

Abstract
Ligand-induced disorder-to-order transition plays a key role in the biological functions of many proteins that contain intrinsically disordered regions. This trait is exhibited by so-called RTX (repeat-in-toxin) motifs found in many virulence factors secreted by numerous Gram-negative pathogenic bacteria: RTX proteins are natively disordered in the absence of calcium but fold upon calcium binding. The adenylate cyclase toxin (CyaA) produced by Bordetella pertussis, the causative agent of whooping cough, contains ∼40 RTX motifs organized in five successive blocks separated by non-RTX flanking regions. This RTX domain mediates toxin binding to its eukaryotic cell receptor. We previously showed that the last block of the RTX domain, block V, which is critical for CyaA toxicity, exhibits the hallmarks of intrinsically disordered proteins in the absence of calcium. Moreover, the C-terminal flanking region of CyaA block V is required for its calcium-induced folding. Here, we describe a comprehensive analysis of the hydrodynamic and electrophoretic properties of several block V RTX polypeptides that differ in the presence and/or length of the flanking regions. Our results indicate that the length of the C-terminal flanking region not only controls the calcium-induced folding but also the calcium-induced multimerization of the RTX polypeptides. Moreover, we showed that calcium binding is accompanied by a strong reduction of the net charge of the RTX polypeptides. These data indicate that the disorder-to-order transition in RTX proteins is controlled by a calcium-induced change of the polypeptide charges and stabilized by multimerization.
Keywords: Adenylate Cyclase, Bacterial Toxins, Biophysics, Calcium-binding Proteins, Spectroscopy, Repeat in Toxin (RTX), Calcium-induced Protein Folding, Intrinsically Disordered Proteins, Multimerization, Protein Charge
Introduction
The past decade has seen a fundamental reappraisal of the protein structure-to-function paradigm because it became evident that a significant fraction of polypeptide sequences code for proteins, commonly designated as natively or intrinsically disordered proteins, that are lacking ordered structures under physiological conditions (1–8). Numerous intrinsically disordered proteins are nevertheless able to acquire ordered conformations upon binding to specific ligands, and in most cases, these disorder-to-order transitions are linked to key functional properties of these proteins. Elucidation of the biochemical factors and physicochemical parameters that control the transition between these different states of the polypeptides is therefore essential to understand the biological functions of intrinsically disordered proteins.
We have recently begun to explore the biophysical mechanisms at work in this process in a newly described class of intrinsically disordered protein that contains so-called RTX repeated motifs and in which the disorder-to-order transition is triggered by the binding of calcium ions (9–11). These motifs are found in many virulence factors secreted by numerous Gram-negative pathogenic bacteria (12, 13). The original name (repeat in toxin) was first coined to group four virulence factors secreted by Escherichia coli, Bordetella pertussis, Pasteurella hemolytica, and Actinobacillus sp., that contain a series of tandemly repeated RTX sequences at their C-terminal end. Since then, many other RTX proteins have been identified in more than 250 bacterial strains (14), and they cover a vast array of biological activities, many of them involved in host-pathogen interactions (13, 14). In most cases, the RTX sequences are located at the C-terminal end of the polypeptides, appended to functional domains endowed with specific biochemical activities such as pore-forming cytolytic activity, proteases, lipases, etc. (13, 14). Despite this functional diversity, most known RTX proteins are thought to be secreted by a dedicated type I secretion system and to require calcium to exert their biological activities.
The prototype sequence of the RTX motif is GGXG(N/D)DX(U)X, where X represents any amino acid, and (U) represents any large hydrophobic residue such as Ile, Leu, Val, Phe, and Tyr (13). The number of RTX motifs in RTX proteins may vary from 5 to more than 50. Structural data available on some RTX-containing proteins showed that these sequences fold in the presence of calcium into an idiosyncratic parallel β-helix structure, where the first six residues, GGXGXD, of each RTX motif form a calcium-binding turn and the last three residues XUX make a short β-strand (15, 16). The assembly of consecutive turns and β-strands builds up a right-handed parallel β-helix that is stabilized by calcium binding. In contrast, in the absence of calcium, these RTX sequences appeared to be largely disordered as revealed in particular by our biophysical studies of the adenylate cyclase (CyaA) toxin from B. pertussis, the causative agent of the whooping cough (17, 18). CyaA contains ∼40 RTX motifs grouped in five consecutive blocks (named blocks I–V) separated by non-RTX-flanking regions. This RTX-containing domain (RD)4 is located at the C-terminal moiety of CyaA (residues 1006–1706) and is implicated in the calcium-dependent binding of the toxin to a specific receptor, the CD11b/CD18 integrin, at the surface of the host target cells. We indeed showed that RD is intrinsically disordered in the absence of calcium but acquires a stable β-rich fold upon calcium binding (9).
It has been proposed that the calcium-induced disorder-to-order transition in RTX motifs may be essential for the secretion of the corresponding polypeptides by their dedicated type I secretion system. The disordered conformations of RTX motifs in the low calcium environment of the bacterial cytosol may facilitate the protein secretion through the type I secretion system machinery, whereas in the external medium, calcium binding could trigger the folding of the proteins into their active conformations for host-pathogen interactions (9, 19–21).
To gain further insights into the mechanisms of calcium-induced folding of the CyaA RTX domain, we further characterized a region of RD that encompasses the last block (block V) of RTX motifs and that was found to be essential for the cytotoxic activity of CyaA (22–24). We demonstrated that the RTX motifs per se were not able to fold in the presence of calcium and that the polypeptide segments at the C termini of the tandemly repeated RTX motifs were essential for the calcium-induced folding (10), yet the molecular basis allowing this C-terminal flanking region to induce the folding of the RTX motifs remained elusive.
Here, we have investigated the electrokinetic and hydrodynamic properties of several RTX polypeptides derived from block V of RD by a combination of complementary biophysical approaches. Our results indicate that the C terminus-flanking region is not only involved in the calcium-induced folding but can also modulate the multimerization of the RTX polypeptides.
Moreover, we showed that calcium binding induced a strong reduction of the mean net charges of the RTX polypeptides. This indicated that the disorder-to-order transition of RTX proteins could be controlled by calcium-induced changes in protein valence.
EXPERIMENTAL PROCEDURES
Reagents
The experiments were done at 25 °C in 20 mm Hepes, 20 mm NaCl, pH 7.4 (Buffer A). All reagents were of the highest purity grade.
Polypeptides Production and Purification
RTX polypeptide production and purification have been described elsewhere (10). Protein preparation is further described in the supplemental materials.
Spectroscopic Methods
Quasi-elastic light scattering (QELS) experiments (11) and electrophoretic mobility measurements were done using a NanoZS instrument (Malvern). Fluorescence measurements were performed with a FP-6200 spectrofluorimeter (Jasco), and CD spectra were recorded on an Aviv model 215 circular dichroism spectropolarimeter, as described previously (10). The estimation of protein shape and hydration using size exclusion chromatography (SEC) connected on-line to a Tetra detector array (TDA) (Viscotek Ltd., a Malvern Company) in conjunction with analytical ultracentrifugation (Beckman XL-I; Beckman Coulter) is described elsewhere (9, 10, 25, 26). A further description of CD, anisotropy, QELS, and electrophoretic mobility experiments is provided in the supplemental materials.
RESULTS
The polypeptides studied in this work have been described previously (10) and are schematized in Fig. 1. They all encompass the core of the last block (block V) of RTX motifs (residues 1530–1630, noted R) from CyaA but differ in the presence of either the N-terminal (residues 1487–1529, noted N) or the C-terminal (C) flanking region, the latter being further subdivided into short CS (residues 1631–1652) and long CL (residues 1631–1680) regions. These polypeptides were produced and purified as described previously (see “Experimental Procedures” and Ref. 10 for details).
FIGURE 1.

Scheme of the six block V polypeptides used in this study. Several regions can be distinguished along the primary sequence of CyaA block V (residues 1487–1680). From left to right on the scheme are: the N-terminal flanking region (N) for residues 1487–1529 (white); the RTX motif region (R; each of the nine RTX motifs is represented by a black box) for residues 1530–1630; the proximal C-terminal flanking region for residues 1631–1652 (the short C region, CS, gray); and the long C-terminal flanking region (CL, hatched) for residues 1631–1680.
Calcium-induced Changes of RTX Protein Hydrodynamic Radii
We first measured the hydrodynamic radii (RH) of the six RTX polypeptides by QELS. In the apo-state (i.e. in the absence of calcium), all RTX polypeptides showed hydrodynamic radii higher than expected for folded proteins of similar molecular masses (compare anhydrous to hydrodynamic radii in Table 1), in agreement with our previous results, which revealed that all RTX polypeptides are intrinsically disordered in the apo-state (10). We previously showed, and confirmed here, that the polypeptides harboring the C-terminal flanking region CL or CS, i.e. NRCL, RCL, NRCS, and RCS, underwent a calcium-induced folding, whereas the NR and R polypeptides, lacking a C-terminal extension, did not (Ref. 10 and supplemental Fig. S1). Accordingly, the addition of calcium did not change the hydrodynamic radii of NR and R (Table 1), whereas it triggered a large decrease of the RH of NRCL and RCL, indicative of a strong compaction of their hydrodynamic volumes upon acquisition of secondary and tertiary structures.
TABLE 1.
Hydrodynamic radii of RTX polypeptides measured by quasi-elastic light scattering
The experimental conditions were as follows: temperature, 25 °C; apo-states, buffer A; in the presence of calcium, buffer A, 2 mm CaCl2 for holo-NRCL, holo-RCL, and holo-RD; buffer A, 5 mm CaCl2 for NRCS, RCS, NR, and R. NA, not applicable (because of the heterogeneity of the samples). The S.D. of the frictional ratio is within 10% of the value.
| NRCL | RCL | NRCS | RCS | NR | R | RD | |
|---|---|---|---|---|---|---|---|
| RH (nm) apo in the absence of calcium | 3.4 ± 0.1 | 3.2 ± 0.1 | 3.4 ± 0.1 | 2.9 ± 0.1 | 3.0 | 2.7 ± 0.1 | 7.2 ± 0.3 |
| RH (nm) holo in the presence of calcium | 2.5 ± 0.1 | 2.2 ± 0.2 | NA | 3.1 ± 0.1 | 3.0 | 2.7 ± 0.1 | 3.1 ± 0.1 |
| R0 anhydrous (nm) Apo | 1.81 | 1.68 | 1.72 | 1.58 | 1.64 | 1.47 | 2.83 |
| R0 anhydrous (nm) Holo | 1.81 | 1.68 | 1.72 | 1.58 | 1.64 | 1.47 | 2.75 |
| f/f0 Apo | 1.88 | 1.93 | 1.97 | 1.84 | 1.82 | 1.84 | 2.54 |
| f/f0 Holo | 1.38 | 1.32 | NA | 1.97 | 1.82 | 1.84 | 1.13 |
Strikingly, the RH of holo-RCS was not significantly different from that of the apo-form, although the CD spectra clearly indicated that RCS acquired secondary and tertiary structures in the presence of calcium (Table 1 and supplemental Fig. S1). Unfortunately, we could not measure the RH of holo-NRCS as a significant fraction of the protein aggregated in these conditions, precluding an accurate analysis of the QELS data. In contrast, the QELS data of apo- and holo-RCS were robust and good enough to accurately determine their translational diffusion coefficients that were also found (see below) to be in agreement with AUC data. It was therefore intriguing that the intrinsically disordered apo-RCS state could fold in the presence of calcium (supplemental Fig. S1) without a concomitant decrease of its hydrodynamic radius (Table 1). To better describe the calcium-induced folding reaction of RCS, we further investigated its hydrodynamic properties by SEC-TDA, by analytical ultracentrifugation (AUC) in velocity mode, by fluorescence anisotropy, and by electrophoretic mobility.
The SEC-TDA experiments were performed on a Superdex 200 column at 25 °C. The chromatographs of RCS in the apo-state and in the holo-state (in the presence of 5 mm calcium) are shown in Fig. 2A. The retention volume of RCS was similar in the absence and in the presence of calcium (Fig. 2A and supplemental Table S1), suggesting that both states exhibited similar hydrodynamic volumes, in agreement with the QELS data. The right angle light scattering signals (RALS, green profiles), combined with the protein concentration determined by the deflection refractometer (black profiles), provided on-line measurements of the molecular mass of each eluting species. As shown in Fig. 2B, the molecular mass determined for the apo-state of RCS was 15.1 kDa, as expected for a monomeric species (Fig. 2B, green dashed trace), whereas the molecular mass of the holo-state (Fig. 2B, green solid trace) was 42 kDa. Hence, these data indicated that RCS formed a trimer upon calcium binding (supplemental Table S1). To further estimate the equilibrium between the monomer and the trimer, holo-RCS was loaded on the SEC-TDA at three different concentrations (70, 35, and 7 μm). Irrespective of the protein concentration, holo-RCS eluted as a single peak exhibiting a molecular mass of 42 ± 2 kDa (data not shown). No other oligomeric states (i.e. monomer, dimer, tetramer, etc.) could be detected, suggesting that the trimeric state was the most stable form of holo-RCS, if not the only possible one. To rule out any possible artifact, we analyzed in parallel the RCL protein by SEC-TDA, and as shown in Fig. 2 (C and D), we found that, in agreement with our previous results (10), RCL is monomeric in both states, and its RH decreased dramatically upon calcium binding (Table 1). Finally, the SEC-TDA experiments also provided the intrinsic viscosity (measured by on-line differential pressure transducers) of each eluting species. As shown in Fig. 2B, the intrinsic viscosity of RCS strongly decreased from 14.7 to 6.4 ml·g−1 upon calcium binding (Fig. 2B, red traces, and supplemental Table S1). This change of intrinsic viscosity indicates that RCS underwent a change of hydration and/or shape upon calcium binding (see below).
FIGURE 2.

Hydrodynamic properties of RCS and RCL polypeptides analyzed by SEC-TDA. RCS (A) and RCL (C) polypeptides, in the absence (dashed line) or in the presence (solid line) of 5 mm CaCl2, were analyzed by size exclusion chromatography on a Superdex 200 connected to a Tetra detector array as described under “Experimental Procedures.” The traces correspond to the right angle light scattering (green lines), differential refractometer (black lines), and differential viscometer (red lines). The experimental conditions were: buffer A at 25 °C. The molecular mass (green lines) and intrinsic viscosity (red lines) are given for RCS (B) and RCL (D) in the absence (dashed lines) or in the presence of calcium (solid lines).
Hydrodynamic Parameters of RTX Proteins
We then measured the sedimentation coefficients of the apo- and holo-states of RCS by AUC (i) to independently confirm the results obtained by QELS and SEC-TDA and (ii) to further determine the respective contribution of the viscosity increment and hydration parameters in the intrinsic viscosity values of the different species. Velocity experiments provided sedimentation coefficients of 1.46 and 3.65 S for apo-RCS and holo-RCS, respectively (Fig. 3 and supplemental Table S1). These data, together with those acquired by QELS and SEC-TDA, provided independent measurements of S, RH, and molecular mass (supplemental Table S1) that are related to each other according to the Svedberg equation S = M(1 − ρv̄)/(6πηNARH). All of the three experimental parameters, S, RH, and M, were found to be remarkably consistent for both the apo- and the holo-states of RCS. Indeed, when two of these experimentally determined parameters, e.g. RH and M, were used to calculate (using the Svedberg equation) the third one (S), the theoretical value obtained was very close to the one experimentally measured. This further confirmed the robustness and accuracy of the measured hydrodynamic parameters. Altogether, the hydrodynamic analysis indicated that apo-RCS was monomeric with a RH of 2.9 nm, whereas holo-RCS folded into a stable and compact trimeric conformation exhibiting a RH of 3.1 nm.
FIGURE 3.
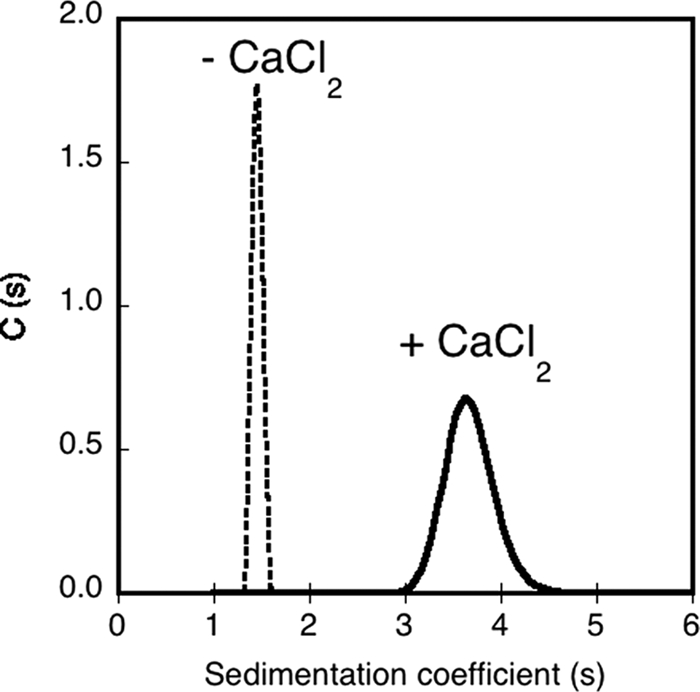
Analytical ultracentrifugation analysis of RCS. Sedimentation coefficient distribution, deduced from the fitted curves, of RCS in the absence (dashed line) or in the presence of 5 mm CaCl2 (solid line). The experimental conditions were: 20 μm RCS and buffer A at 25 °C.
From the intrinsic viscosity of both apo- and holo-RCS, we then determined the respective contribution of the shape factor and the time-averaged apparent hydration. The method to estimate these parameters has been described elsewhere (9, 10, 25, 26). Briefly, the molecular mass, the intrinsic viscosity, and the RH (or alternatively the sedimentation coefficient; see “Experimental Procedures”) are required to calculate the shape factor and the protein hydration using the Einstein viscosity relation. The viscosity increment, v, is a hydrodynamic function (i.e. a shape factor) from which the values of the semi-axes a/b of an ellipsoid can be inferred (27). The time-averaged apparent hydration, δ, defined as the mass of solvent bound to and dragged by the protein along its Brownian motion, is related to the exclusion volume of the protein, i.e. the volume defined by the shearing plane between the protein and the bulk solvent.
As shown in supplemental Table S1, the viscosity increment values, and hence the shape of apo-RCS and holo-RCS, were similar. Therefore, the difference between the intrinsic viscosity of apo-RCS (14.7 ml/g) and holo-RCS (6.4 ml/g) was mainly due to a difference of hydration (see “Experimental Procedures”). Indeed, apo-RCS was characterized by a high time-averaged apparent hydration, close to 3 g/g (supplemental Table S1), a value much higher than the theoretical one (0.43 g/g) computed from the protein sequence using the Sednterp software. RCS underwent a strong dehydration upon calcium binding, although its hydration value (1 g/g) still remained higher than the computed one (supplemental Table S1). The calcium-induced dehydration of RCS was also evidenced by the changes of the frictional ratio (supplemental Table S1). The frictional ratio f/f0 (defined in Table 1 as the ratio of radii, f/f0=RH/R0) is related, through the Perrin equation (see “Experimental Procedures”), to the hydrodynamic shape function (the Perrin factor) and the time-averaged apparent hydration, δ. Interestingly, the decrease of the frictional ratio from 1.8 for apo-RCS to 1.35 for holo-RCS (supplemental Table S1) was essentially assigned to a change of hydration, whereas the shape function was not affected. Similar observations have been reported for the other RTX proteins (Table 1 and Ref. 10), indicating that the strong calcium-dependent dehydration may be a general feature of these polypeptides.
RTX Protein Tryptophan Fluorescence Anisotropy Studies
We further analyzed the properties of RCS and RCL by steady-state tryptophan fluorescence anisotropy, a sensitive tool to explore the flexibility of a fluorescent probe. As shown in Fig. 4A, the anisotropy values of apo-RCL and holo-RCL were similar and quite low yet higher than that of l-tryptophan used as standard. The low anisotropy value of apo-RCL was not unexpected because the polypeptide adopted intrinsically disordered conformations in which the tryptophans may be freely moving. However, the fact that the anisotropy of RCL did not increase upon calcium binding was more surprising, because the tryptophan probes experienced sterical constraints from the surrounding amino-acids in the folded holo-RCL state. We hypothesized that the effects of local constraints on the tryptophan anisotropy may be compensated by the lower hydrodynamic radius, allowing a faster motion (related to the rotational diffusion coefficient of the protein) of holo-RCL as compared with apo-RCL. In other words, the effects of the local constraints on RCL anisotropy (expected increase) may be counterbalanced by the faster rotation of the protein (decreasing anisotropy), thus explaining the absence of a global anisotropy change between the apo- and holo-states. Alternatively, this effect may be due to a nonuniform folding of holo-RCL: a tryptophan-containing region may remain locally partially unfolded even in the presence of calcium. This scenario is possible because one of the RCL tryptophans is located in the C-terminal region.
FIGURE 4.

Tryptophan fluorescence anisotropy of RCL and RCS. The fluorescence anisotropy was recorded at an emission wavelength of 350 nm for RCL (A) and RCS (B) polypeptides in the absence (○) or in the presence (●) of calcium (2 mm for RCL and 5 mm for RCS). In both graphs, l-Trp is represented (×). The experimental conditions were: 2 μm buffer A at 25 °C.
In contrast, the anisotropy of RCS was modulated by calcium binding as shown in Fig. 4B. In the apo-state, the anisotropy of RCS was low likely because of the absence of constraints on the tryptophan residues in this unfolded state, as noted above for apo-RCL. At variance with RCL, calcium binding triggered a significant increase of the anisotropy of RCS. This increase of anisotropy of holo-RCS may reflect the restriction of the tryptophan side chain flexibility as a result of structural constraints within the folded holo-RCS. However, because the hydrodynamic volumes of the trimeric holo-RCS (RH, 3.1 nm) and of the monomeric apo-RCS (RH, 2.9 nm) were similar, the macromolecular dimension of RCS did not change and therefore should not contribute to any variation of anisotropy. Hence, only the tryptophan side chain flexibility (related to the local tertiary contacts) ultimately contributed to the anisotropy changes of RCS. Altogether, these data suggest that the anisotropy of RCL was not modified upon calcium binding due to the compensating effects of local and global changes, whereas that of RCS increased upon calcium binding due to a unique effect of the folding-induced constraints on the tryptophans.
Calcium-induced Changes of the Electrophoretic Mobilities of RTX Proteins
We recently proposed that electrostatic interactions could play a critical role in the folding and stability of RTX proteins (11). To provide a direct experimental evidence of a charge effect on the unfolded-to-folded transition of the RTX proteins, we characterized the electrophoretic mobility (μe), i.e. the velocities in a given electric field, of RCL and RCS in both the apo- and holo-states. Combined with the above-determined hydrodynamic properties, these data allowed us to estimate the mean net charge (i.e. the protein valence) of the polypeptides in the apo- and holo-states. The number of calcium ions bound per protein was then inferred, assuming that the difference in charges between the apo- and the holo-state should correspond to the number of charges brought by the calcium ions bound to the protein in the holo-state.
The approach used to convert the electrophoretic mobility into a number of charges is based on the study of Basak and Ladisch (28) (see “Experimental Procedures”). These authors empirically determined, for a large set of proteins, a relationship between the electrophoretic mobility of a protein (μe), the number of charges (Nc), its molecular mass (M), and its frictional ratio (f/f0) (see the equation under “supplemental materials”). Briefly, this relation indicates that the electrophoretic mobility is proportional to the number of charges and inversely correlated to both the frictional ratio and the molecular mass of the protein. Because we already determined the molecular masses and the frictional ratios of RCL and RCS in both of their apo- and holo-states, the measurement of the electrophoretic mobility values should allow us to estimate the number of charges of each species.
The electrophoretic mobility values measured in fast field reversal mode for RCL and RCS are shown in Fig. 5. These electrophoretic mobility values, together with the molecular masses and frictional ratios determined previously, are also reported in Table 2. As expected for proteins binding several calcium ions (two charges per cation), large variations of the electrophoretic mobility were evidenced between the apo- and the holo-state of each protein. Upon calcium binding, the electrophoretic mobility of RCL changed from −1.75 to −1.06 μm·cm·V−1·s−1 (Fig. 5A). This corresponded to a number of charges of −23.6 for apo-RCL and −9.1 for holo-RCL (Table 2). The charge difference between the holo- and apo-forms (ΔNc = −9.1 − (−23.6) = 14.5) provided an estimation of the number of charges neutralized by the binding of the divalent cation, implicating that approximately seven calcium ions were bound to holo-RCL (NCa2+ = ½*ΔNc = 7.25; Table 2). The electrophoretic mobility of RCS decreased from −1.35 to −0.98 μm·cm·V−1·s−1 upon calcium binding (Fig. 5B). From these values, we estimated a number of charges of −14.9 for the monomeric apo-state and of −15.5 for the trimeric holo-state (Table 2), this latter value corresponding to an averaged charge of −5.2 (i.e. −15.5/3) per monomer of holo-RCS. Therefore the charge difference between holo-RCS and apo-RCS, ΔNc, was 9.7 (ΔNc = −5.2 − (−15.4)) suggesting that approximately five calcium ions were bound per polypeptide in the trimeric holo-RCS (NCa2+ = ½*ΔNc = 4.86 in Table 2).
FIGURE 5.
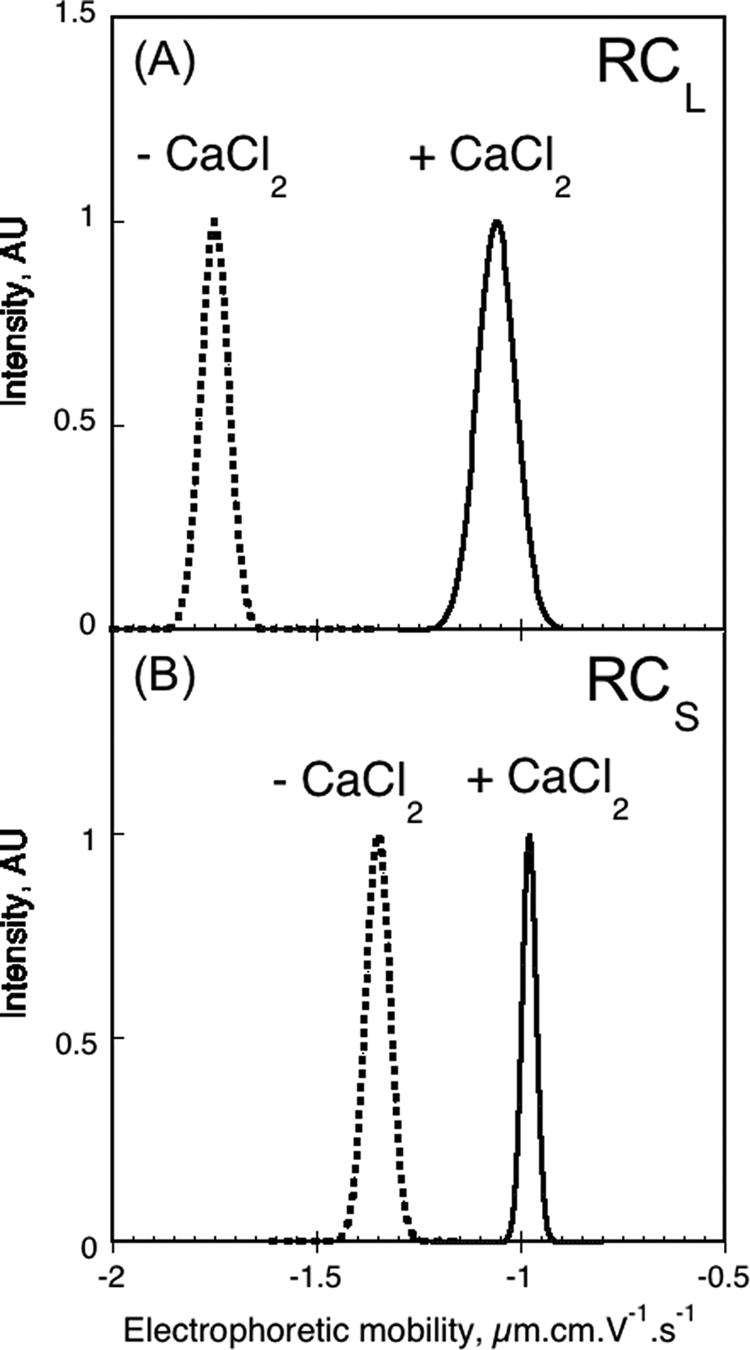
Electrophoretic mobility distribution of RCL and RCS. Electrophoretic mobility of RCL (A) and RCS (B) polypeptides, in the absence (dashed line) or in the presence (solid line) of 2 mm (A), 5 mm (B) CaCl2. The experimental conditions were: 70 μm RCS, 130 μm RCL, and buffer A at 25 °C.
TABLE 2.
Electrophoretic mobility of RCL and RCS polypeptides
The experimental conditions were as follows: temperature, 25 °C; apo-states, buffer A; in the presence of calcium, buffer A, 2 mm CaCl2 for holo-RCL; buffer A, 5 mm CaCl2 for RCS.
| Parameters | RCL | RCL + Calcium | RCS | RCS + Calcium |
|---|---|---|---|---|
| Molecular mass (kDa) | 17.1 | 17.1 | 14.1 | 42.3 |
| Frictional ratio (f/f0)a | 1.93 | 1.32 | 1.83 | 1.35 |
| μe (μm·cm·V−1·s−1)b | −1.75 ± 0.08 | −1.06 ± 0.11 | −1.35 ± 0.07 | −0.98 ± 0.04 |
| Nc | −23.6 ± 1.1 | −9.1 ± 0.4 | −14.9 ± 0.7 | −15.5 ± 1.1 |
| Nc/monomer | −23.6 ± 1.1 | −9.1 ± 0.4 | −14.9 ± 0.7 | −5.2 ± 0.3 |
| ΔNc = NcHolo − NcApoc | 14.5 ± 1.2 | 9.7 ± 0.7 | ||
| Number of calcium (Nca2+)c,d | 7.25 ± 0.6 | 4.86 ± 0.4 | ||
| Nc from the amino acid sequencee | −23 | −19 |
a Experimental data from analytical ultracentrifugation.
b Experimental data from electrophoretic mobility.
c Parameters calculated as described under “Experimental Procedures.”
d Number of calcium, NCa2+ = (ΔNc)/2.
e The number of charges, Nc, is calculated as follows: Nc = |nK + nR| − |nD + nE|.
DISCUSSION
Previous studies have shown that RTX-containing polypeptides are remarkable models to explore the biophysical principles underlying the ligand-induced disorder-to-order transition in proteins (9–11, 24, 29–31). We indeed showed that a 700-residue-long polypeptide, corresponding to the RD of the CyaA toxin, is natively disordered (yet fully soluble) in the absence of calcium and folds upon calcium binding into a stable, β-rich structure in a highly cooperative manner (9, 11). Further biophysical studies of a smaller region from the CyaA RTX domain, encompassing a single block of RTX repeats (block V), have shown that the folding of RTX motifs in the presence of calcium is critically dependent upon the adjacent polypeptide segment immediately downstream to the RTX sequences (10, 31). How this C-terminal flanking region and calcium binding could contribute to the folding of the RTX motifs remains to be elucidated.
To address these questions, we have performed here a comprehensive analysis of the hydrodynamic properties of several RTX polypeptides, all containing the RTX core (R) of block V of repeats but differing in their N- or C-terminal flanking regions. We examined more thoroughly the two polypeptides, RCS and RCL, made of the RTX core (R) followed by either a short (CS) or a long (CL) C-terminal flanking region. Our prior studies have shown that both polypeptides fold into a β-rich structure in the presence of calcium but with a significant difference in affinity for this cation (10).
In the absence of calcium, all of the RTX polypeptides were found to be monomeric and highly hydrated, and they exhibited high hydrodynamic radii, as expected for intrinsically disordered proteins. Upon calcium-induced folding, RCL experienced a strong compaction as shown by a large decrease of its hydrodynamic radius (Table 1), whereas it remained monomeric (Fig. 2, C and D, and Ref. 10). In contrast, the RCS polypeptide folded in the presence of calcium into a trimeric state that exhibited roughly the same hydrodynamic radius as the unfolded monomeric apo-form. Hence, these results indicated that the C-terminal flanking region is not only implicated in the calcium-induced folding of the RTX β-helix, but that it also affects the quaternary structure of the polypeptide. Importantly, we could not detect any monomeric or dimeric forms of RCS, in the presence of calcium, suggesting that holo-RCS is only stable as a trimer.
The striking difference in behavior of these two polypeptides has important implications for the folding reaction of intrinsically disordered RTX motifs into calcium-loaded β-structures. It has been previously reported that β-helices have the propensity to multimerize (29, 32–36). It has been suggested that intramolecular interactions within the β-helices are too weak to stabilize the structure, whereas additional intermolecular interactions may provide further energy to stabilize the assembly of β-helix proteins (16, 32). Moreover, Lilie et al. (29) showed that a synthetic polypeptide made of several consensus RTX motifs was able to form a β-helix in the presence of calcium and polyethylene glycol but only upon polymerization, suggesting that intermolecular interactions were indeed essential to stabilize the putative β-roll structure of these synthetic RTX peptides (29).
Our present results strongly support the idea that a β-helix structure made of RTX motifs is thermodynamically weakly stable per se. In the RCS polypeptide, the short C-terminal flanking region may not be sufficient to stabilize the β-helix fold within a monomeric species, whereas the thermodynamic contribution of intermolecular interactions within a trimer of RCS may provide additional energy to maintain a β-helix conformation. In other words, by restricting the degrees of freedom of the polypeptides, the trimeric state may provide a significant entropic contribution to stabilize the β-helix structure. This may explain why we could not detect any monomeric or dimeric holo-RCS by SEC-TDA even at the lowest protein concentration tested. The holo-state may only exist as a trimer, which provides the favorable environment to stabilize the calcium-loaded structure. We propose that monomeric RCS mainly populates the intrinsically disordered state. In the presence of calcium, monomers may experience transient exchanges between calcium-loaded partially folded and unfolded states. Upon diffusion/collision, intermolecular interactions between partially folded RCS could take place and lead to the formation of a stable trimeric complex. At variance, in RCL the long C terminus-flanking region may provide enough intramolecular contacts to stabilize the holo-protein in a monomeric form.
The propensity of the RTX β-helices to be stabilized via intermolecular interactions may be pertinent as well for the folding of the whole RD domain that contains five blocks of RTX motifs. Indeed, Iwaki et al. (23) initially reported that the C-terminal region of RD encompassing the RTX block V could functionally complement in vitro a truncated CyaA (lacking the last 75 residues) to restore its cytotoxic activities. Later, Bejerano et al. (22) showed that this C-terminal region of RD could associate in a calcium-dependent manner with the truncated CyaA. Our present results suggest that block V, which can fold as an autonomous domain in the presence of calcium, might provide stabilizing contacts to the upstream blocks (I–IV) of RTX motifs and trigger the calcium-dependent folding of the whole RD domain. Interestingly, we recently found that calcium-induced folding of RD is a highly cooperative process (11). This indicates that, in vitro, the loading of the different calcium-binding sites (many of them deviating from the canonical RTX sequences) occurs in a tightly concerted manner. Hence, block V may serve as a nucleus to trigger the folding of the whole RD domain, assuming that, when secreted by the type I secretion machinery, the C-terminal extremity of CyaA is reaching first the calcium-rich extracellular medium.
Our present study also highlighted some important biophysical properties of the RTX polypeptides regarding, in particular, their calcium-induced folding process, which is pertinent for the disorder-order equilibrium of proteins in general. We first compared and described quantitatively the hydrodynamic properties of the apo- and holo-states of both RCL and RCS by using the expression that relates the hydrodynamic volume to the molecular mass, the partial specific volume, the time-averaged hydration, and the buffer density.
As previously reported (10), apo- and holo-RCL exhibited a large difference in their retention volumes on SEC. This variation resulted from a decrease of both the hydrodynamic volume (VH in the left part of the equation) and the hydration (δ in the right part of the equation) of the protein upon calcium binding. RCL yet remained a monomer in both the apo- and the holo-state. The response of RCS to calcium binding was found here to be drastically different. RCs also experienced a large reduction of its hydration upon calcium binding, but at variance with RCL, it was essentially compensated by an increase in molecular mass, from monomer to trimer, whereas the overall hydrodynamic volume remained unchanged. A plot of the hydrodynamic radii of the RTX polypeptides as a function of their molecular masses, according to Uversky (37), revealed that all of the apo-forms lie between premolten globule conformations and natively unfolded coils, whereas the holo-forms adopt compact conformations (Fig. 6). Also, whereas NRCL, RCL, and RD proteins experienced a calcium-induced decrease of their hydrodynamic radius (i.e. change along the y axis to reach the region of compact proteins), RCS responded to calcium by a change of its apparent molecular mass (horizontal shift on the x axis).
FIGURE 6.

Dependences of the hydrodynamic radius on protein molecular mass. The plot shows the hydrodynamic radii (RH) of the apo-state (closed symbols) and the holo-state (open symbols) of RD (◣), NRCL (♦), RCL (■), NRCS (▴), RCS (●), NR (+), and R (▾) polypeptides as a function of their respective molecular masses. The logarithmic dependences of the RH on protein molecular mass for native (bold dashed line), molten globule (thin dashed line), premolten globule (thin solid line), and natively unfolded proteins (bold solid line) conformations were computed according to the equations described by Uversky (Ref. 37 and the references therein).
Finally, we attempted to further define the biophysical principles that might control the calcium-induced folding of RTX proteins. We suggested recently that electrostatic interactions might play an important role in the disorder-to-order transition of RD (11). We proposed that, in the apo-form, electrostatic repulsions between the charged Asp residues of the RTX motifs might force the polypeptide chain to adopt expanded and disordered conformations. Calcium binding to the RTX motifs could screen the Asp negative charges, thus allowing the polypeptide chain to collapse and fold into a compact structure.
To directly test this model, we experimentally measured here the changes of the mean net charge induced by the binding of calcium to the RTX proteins. Electrophoretic mobility measurements, in conjunction with AUC, QELS, and SEC-TDA (see “Experimental Procedures” for details), allowed us to estimate the mean net charge of both the apo- and the holo-state of RCS and RCL. From the mean net charge values, we calculated that the RCL polypeptide binds ∼7 calcium ions, whereas the trimeric holo-RCS binds ∼15 calcium ions, corresponding to roughly 5 calcium ions/monomer on average. These results are in good agreement with our prior calcium binding measurements (10, 24). Interestingly, the somewhat lower number of calcium-binding sites in RCS may indicate that in the trimeric form of holo-RCS, the packing constraints may distort or prevent the folding of certain RTX motifs (as revealed by the lowest n-π band intensity of RCS as compared with RCL in supplemental Fig. S1). The large changes in the mean net charge of the RTX polypeptides upon calcium binding are interesting to compare in terms of a charge-hydrophobicity phase plot (38). As shown in Fig. 7, the apo-proteins range in the intrinsically disordered region of the charge-hydrophobicity phase space, whereas the strong decrease of the mean net charge upon calcium binding shifts the holo-proteins well below the boundary between unfolded and folded proteins (Fig. 7). To our knowledge, these are the first experimental measurements of ligand-induced changes of polypeptide charges associated with a disorder-to-order transition.
FIGURE 7.

Dependence of the mean net charge and the mean hydrophobicity of the block V polypeptides. This plot presents the charge hydrophobicity phase space for both the apo-state (circles) and the holo-state (squares) of RD (black), NRCL (blue), RCL (red), NRCS (green), RCS (purple), NR (gray), and R (orange) polypeptides. The filled symbols correspond to the mean net charge values of RCS and RCL that were experimentally determined herein. The open symbols correspond to the mean net charge values of the RTX polypeptides that were theoretically computed from the amino acid sequence and considering that block V polypeptides and the full RD protein should bind 7 and 35 calcium ions, respectively (24). The theoretically computed charge value of holo-RCL (open red square) is hidden by the experimental measurement of the protein charge (closed red square). The oblique line represents the boundary between intrinsically disordered and folded proteins according to Uversky et al. (38).
Concluding Remarks
We previously proposed that the intrinsically disordered state of the CyaA RTX motifs in the bacterial cytoplasm may favor the secretion of the toxin by the type I secretion system (9, 10), whereas the calcium-induced folding of the RD domain in the extracellular milieu is critical for the CyaA cytotoxic activity. We showed here that RTX blocks could fold and stabilize upon multimerization, a cooperative process that might contribute to improve the secretion efficiency. We further showed that the apo-forms of RTX block V polypeptides adopt a premolten globule state that switches to a native state in the presence of calcium. This transition appeared finely tuned from a thermodynamic point of view to respond to calcium binding in physiological conditions. Moreover, we showed that the high number of calcium bound per protein allows switching from an intrinsically disordered, highly charged apo-form to a folded holo-state with a reduced mean net charge. Our data suggest that the folding of the RTX proteins is controlled by a charge change effect caused by the variations of physiological concentrations of calcium. The simplicity of this calcium-induced, charge-driven disorder-to-order transition may favor a robust and chaperone-independent protein folding process adapted to the biogenesis of the RTX proteins.
Supplementary Material
Acknowledgments
We thank Bertrand Raynal for expertise and help on AUC and for the critical reading of the manuscript. We also thank J. D'Alayer for the N-terminal sequencing of the proteins and Johanna C. Karst for fruitful comments on this manuscript.
This work was supported by CNRS Institut Pasteur Projet Transversal de Recherche Grant PTR#374 and the Agence Nationale de la Recherche Programme Jeunes Chercheurs Grant ANR-09-JCJC-0012.

The on-line version of this article (available at http://www.jbc.org) contains supplemental text, Table S1, and Fig. S1.
- RD
- RTX-containing domain
- QELS
- quasi-elastic light scattering
- SEC
- size exclusion chromatography
- TDA
- Tetra detector array
- AUC
- analytical ultracentrifugation.
REFERENCES
- 1. Dunker A. K., Lawson J. D., Brown C. J., Williams R. M., Romero P., Oh J. S., Oldfield C. J., Campen A. M., Ratliff C. M., Hipps K. W., Ausio J., Nissen M. S., Reeves R., Kang C., Kissinger C. R., Bailey R. W., Griswold M. D., Chiu W., Garner E. C., Obradovic Z. (2001) J. Mol. Graph. Model. 19, 26–59 [DOI] [PubMed] [Google Scholar]
- 2. Dyson H. J., Wright P. E. (2005) Nat. Rev. Mol. Cell Biol. 6, 197–208 [DOI] [PubMed] [Google Scholar]
- 3. Fink A. L. (2005) Curr. Opin. Struct. Biol. 15, 35–41 [DOI] [PubMed] [Google Scholar]
- 4. Receveur-Bréchot V., Bourhis J. M., Uversky V. N., Canard B., Longhi S. (2006) Proteins 62, 24–45 [DOI] [PubMed] [Google Scholar]
- 5. Eliezer D. (2009) Curr. Opin. Struct. Biol. 19, 23–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Midic U., Oldfield C. J., Dunker A. K., Obradovic Z., Uversky V. N. (2009) Protein Pept. Lett. 16, 1533–1547 [DOI] [PubMed] [Google Scholar]
- 7. Wright P. E., Dyson H. J. (2009) Curr. Opin. Struct. Biol. 19, 31–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tompa P., Kovacs D. (2010) Biochem. Cell Biol. 88, 167–174 [DOI] [PubMed] [Google Scholar]
- 9. Chenal A., Guijarro J. I., Raynal B., Delepierre M., Ladant D. (2009) J. Biol. Chem. 284, 1781–1789 [DOI] [PubMed] [Google Scholar]
- 10. Sotomayor Pérez A. C., Karst J. C., Davi M., Guijarro J. I., Ladant D., Chenal A. (2010) J. Mol. Biol. 397, 534–549 [DOI] [PubMed] [Google Scholar]
- 11. Chenal A., Karst J. C., Peréz A. C., Wozniak A. K., Baron B., England P., Ladant D. (2010) Biophys. J. 99, 3744–3753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Coote J. G. (1992) FEMS Microbiol. Rev. 8, 137–161 [DOI] [PubMed] [Google Scholar]
- 13. Welch R. A. (2001) Curr. Topics Microbiol. Immunol. 257, 85–111 [DOI] [PubMed] [Google Scholar]
- 14. Linhartova I., Bumba L., Masin J., Basler M., Osicka R., Kamanova J., Prochazkova K., Adkins I., Hejnova-Holubova J., Sadilkova L., Morova J., Sebo P. (2010) FEMS Microbiol. Rev. 34, 1076–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Baumann U., Wu S., Flaherty K. M., McKay D. B. (1993) EMBO J. 12, 3357–3364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Meier R., Drepper T., Svensson V., Jaeger K. E., Baumann U. (2007) J. Biol. Chem. 282, 31477–31483 [DOI] [PubMed] [Google Scholar]
- 17. Ladant D., Ullmann A. (1999) Trends Microbiol. 7, 172–176 [DOI] [PubMed] [Google Scholar]
- 18. Vojtova J., Kamanova J., Sebo P. (2006) Curr. Opin. Microbiol. 9, 69–75 [DOI] [PubMed] [Google Scholar]
- 19. Pimenta A. L., Racher K., Jamieson L., Blight M. A., Holland I. B. (2005) J. Bacteriol. 187, 7471–7480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Holland I. B., Schmitt L., Young J. (2005) Mol. Membr. Biol. 22, 29–39 [DOI] [PubMed] [Google Scholar]
- 21. Delepelaire P. (2004) Biochim. Biophys. Acta 1694, 149–161 [DOI] [PubMed] [Google Scholar]
- 22. Bejerano M., Nisan I., Ludwig A., Goebel W., Hanski E. (1999) Mol. Microbiol. 31, 381–392 [DOI] [PubMed] [Google Scholar]
- 23. Iwaki M., Ullmann A., Sebo P. (1995) Mol. Microbiol. 17, 1015–1024 [DOI] [PubMed] [Google Scholar]
- 24. Bauche C., Chenal A., Knapp O., Bodenreider C., Benz R., Chaffotte A., Ladant D. (2006) J. Biol. Chem. 281, 16914–16926 [DOI] [PubMed] [Google Scholar]
- 25. Bourdeau R. W., Malito E., Chenal A., Bishop B. L., Musch M. W., Villereal M. L., Chang E. B., Mosser E. M., Rest R. F., Tang W. J. (2009) J. Biol. Chem. 284, 14645–14656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Karst J. C., Sotomayor Pérez A. C., Guijarro J. I., Raynal B., Chenal A., Ladant D. (2010) Biochemistry 49, 318–328 [DOI] [PubMed] [Google Scholar]
- 27. Harding S. E., Cölfen H. (1995) Anal. Biochem. 228, 131–142 [DOI] [PubMed] [Google Scholar]
- 28. Basak S. K., Ladisch M. R. (1995) Anal. Biochem. 226, 51–58 [DOI] [PubMed] [Google Scholar]
- 29. Lilie H., Haehnel W., Rudolph R., Baumann U. (2000) FEBS Lett. 470, 173–177 [DOI] [PubMed] [Google Scholar]
- 30. Szilvay G. R., Blenner M. A., Shur O., Cropek D. M., Banta S. (2009) Biochemistry 48, 11273–11282 [DOI] [PubMed] [Google Scholar]
- 31. Blenner M. A., Shur O., Szilvay G. R., Cropek D. M., Banta S. (2010) J. Mol. Biol. 400, 244–256 [DOI] [PubMed] [Google Scholar]
- 32. Steinbacher S., Seckler R., Miller S., Steipe B., Huber R., Reinemer P. (1994) Science 265, 383–386 [DOI] [PubMed] [Google Scholar]
- 33. Raetz C. R., Roderick S. L. (1995) Science 270, 997–1000 [DOI] [PubMed] [Google Scholar]
- 34. Kisker C., Schindelin H., Alber B. E., Ferry J. G., Rees D. C. (1996) EMBO J. 15, 2323–2330 [PMC free article] [PubMed] [Google Scholar]
- 35. Beaman T. W., Binder D. A., Blanchard J. S., Roderick S. L. (1997) Biochemistry 36, 489–494 [DOI] [PubMed] [Google Scholar]
- 36. Govaerts C., Wille H., Prusiner S. B., Cohen F. E. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 8342–8347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Uversky V. N. (2002) Eur. J. Biochem. 269, 2–12 [DOI] [PubMed] [Google Scholar]
- 38. Uversky V. N., Gillespie J. R., Fink A. L. (2000) Proteins 41, 415–427 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.