich D., Nidetzky B., Prillinger H., Kulbe K. D. (1998) Appl. Biochem. Biotechnol. 70–72, 237–248 [DOI] [PubMed] [Google Scholar]
- 28. Tan T. C., Pitsawong W., Wongnate T., Spadiut O., Haltrich D., Chaiyen P., Divne C. (2010) J. Mol. Biol. 402, 578–594 [DOI] [PubMed] [Google Scholar]
- 29. Hallberg B. M., Leitner C., Haltrich D., Divne C. (2004) J. Mol. Biol. 341, 781–796 [DOI] [PubMed] [Google Scholar]
- 30. Kujawa M., Ebner H., Leitner C., Hallberg B. M., Prongjit M., Sucharitakul J., Ludwig R., Rudsander U., Peterbauer C., Chaiyen P., Haltrich D., Divne C. (2006) J. Biol. Chem. 281, 35104–35115 [DOI] [PubMed] [Google Scholar]
- 31. Pitsawong W., Sucharitakul J., Prongjit M., Tan T. C., Spadiut O., Haltrich D., Divne C., Chaiyen P. (2010) J. Biol. Chem. 285, 9697–9705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Spadiut O., Tan T. C., Pisanelli I., Haltrich D., Divne C. (2010) FEBS J. 277, 2892–2909 [DOI] [PubMed] [Google Scholar]
- 33. Danneel H. J., Rössner E., Zeeck A., Giffhorn F. (1993) Eur. J. Biochem. 214, 795–802 [DOI] [PubMed] [Google Scholar]
- 34. Schowen K. B., Schowen R. L. (1982) Methods Enzymol. 87, 551–606 [PubMed] [Google Scholar]
- 35. Venkatasubban K. S., Schowen R. L. (1984) CRC Crit. Rev. Biochem. 17, 1–44 [DOI] [PubMed] [Google Scholar]
- 36. Francisco W. A., Abu-Soud H. M., DelMonte A. J., Singleton D. A., Baldwin T. O., Raushel F. M. (1998) Biochemistry 37, 2596–2606 [DOI] [PubMed] [Google Scholar]
- 37. Cook P. F. (1991) Enzyme Mechanism from Isotope Effects: Theoretical Basis and Mechanistic Utility of Solvent Isotope Effect, pp. 74–123, CRC Press, Boca Raton, FL [Google Scholar]
- 38. Schowen K. B., Limbach H. H., Denisov G. S., Schowen R. L. (2000) Biochim. Biophys. Acta 1458, 43–62 [DOI] [PubMed] [Google Scholar]
- 39. Macheroux P., Ghisla S., Sanner C., Rüterjans H., Müller F. (2005) BMC Biochem. 6, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Orville A. M., Lountos G. T., Finnegan S., Gadda G., Prabhakar R. (2009) Biochemistry 48, 720–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Orru R., Pazmiño D. E., Fraaije M. W., Mattevi A. (2010) J. Biol. Chem. 285, 35021–35028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Evans M. G., Uri N. (1949) Trans. Faraday Soc. 45, 224–230 [Google Scholar]
- 43. Sheng D., Ballou D. P., Massey V. (2001) Biochemistry 40, 11156–11167 [DOI] [PubMed] [Google Scholar]
- 44. Ruangchan N., Tongsook C., Sucharitakul J., Chaiyen P. (2011) J. Biol. Chem. 286, 223–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Merenyi G., Lind J. (1991) J. Am. Chem. Soc. 113, 3146–3153 [Google Scholar]