

Abstract
Whether RET is able to directly phosphorylate and activate downstream targets independently of the binding of proteins that contain Src homology 2 or phosphotyrosine binding domains and whether mechanisms in trans by cytoplasmic kinases can modulate RET function and signaling remain largely unexplored. In this study, oligopeptide arrays were used to screen substrates directly phosphorylated by purified recombinant wild-type and oncogenic RET kinase domain in the presence or absence of small molecule inhibitors. The results of the peptide array were validated by enzyme kinetics, in vitro kinase, and cell-based experiments. The identification of focal adhesion kinase (FAK) as a direct substrate for RET kinase revealed (i) a RET-FAK transactivation mechanism consisting of direct phosphorylation of FAK Tyr-576/577 by RET and a reciprocal phosphorylation of RET by FAK, which crucially is able to rescue the kinase-impaired RET K758M mutant and (ii) that FAK binds RET via its FERM domain. Interestingly, this interaction is abolished upon RET phosphorylation, indicating that RET binding to the FERM domain of FAK is a priming step for RET-FAK transactivation. Finally, our data indicate that FAK inhibitors could be used as potential therapeutic agents for patients with multiple endocrine neoplasia type 2 tumors because both, treatment with the FAK kinase inhibitor NVP-TAE226 and FAK down-regulation by siRNA reduced RET phosphorylation and signaling as well as the proliferation and survival of tumor and transfected cell lines expressing oncogenic RET.
Keywords: Anticancer Drug, Protein Phosphorylation, Protein-Protein Interactions, Receptor Tyrosine Kinase, Signal Transduction
Introduction
Receptor tyrosine kinases (RTKs)2 are key molecules for the recognition and transduction of external signals across the plasma membrane. Thus, RTKs play an essential role in regulating fundamental cellular and biological processes in the cell, such as cell proliferation, cell cycle, cell migration, metabolism, and survival (1). Once stimulated by their cognate ligands, activation of the kinase domain leads to trans-phosphorylation of specific tyrosine residues within the activation segment of the kinase domain and the C-terminal tail. These phosphotyrosine residues serve as docking sites for adaptor and signaling proteins that contain Src homology domain 2 (SH2) or phosphotyrosine binding (PTB) domains triggering the activation of intracellular signaling cascades (1, 2). The selective and specific recognition of short peptide motifs in RTKs by proteins containing modular interaction domains has been an area of intense study over recent years in the kinase signaling field (3). However, whether RTKs are able to directly phosphorylate and activate downstream targets and whether mechanisms in trans by cytoplasmic kinases can regulate activation and signaling by RTKs remain to be evaluated.
RET (rearranged during transfection) is an RTK expressed and required during early development for the formation of neural crest-derived lineages, kidney organogenesis, and spermatogenesis (4–6). To date, a family of glial cell-derived neurotrophic factor (GDNF) ligands, which includes GDNF, ARTN, NRTN, and PSPN, and a family of glycosylphosphatidylinositol-linked co-receptors, GFRα1 to -4, have been identified (7). A large number of studies have established a canonical model for the mechanism of RET activation and signaling. Ligand·co-receptor·RET complex formation results in transient RET dimerization, activation of the RET tyrosine kinase domain, and autophosphorylation of intracellular tyrosine residues. These phosphotyrosine residues act as docking sites for SH2 and PTB domain-containing proteins that, in turn, trigger the activation of downstream signaling pathways, such as the Ras/MAPK/ERK1/2, PI3K/AKT, JNK, p38SAPK, STAT3, SRC, ERK5, and phospholipase Cγ pathways (6, 8, 9). Gene amplification, overexpression, or mutations that lead to aberrant signaling are responsible for the acquired transforming potential of oncogenic RTKs (8). In the case of RET, germ line missense-activating point mutations cause the cancer syndrome multiple endocrine neoplasia type 2 (MEN2). Three mutation-specific disease phenotypes can be recognized: MEN2A, MEN2B, and a familial form of medullary thyroid carcinoma (9). Despite a clear correlation between the phenotype of these diseases and the different RET oncogenic mutations, the molecular mechanisms that connect the different RET mutants with their phenotype are not well understood.
In this study, we have used peptide arrays to screen for substrates directly phosphorylated by purified recombinant wild-type (WT) RET kinase domain and a kinase domain containing the oncogenic V804M gatekeeper mutation (10) to address the following questions: (i) Can RET directly phosphorylate and activate downstream targets independent of the binding of SH2 or PTB domain-containing docking proteins? (ii) Can feedback mechanisms by cytoplasmic kinases modulate RET activation and function? (iii) Are these phosphorylation events and feedback mechanisms potentiated by oncogenic mutations in RET and hence required for the malignant phenotype of oncogenic RET-driven tumor cells?
EXPERIMENTAL PROCEDURES
Oligopeptide Kinase Array
Experiments were carried out using a 96-array semiautomated system (PamStation 96, PamGene, 's-Hertogenbosch, The Netherlands). The PamChip array contains 144 oligopeptide-tyrosine kinase target sequences per slide, representing SH2 and PTB domain-containing proteins, non-SH2 and PTB domain-containing proteins, and RTKs, immobilized on a porous microarray surface through the peptide N terminus, representing a platform of tyrosine kinase substrates. Each array was blocked with 0.2% bovine serum albumin (BSA), fraction V (Calbiochem Immunochemicals, Merck KGaA, Darmstadt, Germany), by pumping it through the porous microarray for 30 cycles of 60 s each and then washed twice with ABL protein-tyrosine kinase reaction buffer solution (New England Biolabs, Ipswich, MA). Incubation with kinases was carried out as described previously (11–13) using 80 ng of soluble recombinant protein/array at 30 °C. Samples were pulsed back and forth through the porous material for 60 cycles to maximize reaction kinetics and to reduce analysis time. Phosphorylated peptides on the PamChip were recognized using a phosphotyrosine FITC-conjugated antibody. At every fifth pump cycle, a 16-bit TIFF image was taken with a built-in CCD camera (PamGene). Inhibitory experiments were carried out with the indicated small molecule inhibitors (see “Cell Lines and Reagents”). Each inhibitor was mixed with the reaction mixture to the indicated final concentrations just before incubation on the array. Acquired data from the PamStation 96 were captured with the supplied software package BioNavigator (version 0.3.1; PamGene) and exported to Excel for further analysis.
In Vitro Kinase Assays and Enzyme Kinetics Experiments
Baculovirus recombinant RET WT and V804M kinase domains were expressed in Sf9 cells, purified, and assayed using an ELISA-based kinase test, as described previously (14–16). Recombinant full-length focal adhesion kinase (FAK) was purchased from Cell Signaling Technology. Unless otherwise stated, in vitro kinase assays were performed by incubating RET and FAK at 30 °C for 15 min in reaction buffer (25 mm Hepes, pH 7, 1 mm MnCl2, 5 mm MgCl2) with or without FAK inhibitor NVP-TAE226 and 150 μm ATP. The kinase reaction was stopped by the addition of Laemmli buffer, and the sample was then heated for 5 min at 95 °C and loaded onto SDS-PAGE, transferred to a nitrocellulose membrane, and probed with the indicated antibodies. Enzyme kinetic experiments were performed as described previously (16). Briefly, rates of phosphorylation by RET kinase were determined using a continuous ADP-coupled kinase assay with a FAK (FAK(569–581)_Y576/7; RYMEDSTYYKASK) peptide.
GST Pull-down Experiments
Wild-type and mutant GST-FAK N-terminal domain proteins (amino acids 1–405), containing the FERM domain of FAK (hereafter called GST-FERM-FAK), were expressed as described previously (17). For binding assays, recombinant WT RET kinase domain (300 μg) was incubated with GST-FERM-FAK (200 μg) immobilized on glutathione-agarose beads in 20 mm Tris-HCl, 100 mm NaCl, and 1 mm DTT (buffer A) for 90 min rotating at 4 °C. Beads were spun down at 500 rpm for 5 min and washed five times with buffer A. Samples (5 μl) were loaded onto SDS-polyacrylamide gels and stained with Coomassie dye.
Cell Lines and Reagents
HEK293 (human embryonic kidney) parental and transfected cell lines were cultured in DMEM (Invitrogen) supplemented with 10% fetal bovine serum (FBS; Invitrogen) and 500 μg/ml G418 (Invitrogen) for selection of transfected clones. MZ-CRC-1 cells were grown in DMEM supplemented with 20% FBS. Sorafenib (BAY 43-9006) was provided by Bayer HealthCare Pharmaceuticals (West Haven, CT). The FAK inhibitor NVP-TAE226 and the tyrosine kinase inhibitor STI571 were provided by Novartis (Basel, Switzerland), AG490 and PP1 were from Promega (Madison, WI). The following antibodies were used: phospho-Tyr-1062 RET (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), ERK1/2, phospho-ERK1/2, FAK, phospho-Tyr-576/577 FAK, phospho-Tyr-397 FAK, phospho-Tyr-925 FAK, SRC, phospho-Tyr-416 SRC, p38 and phospho-p38, JNK and phospho-JNK, RET (C31B4) (Cell Signaling Technology, New England Biolabs), anti-HisG (Invitrogen), Alexa488 anti-rabbit Ig, Alexa555 anti-mouse, and Alexa647-conjugated phalloidin (Molecular Probes, Inc., Eugene, OR).
Cell Assays
For cell proliferation assays, 2.5 × 105 of MZ-CRC-1 cells were plated in 12-well plates in the presence of increasing concentrations of NVP-TAE226 or vehicle (DMSO, 1:1000). Cell proliferation was determined by counting cells with a hemocytometer at days 1, 2, 4, 8, and 10. For three-dimensional culture, we followed the same protocol as described previously (18). Briefly, 0.2 ml of growth factor-enriched Matrigel (BD Biosciences) diluted 1:1 in PBS per well was plated into 24-well tissue culture dishes and left at 37 °C for 2–3 h. Next, 2.5 × 104 cells/well were single cell-seeded and cultured under the indicated conditions for 15 days. Medium was replaced every 3 days. Cell Titer Glow experiments, siRNA transfections, and Western blot analysis were performed as described previously (18).
Confocal Microscopy
Cells cultured on glass coverslips were fixed in 4% formaldehyde for 1 h at room temperature, followed by permeabilization in 0.5% Triton X-100 in PBS for 10 min. Immunostaining with primary antibodies for 1 h was followed by incubation with Alexa486 anti-rabbit Ig and Alexa555 anti-mouse Ig for 45 min. Alexa647-phalloidin was used to stain actin filaments. Fluorescent images were captured sequentially in three channels on a Leica Microsystems TCS-SP2 confocal microscope.
Statistical Analysis
Statistical and enzyme kinetics analyses were performed using Prism software (GraphPad Software, La Jolla, CA). Unless otherwise stated, cell number assays were compared using one-way analysis of variance for repeating measures and Dunnett's test. Differences were considered statistically significant when p < 0.05.
RESULTS
FAK Is a Direct Substrate of RET
To determine whether RET kinase is able to directly phosphorylate downstream targets, the PamChip tyrosine kinase oligopeptide array was employed (11–13). Purified recombinant WT RET kinase domain was incubated on the PamChip in the absence or presence of ATP. RET kinase activation, as indicated by the increase in phosphorylation upon the addition of ATP, was monitored by a RET Tyr-1029-containing peptide as a positive control. RET Tyr-1029 is an autophosphorylation site shown by mass spectrometry analysis (supplemental Fig. 1). Upon RET kinase activation, a considerable number of substrates were phosphorylated, ranging from low to high levels of intensity (Fig. 1, A and B). A close examination of the targets showed that many of them contained SH2 or PTB domains, such as CBL, phospholipase Cγ1, STAT3, and STAT4 (Fig. 1C). One of the substrates phosphorylated by RET kinase was a FAK activation loop peptide containing Tyr-576 and Tyr-577 (Fig. 1C). RET-mediated phosphorylation of FAK was validated in a cell-based experiment by Western blotting using the T47D cell line (a human breast cancer cell line expressing wild type RET) stimulated with 20 ng/ml GDNF in the presence of 100 ng/ml soluble GFRα1 co-receptor (Fig. 1D). Following GDNF stimulation, increased phosphorylation of FAK at Tyr-576/577 as well as phosphorylation of RET at Tyr-905 was observed. The fact that FAK does not contain any SH2 or PTB domains (19) prompted us to hypothesize that a direct phosphorylation of FAK by RET may be an important step in the RET-FAK signaling pathway.
FIGURE 1.

FAK is a direct substrate of RET kinase. A, recombinant WT RET kinase domain (80 ng) was incubated on a PamChip oligopeptide array as described under “Experimental Procedures” in the presence or absence of ATP (100 μm). Data represent signal increase (arbitrary units) between ATP-treated and untreated samples of the whole oligopeptide substrate library from two experiments. B, representative images of the array captured at 5 min using a CCD camera are shown. C, signal increases of control (no peptide) and STAT3(698–710)_Y705 (DPGSAAPYLKTKF), STAT4(686–698)_Y693 (TERGDKGYVPSVF), PLCG1(1246–1258)_S1248/Y1253 (EGSFESRYQQPFE), FAK(569–581)_Y576/7 (RYMEDSTYYKASK), RET(1022–1034)_Y1029 (TPSDSLIYDDGLS), EGFR(862–874)_Y869 (LGAEEKEYHAEGG), and CBL(693–705)_Y700 (EGEEDTEYMTPSS) peptides are shown. Data represent the average increased signal ± S.D. (error bars) of two experiments. D, Western blot analyses of whole cell extracts from T47D cells treated with soluble GFRα-1 (100 ng/ml) in the presence or absence of GDNF (20 ng/ml) for 10 min using the indicated antibodies (phosphospecific Tyr-905 RET and Tyr-576/577 FAK antibodies were used simultaneously to detect RET and FAK phosphorylation status). E, enzymatic assay performed with soluble RET WT kinase (residues 705–1013; 2.5 μg) incubated with the indicated concentrations of FAK(569–581)_Y576/7 peptide. Data are representative of six independent experiments in triplicate using different protein batches. F, enzymatic assay performed with increasing amounts of recombinant RET WT kinase domain (0, 2.5, 5, and 10 μg) incubated with the FAK(569–581)_Y576/7 peptide. Data are representative of two independent experiments in triplicate using different protein batches.
Kinetic Characterization of FAK Peptide Substrate Phosphorylation
To further characterize the RET-mediated phosphorylation of the FAK peptide, enzyme kinetic constants were measured using a previously described continuous coupled spectrophotometric assay to detect ADP generation (16). The FAK(569–581)_Y576/7 peptide could be efficiently phosphorylated with good Michaelis-Menten saturation kinetics (Fig. 1E) with Kcat = 3.14 ± 0.327 min−1 and Km = 1.72 ± 0.3387 mg/ml. Further, increasing the amount of RET kinase domain enhanced the specificity for the FAK substrate, as indicated by the proportional increase in the catalytic efficiency (Kcat/Km) constant (Fig. 1F).
A Direct and Reciprocal RET-FAK Transactivation Mechanism
The observation that RET can phosphorylate a FAK-derived peptide containing activation loop tyrosines 576 and 577 and that these two tyrosines are SRC phosphorylation sites required for full FAK kinase activity (20) raised the question as to whether activated RET could modulate FAK activation and signaling. To address this, we evaluated first the effect of WT RET kinase domain activation on full-length FAK tyrosine phosphorylation in vitro (Fig. 2A). RET WT kinase domain was incubated together with full-length recombinant FAK in in vitro kinase assays, and the level of tyrosine phosphorylation was detected using phospho-specific antibodies. In the absence of ATP, there was residual background phosphorylation on FAK Tyr-925, and very low basal phosphorylation was observed on FAK Tyr-576/577. Strikingly, when FAK and RET kinases were co-incubated in the presence of ATP, a marked increased in FAK Tyr-576/577 and Tyr-925 phosphorylation was observed together with a shift in mobility of FAK, indicating conversion to an activated state. Full FAK kinase activity is achieved upon FAK Tyr-576 and Tyr-577 phosphorylation (19, 20). Further, the activation of both kinases also resulted in a substantial increase in RET Tyr-905 phosphorylation (Fig. 2A, bottom), suggesting that activated FAK could in turn modulate RET activation and signaling in trans. To address this, the co-incubation experiment was repeated in the presence of the FAK inhibitor NVP-TAE226 (Fig. 2B). In the presence of both RET kinase domain and FAK, NVP-TAE226 treatment blocked the enhanced levels of FAK Tyr-576/577 phosphorylation induced by RET and reduced the level of RET Tyr-905 phosphorylation equivalent to that observed when the RET kinase domain was incubated in the absence of FAK. The inability of NVP-TAE226 to completely block RET phosphorylation indicates that it has no effect on RET kinase activity and that the increased levels of RET phosphorylation observed upon co-incubation with FAK kinase result from a FAK-mediated transphosphorylation event. Finally, the co-incubation experiment was repeated with the kinase-impaired RET K758M mutant (Fig. 2C). As expected, incubation of RET K758M alone in the presence of ATP did not result in RET autophosphorylation; however, phosphorylation of this mutant was observed when co-incubated with FAK kinase. Together, these data demonstrate that FAK can directly phosphorylate RET and that there is a direct and reciprocal RET-FAK transactivation mechanism.
FIGURE 2.

A direct and reciprocal RET-FAK transactivation mechanism. A, recombinant RET WT kinase domain and recombinant full-length FAK were incubated as indicated, in the absence (−) or presence (+) of ATP (150 μm), and kinase activity was monitored by Western blot analysis using the indicated antibodies. Data shown are representative of three independent experiments. B, recombinant RET WT kinase domain and recombinant full-length FAK were incubated under the indicated conditions, with or without ATP (150 μm) and NVP-TAE226 (5 μm). Kinase activity was monitored by Western blot analysis using the indicated antibodies. Data are representative of three independent experiments. C, recombinant RET K758M kinase domain mutant and recombinant full-length FAK were incubated under the indicated conditions, and kinase activity was monitored by Western blot analysis using the indicated antibodies. Data are representative of two independent experiments.
RET Activation Loop Tyr-905 Is Required for RET-induced FAK Tyr-576/577 Phosphorylation
To explore the mechanism by which RET mediates FAK phosphorylation, we investigated next which residues within the RET kinase domain were required for RET-induced FAK Tyr-576/577 phosphorylation. We generated a series of RET kinase domain tyrosine to phenylalanine point mutants (Y826F, Y900F, Y905F, and Y981F) as well as the double mutant Y900F/Y905F. First, we tested the kinetic parameters of the FAK(569–581)_Y576/7 peptide phosphorylation by WT and mutant RET kinase domains (Fig. 3, A and B). Strikingly, the RET Y905F mutant, but not Y900F, showed a significant reduction in catalytic efficiency constant (Kcat/Km) compared with WT RET. An equivalent reduction in activity was observed with the double Y900F/Y905F mutant. These data demonstrate that Tyr-905, and not Tyr-900, in the RET activation loop is required for RET-induced FAK Tyr-576/577 phosphorylation. Interestingly, we also observed a marked effect of the Y981F mutant, suggesting a potential role of Tyr-981 on RET-induced FAK phosphorylation. In control experiments using a nonspecific poly-Glu-Tyr (pEY) peptide, none of the RET mutants showed any impairment in kinase activity (supplemental Fig. 2). This finding indicates that the enzymatic properties of RET kinase with the different Tyr to Phe mutations do not impact on the ability of RET kinase to phosphorylate a nonspecific exogenous substrate in vitro. To further investigate the effect of these mutations on RET kinase function, we analyzed their tyrosine autophosphorylation status upon ATP stimulation. Strikingly, most of the mutants were not severely compromised in terms of autophosphorylation. The exception was the double mutant Y900F/Y905F, which displayed significantly reduced levels of RET phosphorylation (Fig. 3C). From this finding, we conclude that RET Tyr-905 is required by RET-induced FAK Tyr-576/577 phosphorylation.
FIGURE 3.

RET Tyr-905 is required for RET-induced FAK Tyr-576/577 peptide phosphorylation. A, enzymatic assay performed with 5 μg of soluble WT RET kinase domain (residues 705–1013) or the indicated mutants incubated with increasing concentrations of the FAK(569–581)_Y576/7 peptide. Data shown represent mean ± S.E. (error bars) from six replicate samples. Equivalent results were obtained in two independent experiments using different RET kinase batches. B, catalytic efficiency constants (Kcat/Km) of A. Data represent mean ± S.E. of two independent experiments. *, p < 0.05, two-tailed Student's t test. C, Western blot analysis of RET WT kinase domain and the indicated mutants stimulated with ATP (5 mm) and MgCl2 (10 mm) for 5, 30, and 90 min using the indicated antibodies. Total levels of RET were monitored by Coomassie staining. Equivalent results were obtained in two independent experiments using different protein batches.
FAK Binds RET via Its FERM Domain
Despite the direct and reciprocal RET-FAK phosphorylation shown in this study (Figs. 1–3), FAK does not contain an SH2 or PTB domain. Hence, the mechanism by which RET interacts with FAK remains to be elucidated. We hypothesized that appropriate RET-FAK signaling will depend on coordinated coupling of a protein-protein interaction to prime the catalytic activation and subsequent reciprocal phosphorylation. To test this hypothesis, we performed pull-down assays using the FERM domain of FAK (GST-FERM-FAK). As demonstrated in Fig. 4A, the RET kinase domain bound to GST-FERM-FAK and not to GST alone control. Interestingly, this interaction was prevented by phosphorylation of the RET kinase domain (Fig. 4B). These data suggest two potential mechanisms: (i) the FERM domain of FAK could repress RET kinase activity, and/or (ii) RET binding to the FERM domain of FAK could relieve the FAK kinase domain from its autoinhibitory state and thereby make the FAK activation loop Tyr-576/577 accessible for RET phosphorylation (see “Discussion”). Finally, we investigated which residues in the FERM-FAK domain are required for the interaction with RET. One of the striking features of the FERM structure is a large patch of basic residues on the surface at the tip of the F2 subdomain (21). Analysis of the x-ray crystal structure (21) shows that this basic patch is solvent-exposed. Therefore, this region was considered a candidate for the FERM binding site to the RET kinase domain. We tested two sets of mutations within this basic patch, one with alanine substitutions at lysines 190 and 191 (KK) and one with alanine substitutions at lysines 216 and 218 and at arginine 221 (KKR). These mutations were engineered into the GST-FERM-FAK domain fusion protein construct, expressed, and tested for their ability to associate with RET kinase in vitro by GST pull-down assays. Both mutants resulted in loss of RET kinase binding to the FERM-FAK domain (Fig. 4C). From these results, we conclude that basic and exposed residues on the F2 FERM subdomain of FAK are important for binding to the RET kinase domain.
FIGURE 4.

The FERM-FAK domain binds the RET kinase. GST pull-down assays were performed as indicated under “Experimental Procedures.” A, pull-down experiments performed with GST-protein control or GST-FERM-FAK incubated with buffer A (control) or recombinant RET kinase domain (RET KD) (*). Input samples were run on the same gel. Data are representative of three independent experiments. B, pull-down experiments performed with GST-protein control or GST-FERM-FAK incubated with buffer A (control), recombinant RET kinase domain, or phosphorylated recombinant RET kinase (90 min). Input samples were run on the same gel. Data are representative of three independent experiments. C, pull-down experiments performed with GST-protein control, GST-FERM-FAK wild type (WT), GST-FERM-FAK KK, and GST-FERM-FAK KKR mutants incubated with recombinant RET kinase domain, respectively. Input samples were also analyzed on the same gel. Data are representative of three independent experiments.
Increased FAK Peptide Phosphorylation by the RET V804M Kinase Domain
To examine the effect of oncogenic RET kinase domain mutations on the catalytic activity and substrate phosphorylation, with particular attention to the FAK Tyr-576/577 peptide, recombinant RET V804M kinase domain was compared with WT RET kinase using the PamChip array. RET V804M is an oncogenic “gatekeeper” mutation within the kinase domain, which also renders RET resistant to several small molecule inhibitors (10). As expected, the RET V804M mutant showed increased kinase activity compared with WT RET kinase domain as indicated, in a normal distribution, by the -fold increase of peptide phosphorylation (Fig. 5, A and B). Intrinsic kinase activity, monitored by RET Tyr-1029 peptide phosphorylation, revealed a 6.2-fold increase in RET V804M kinase activity (Fig. 5B, inset). Importantly, a close examination of targets revealed that RET V804M triggered an increased phosphorylation of FAK, SRC, STAT3, and STAT4 peptides (Fig. 5B). Moreover, RET V804M kinase mutant displayed altered substrate specificity, as indicated by those highly phosphorylated targets, which were not preferentially phosphorylated by WT RET kinase, and those peptides that were preferentially phosphorylated by WT RET WT kinase compared with the V804M oncogenic mutant (Figs. 1C and 5B).
FIGURE 5.
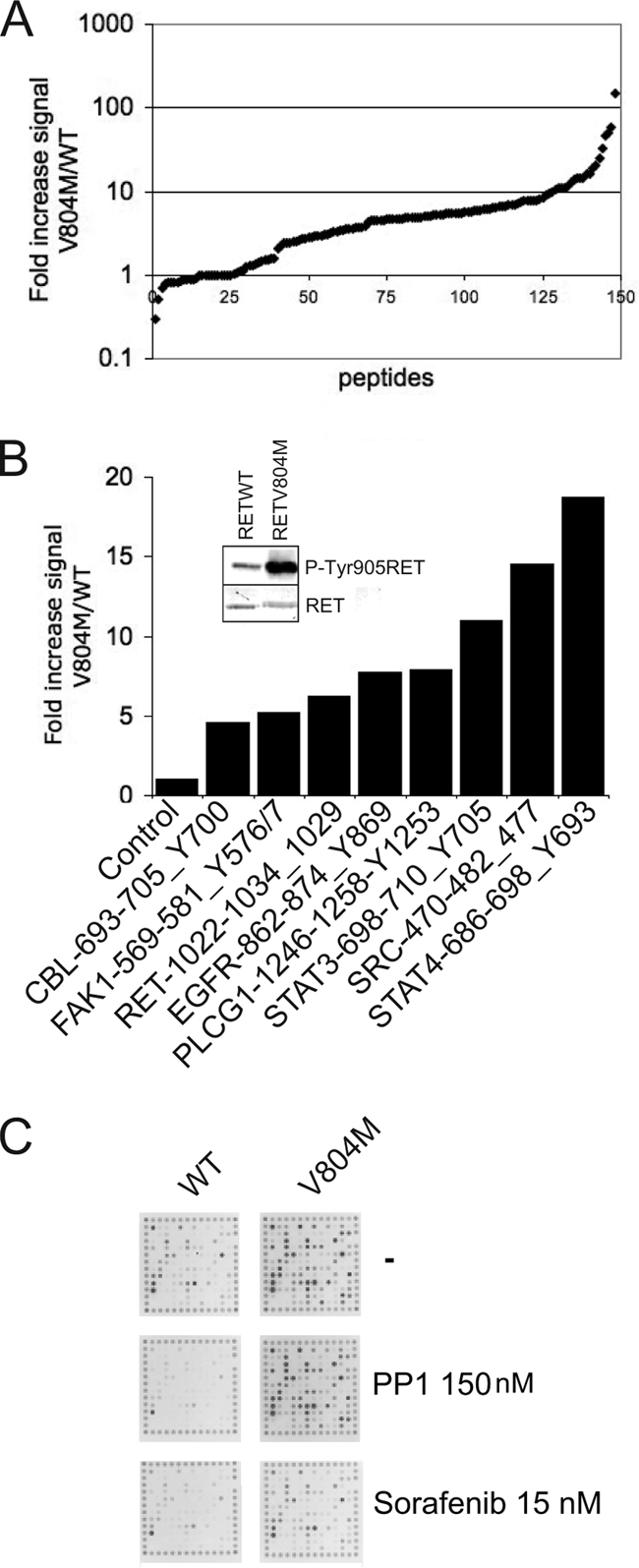
Increased catalytic activity and FAK peptide phosphorylation by the gatekeeper mutant and drug-resistant RET V804M kinase domain. A, equal amounts (80 ng) of RET WT or RET V804M kinase domains were incubated separately on PamChips in the presence of ATP (100 μm). Data represent -fold increase in signal intensity of the whole oligopeptide library induced by RET V804M kinase mutant versus RET WT kinase from two experiments with multiple replicates (n > 15). Peptides are depicted by signal, not position on the array. B, -fold increase in signal intensity (RET V804M/RET WT) of individual peptide targets: STAT4(686–698)_Y693, SRC(470–482)_Y477, STAT3(698–710)_Y705, RET(1022–1034)_Y1029, FAK(569–581)_Y576/7, EGFR(862–874)_Y869, and CBL(693–705)_Y700 peptides induced by RET V804M versus RET WT kinase in the presence of ATP (100 μm). C, RET WT or RET V804M kinase domains were incubated separately on a PamChip in the presence of the tyrosine kinase inhibitors PP1 (150 nm) and sorafenib (15 nm). Representative CCD images taken at 5 min are shown.
Next, small molecule inhibitors were used to verify the drug-resistant status of RET V804M compared with WT RET kinase on the PamChip array (Fig. 5C). PP1 (150 nm) inhibited WT RET kinase but had no effect on the drug-resistant V804M mutant. Interestingly, sorafenib was able to inhibit tyrosine kinase activity of both RET V804M and WT RET kinase at 15 nm concentration (Fig. 5C). These results validate the use of small molecule inhibitors on the PamChip peptide array and support and extend previous studies where the capacity of sorafenib to inhibit RET (both WT and the gate-keeper mutant V804M) was evaluated in other experimental systems (10, 22, 23).
Validation of the Oligopeptide Kinase Array in Cell-based Experiments Revealed Increased FAK Tyr-576/577 Phosphorylation Induced by RET V804M
To validate the results obtained with the PamChip array in cell-based experiments, human embryonic kidney HEK293 cells were transiently transfected with empty vector (pCMV), full-length RET V804M, or RET C634R. RET C634R contains a wild-type kinase domain but a mutated cysteine in the extracellular domain, which results in constitutive receptor dimerization and activation independent of ligand. Levels of RET phosphorylation at three specific tyrosines (positions 905, 981, and 1062) together with the tyrosine phosphorylation status of downstream targets were evaluated in the presence of PP1, Glivec (STI571), sorafenib, or vehicle (DMSO) (Fig. 6). Despite showing an efficient inhibitory effect at nanomolar concentrations against RET, PP1 and sorafenib target kinases other than RET (10, 22, 23). In vehicle-treated cells, RET V804M displayed increased levels of RET phosphorylation at Tyr-1062, Tyr-981, and Tyr-905 when compared with RET C634R that contains a wild-type kinase domain. PP1 inhibited RET C634R phosphorylation, whereas RET V804R was insensitive to the drug. STI571 was a poor RET inhibitor at physiologically relevant concentrations, whereas sorafenib was able to block tyrosine phosphorylation of both RET C634R and RET V804M.
FIGURE 6.

Validation of the oligopeptide kinase arrays in cell-based experiments. HEK293 cells transfected with empty vector (pCMV), RET C634R, or RET V804M were serum-starved (overnight) and then treated for 90 min with the inhibitors PP1 (1 μm), Glivec (10 μm), sorafenib (500 nm), or vehicle control (DMSO, 1:1000). Whole cell extracts were subject to Western blot analysis using the indicated antibodies. Data are representative of two independent experiments.
Levels of phosphorylated RET downstream targets were also evaluated. First, FAK Tyr-576/577 phosphorylation induced by RET C634R or RET V804M mutants was investigated. In vehicle-treated cells, FAK phosphorylation was induced by both mutants, but in the case of RET V804M, FAK Tyr-576/577 phosphorylation was enhanced (Fig. 6). PP1 treatment only affected FAK Tyr-576/577 phosphorylation induced by RET C634R, whereas no effect was seen on FAK Tyr-576/577 phosphorylation induced by RET V804M. Again, sorafenib blocked FAK Tyr-576/577 phosphorylation induced by both RET C634R and RET V804M. Next, we examined the STAT3 pathway. In vehicle-treated cells, levels of STAT3 Tyr-705 phosphorylation were higher in cells expressing RET V804M compared with RET C634R. PP1 treatment reduced STAT3 Tyr-705 phosphorylation induced by RET C634R but not by the gatekeeper mutant RET V804M. Glivec (STI571) had no effect; however, sorafenib treatment inhibited STAT3 Tyr-705 phosphorylation regardless of the RET mutation status. Finally, the SRC pathway was also analyzed. In vehicle-treated cells, phosphorylation of SRC Tyr-416 was only observed by RET V804M mutant and not by RET C634R. Again, PP1 had little effect on SRC phosphorylation induced by RET V804M, whereas sorafenib reduced SRC Tyr-416 phosphorylation to basal levels. As controls, we also analyzed the activation of the MAPK-ERK1/2, SAPK-p38, and JNK pathways and found that levels of ERK1/2 and p38 phosphorylation were equal between RET C634R and RET V804M mutants, whereas higher levels of JNK1 (lower 47 kDa band) phosphorylation correlated with RET V804M.
FAK Phosphorylation Induced by Specific MEN2-RET Mutants in HEK293 Cells
The molecular mechanisms that connect the different MEN2-associated RET mutants with their oncogenic phenotype are not well understood. Here, we investigated if the ability of RET V804M kinase mutant to activate FAK was recapitulated by other oncogenic mutations. First, to have a global view of the phosphotyrosine signaling status induced by WT RET and the three different RET oncogenic mutants (RET C634R, RET V804M, and RET M918T), total phosphotyrosine levels in whole cell extracts were monitored using the panphosphotyrosine antibody 4G10 (Fig. 7A). The MEN2B RET M918T mutant induced very high levels of tyrosine phosphorylation-driven signaling. These data correlate with the higher transformation potential and more aggressive phenotype seen in MEN2B patients (9). RET V804M (a gatekeeper and oncogenic mutant, associated with familial form medullary thyroid carcinoma) and RET C634R (a MEN2A-associated mutant) followed in decreasing order. Next, we analyzed the downstream pathways using phospho-specific antibodies (Fig. 7B). In the absence of GDNF and GFRα1, transfection of WT RET induced low levels of FAK Tyr-576/577 phosphorylation, which were increased by covalent dimerization induced by RET C634R. The kinase-activated mutants RET V804M and RET M918T displayed the highest levels of FAK phosphorylation. SRC Tyr-416 phosphorylation was only induced by kinase-activated RET mutants, the level of induced phosphorylation being higher by the MEN2B RET M918T. Finally, all of the oncogenic RET mutants induced higher levels of ERK1/2 phosphorylation compared with WT RET. Together, these results indicate that phosphorylation of FAK at Tyr-576/577 and SRC on Tyr-416 are potentiated by oncogenic mutations and in particular the MEN2B-RET M918T mutant.
FIGURE 7.

FAK phosphorylation induced by specific MEN2-RET mutants in HEK293 cells. HEK293 cells transfected with empty vector (pCMV), RET WT, RET C634R, RET V804M, or RET M918T (full-length cDNAs) were serum-starved overnight. Whole cell extracts were subject to Western blot analysis using total phosphotyrosine antibody 4G10 (A) or the indicated antibodies (B). Data are representative of two independent experiments.
FAK Inhibition or Silencing Impairs RET Phosphorylation and RET-dependent Cell Proliferation
To test the biological importance of the reciprocal RET-FAK transactivation, established HEK293 cells expressing a constitutively active RET mutant (RET C634R) were treated with the FAK kinase inhibitor NVP-TAE226 (Fig. 8A). FAK Tyr-576/577 phosphorylation was inhibited at nanomolar concentrations. Importantly, this compound also inhibited RET tyrosine phosphorylation, as indicated by decreased levels of phosphorylation of RET Tyr-905 and RET Tyr-1062 (Fig. 8A) at concentrations that do not affect RET kinase activity directly (Fig. 2B). In addition, down-regulation of FAK expression by siRNA oligonucleotides resulted in a decrease of RET phosphorylation at Tyr-905 and Tyr-1062 (Fig. 8B) and, crucially, in a significant reduction of cell survival (Fig. 8, C and D). Together, these data demonstrate that the reciprocal RET-FAK transphosphorylation mechanism is functional in cells expressing full-length RET and FAK.
FIGURE 8.

FAK inhibition or siRNA-mediated silencing impacts on RET phosphorylation and in RET-mediated cell proliferation. A, whole cell extracts from serum-starved (overnight) HEK293 cells expressing oncogenic RET (RET C634R) treated with increasing amounts of NVP-TAE226 for 90 min or vehicle-treated cells (DMSO control, 1:1000) were analyzed by Western blotting using the indicated antibodies. Data shown are representative of three independent experiments. B, HEK293 cells expressing oncogenic RET (RET C634R) were mock-transfected or transfected with non-targeting control siRNA or FAK siRNA. The following day, cells were serum-starved, and total cell extracts (72 h) were subjected to Western blot analysis using the indicated antibodies. Data are representative of two independent experiments. C, parental HEK293 cells or HEK293 cells stably expressing empty vector (pCMV) or oncogenic RET (RET C634R) were transfected with control (siCON) or FAK siRNA (siFAK). Survival fractions (ratio between siFAK- versus siCON-transfected cells) after 3 days were calculated using Cell Titer Glow. Data shown (mean ± S.E. (error bars)) are representative of four independent experiments. ***, p < 0.001. D, representative phase-contrast images of cells analyzed in C are depicted.
FAK Inhibition Impacts on RET Phosphorylation and the Phenotype of MZ-CRC-1 Cancer Cells
The medullary thyroid tumor cell line MZ-CRC-1 was derived from a patient with a RET M918T mutant allele. In these cells, the FAK kinase inhibitor NVP-TAE226 blocked FAK Tyr-576/577 phosphorylation at nanomolar concentrations. Importantly, at concentrations at which this inhibitor has no effect on RET M918T (24), this compound also inhibited RET tyrosine phosphorylation and downstream signaling, as indicated by decreased levels of phosphorylation of RET Tyr-1062, RET Tyr-905, and ERK1/2 (Fig. 9A), demonstrating that FAK can regulate RET phosphorylation and signaling in tumor cells as well as transfected HEK293 cells.
FIGURE 9.

The FAK inhibitor NVP-TAE226 impacts on RET signaling and the phenotype of MZ-CRC-1 tumor cells. A, whole cell extracts from serum-starved (overnight) MZ-CRC-1 cells treated with increasing amounts of NVP-TAE226 for 90 min or vehicle-treated cells (DMSO control, 1:1000) were analyzed by Western blotting using the indicated antibodies. Data shown are representative of three independent experiments. B, 2 × 105 MZ-CRC-1 cells were plated in 6-well plates and treated with increasing amounts of NVP-TAE226 or vehicle (DMSO, 1:1000) for 8 days. Data represent the number of cells (× 104) at the indicated time points ± S.E. (error bars) from two experiments in triplicate. C, MZ-CRC-1 cells were plated subconfluently onto plastic or glass coverslips in the presence of NVP-TAE226 (1 μm) or vehicle (DMSO, 1:1000) for 3 days, and phase-contrast or confocal microscopy images were captured, respectively. For confocal microscopy images of MZ-CRC-1 cells, actin filaments were stained with Alexa647-phalloidin (green), and nuclei were counterstained with DAPI (blue). The mean number of cells with elongated tails per field (10 fields from two independent experiments) was quantified ± S.E. Scale bar, 150 μm. D, MZ-CRC-1 cells were plated subconfluently on glass coverslips and cultured in the presence of NVP-TAE226 (1 μm) or vehicle (DMSO, 1:1000) for 3 days, after which cells were stained with an anti-vinculin antibody followed by Alexa555 anti-mouse Ig (red). Actin filaments were stained with Alexa647-phalloidin (green). Scale bar, 75 μm (left) or 37.5 μm (right). E, MZ-CRC-1 cells were plated as single cells on a Matrigel bed in the presence of NVP-TAE226 or vehicle (DMSO, 1:1000) and cultured for 15 days. Data represent -fold increase in colony number at day 15 of vehicle versus NVP-TAE226-treated cells from two experiments performed in duplicate. Representative phase-contrast images of cells taken at days 1, 7, and 15 are shown. Scale bar, 150 μm.
In cell proliferation assays, treatment with NVP-TAE226 at low micromolar concentrations inhibited MZ-CRC-1 cell proliferation (Fig. 9B). Closer examination of the cells revealed that the NVP-TAE226-treated cells had a more elongated phenotype and an increased number of long cellular processes (Fig. 9C). To examine whether this phenotype was associated with a defect in focal adhesion formation, cells were treated with NVP-TAE226 or vehicle for 72 h and then analyzed by confocal microscopy following staining with an anti-vinculin antibody and phalloidin (Fig. 9D). An increase in focal adhesion structures (enriched in vinculin and F-actin) was observed in NVP-TAE226-treated cells. This most likely reflects the decreased level of FAK protein following NVP-TAE226 treatment for 72 h (supplemental Fig. 3) and the consequent lack of efficient focal adhesion turnover, as was previously reported with the FAK-deficient mice (25). Finally, we analyzed the effect of NVP-TAE226 on MZ-CRC-1 cells grown on a thick bed of growth factor-enriched Matrigel (Fig. 9E). NVP-TAE226 decreased the number of colonies formed on this three-dimensional matrix (Fig. 9E) as well as the number of invasive fronts penetrating into the Matrigel (data not shown). Together, these data indicate that FAK kinase inhibitor NVP-TAE226 impacts on RET phosphorylation and the proliferation and phenotype of MZ-CRC-1 cancer cells.
DISCUSSION
Specific protein-protein interactions underlie cellular signal transduction and consequently many essential biological processes in the cell. The recognition of a short linear peptide sequence in one protein by a modular domain in another represents a common feature of macromolecular signaling complexes in cells. The importance of this mode of protein-protein interaction is reinforced by the large number of peptide-binding domains encoded by the human genome (26). Appropriate tyrosine kinase signaling depends on coordinated coupling of protein-protein interactions with catalytic activation (2, 26). Focal adhesion kinase (FAK) integrates signals from integrins and growth factor receptors to regulate cellular responses, including cell adhesion, migration, and survival (27). The precise mechanisms by which growth factor receptors initiate FAK activation are not fully understood, but integrin activation is known to promote FAK Tyr-397 phosphorylation (28). The phosphorylated Tyr-397 site and a proximal PXXP motif recruit and activate SRC via binding to its SH2 and SH3 domains, forming a signaling complex that phosphorylates p130Cas, paxillin, and other focal adhesion-associated proteins (19). SRC in turn phosphorylates Tyr-576 and Tyr-577 in the activation loop of the FAK kinase domain, resulting in fully activated FAK kinase. In addition, phosphorylation of FAK at Tyr-925 promotes Grb2 adaptor binding and is linked to the activation of the Ras-ERK2 mitogen-activated protein kinase cascade (29).
High density peptide arrays have emerged as useful tools to map domain-mediated protein-protein interaction networks (30, 31). In this study, we used peptides arrays to screen for substrates directly phosphorylated by WT and V804M mutant RET kinase in the presence or absence of several small molecule inhibitors. In this system, we identified FAK as a direct target of the RET kinase domain and demonstrated that FAK Tyr-576/577 phosphorylation was enhanced by the V804M RET oncogenic kinase. Several studies have previously suggested a potential RET-FAK functional interaction. In neuroblastoma cells, GDNF-stimulated RET resulted in tyrosine phosphorylation of paxillin, p130Cas, and FAK, all proteins associated with focal adhesions (32). In MTC-TT tumor cells, it was reported that treatment with the kinase inhibitor PP2 or with RET siRNA decreased the levels of RET phosphorylation and protein levels and decreased FAK phosphorylation (33). Further, immunoprecipitated RET from MTC-TT tumor cells and recombinant RET kinase domain have been shown to directly phosphorylate FAK on Tyr-576/577 and Tyr-925 but not on Tyr-397 (34). Our data extend these studies and provide important insights into the RET-FAK activation mechanism. We provide evidence of a direct and reciprocal 2-fold RET-FAK transactivation mechanism. First, RET directly phosphorylates FAK Tyr-576/577 in the activation loop but also phosphorylates Tyr-925 in the focal adhesion targeting domain. However, RET does not phosphorylate FAK on Tyr-397 in the linker region, which is the canonical priming site required for SRC kinase phosphorylation (19, 34). Second, a reciprocal phosphorylation of RET in trans by FAK, which is able to rescue the kinase-impaired RET K758M mutant, regulates RET tyrosine phosphorylation and its downstream signaling. Strikingly, FAK does not contain an SH2 or a PTB domain, and, despite the evidence of an interaction shown in this study and others (32–34), the nature of this protein-protein interaction mechanism was not known. Here we demonstrate that FAK binds RET kinase via its FERM domain. This is the first study to show a direct interaction of a FERM domain-containing protein with the RET receptor. Structural analysis reveals that the FERM domain of FAK binds to its own kinase domain, blocking access to the catalytic cleft and protecting the FAK activation loop from SRC phosphorylation (27). We hypothesize that conformational changes would be required to render FAK Tyr-576/Tyr-577 accessible for efficient phosphorylation by RET. It is certainly attractive to speculate that RET binding relieves FAK from its autoinhibitory state and thereby induces conformational changes that make activation loop Tyr-576/577 accessible for RET phosphorylation. Consistent with this, we show that RET Tyr-905, but not Tyr-900, is required for efficient RET-induced FAK Tyr-576/577 phosphorylation, indicating that phosphorylation events between both kinases could take place via activation loop phosphorylation in trans. This would represent the second step in the transactivation mechanism following the FAK FERM domain binding to RET kinase in the unphosphorylated state (priming).
In this study, we have described a direct and reciprocal RET-FAK transactivation mechanism in vitro and in tumor and transfected cell lines. In HEK293 cells expressing oncogenic RET, treatment with the FAK kinase inhibitor NVP-TAE226 and siRNA-mediated FAK down-regulation resulted in reduced RET phosphorylation levels and signaling as well as cell survival and proliferation. To further explore the role of this interaction in tumorigenesis, an experimental model based on the MZ-CRC-1 tumor cell line was used. In vitro experiments showed that treatment with the FAK inhibitor NVP-TAE226 inhibited FAK Tyr-576/577 phosphorylation and, importantly, RET tyrosine phosphorylation, downstream signaling, and cell proliferation. Because the inhibition of RET phosphorylation occurred at a concentration of NVP-TAE226 that does not affect RET kinase activity in in vitro kinase assays, these data support the hypothesis that RET phosphorylation by FAK can regulate RET function. NVP-TAE226-treated MZ-CRC-1 tumor cells showed a defect in migration and invasion that was associated with an increase in vinculin-rich focal adhesions. This is consistent with previous reports where cells derived from FAK-deficient mice were shown to have a migratory defect and an increased number of focal adhesions (35). These data suggest that the antiproliferative properties of NVP-TAE on MZ-CRC-1 tumor cells could be due to NVP-TAE disrupting the RET-FAK-mediated regulation of focal adhesion turnover and cell junction maintenance that is required for optimal epithelial cell growth and proliferation.
Importantly, these data also raise the question as to whether NVP-TAE226 could be used in the treatment of MEN2 patients, in particular for the MEN2B phenotype. FAK expression and levels of FAK tyrosine phosphorylation are up-regulated in several cancers, including those of the brain, ovary, colon, breast, prostate, liver, and thyroid (36–42). In the present study, the signaling profile of several MEN2-associated RET mutants showed that increased levels of phosphorylated FAK Tyr-576/577 are associated with oncogenic RET activity and that increased levels of both FAK Tyr-576/577 and SRC Tyr-416 phosphorylation correlated mainly with the MEN2B-associated RET M918T mutant, linking the hyperactivation of FAK and SRC to the more aggressive disease phenotype. Recently, NVP-TAE226 was shown to efficiently inhibit growth and invasion of glioma and ovarian cancer cells (24, 37, 43) and to induce apoptosis in breast cancer cell lines (44). In addition, treatment with this compound increased survival rates of animals with glioma xenografts (24) or ovarian tumor cell implants (37).
In conclusion, using a proteomic approach together with functional and biochemical analysis, we show that RET kinase can signal independently of proteins that contain SH2 or PTB domains, by binding to the FERM domain of FAK. This is followed by direct phosphorylation of the FAK activation loop Tyr-576 and Tyr-577, which in turn leads to reciprocal phosphorylation of RET in trans by FAK. RET-driven tumor cells are dependent on this mechanism because the FAK inhibitor NVP-TAE226 and siRNA-mediated FAK silencing impair RET phosphorylation and signaling and the survival of tumor and transfected cells expressing oncogenic RET. These data indicate that targeting oncogenic RET signaling with FAK inhibitors may be a useful alternative therapeutic treatment for MEN2 patients.
Supplementary Material
Acknowledgments
We thank Novartis for the NVP-TAE226 inhibitor, Rob Ruijtenbeek (PamGene) for helpful comments on the manuscript, Prof. M. Schaller for providing the GST-FERM-FAK plasmids, and Roger Georges and the Protein Production Facility (London Research Institute, Cancer Research UK) for helping with baculovirus expression of recombinant protein. The National Institute for Health Research Biomedical Research Centre is supported by the National Health Service.
This work was supported by grants from Breakthrough Breast Cancer (to C. M. I.), Ministerio de Educación y Ciencia of Spain Grant EX2006_1341 (to I. P.-M.), and Cancer Research UK (to I. P.-M. and N. Q. M.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–3.
- RTK
- receptor tyrosine kinase
- PTB
- phosphotyrosine binding
- SH2
- Src homology 2
- FAK
- focal adhesion kinase
- MEN2
- multiple endocrine neoplasia type 2
- MTC
- medullary thyroid carcinoma
- GDNF
- glial cell-derived neurotrophic factor.
REFERENCES
- 1. Schlessinger J. (2000) Cell 103, 211–225 [DOI] [PubMed] [Google Scholar]
- 2. Pawson T., Kofler M. (2009) Curr. Opin. Cell Biol. 21, 147–153 [DOI] [PubMed] [Google Scholar]
- 3. Smith M. J., Hardy W. R., Murphy J. M., Jones N., Pawson T. (2006) Mol. Cell. Biol. 26, 8461–8474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. de Groot J. W., Links T. P., Plukker J. T., Lips C. J., Hofstra R. M. (2006) Endocr. Rev. 27, 535–560 [DOI] [PubMed] [Google Scholar]
- 5. Schuchardt A., D'Agati V., Larsson-Blomberg L., Costantini F., Pachnis V. (1994) Nature 367, 380–383 [DOI] [PubMed] [Google Scholar]
- 6. Manié S., Santoro M., Fusco A., Billaud M. (2001) Trends Genet. 17, 580–589 [DOI] [PubMed] [Google Scholar]
- 7. Airaksinen M. S., Titievsky A., Saarma M. (1999) Mol. Cell Neurosci. 13, 313–325 [DOI] [PubMed] [Google Scholar]
- 8. Zwick E., Bange J., Ullrich A. (2001) Endocr. Relat. Cancer 8, 161–173 [DOI] [PubMed] [Google Scholar]
- 9. Plaza-Menacho I., Burzynski G. M., de Groot J. W., Eggen B. J., Hofstra R. M. (2006) Trends Genet. 22, 627–636 [DOI] [PubMed] [Google Scholar]
- 10. Carlomagno F., Guida T., Anaganti S., Vecchio G., Fusco A., Ryan A. J., Billaud M., Santoro M. (2004) Oncogene 23, 6056–6063 [DOI] [PubMed] [Google Scholar]
- 11. Hilhorst R., Houkes L., van den Berg A., Ruijtenbeek R. (2009) Anal. Biochem. 387, 150–161 [DOI] [PubMed] [Google Scholar]
- 12. Sikkema A. H., Diks S. H., den Dunnen W. F., ter Elst A., Scherpen F. J., Hoving E. W., Ruijtenbeek R., Boender P. J., de Wijn R., Kamps W. A., Peppelenbosch M. P., de Bont E. S. (2009) Cancer Res. 69, 5987–5995 [DOI] [PubMed] [Google Scholar]
- 13. Maat W., el Filali M., Dirks-Mulder A., Luyten G. P., Gruis N. A., Desjardins L., Boender P., Jager M. J., van der Velden P. A. (2009) Br. J. Cancer 101, 312–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mologni L., Sala E., Riva B., Cesaro L., Cazzaniga S., Redaelli S., Marin O., Pasquato N., Donella-Deana A., Gambacorti-Passerini C. (2005) Protein Expr. Purif. 41, 177–185 [DOI] [PubMed] [Google Scholar]
- 15. Sala E., Mologni L., Cazzaniga S., Papinutto E., Gambacorti-Passerini C. (2006) Int. J. Biol. Macromol. 39, 60–65 [DOI] [PubMed] [Google Scholar]
- 16. Knowles P. P., Murray-Rust J., Kjaer S., Scott R. P., Hanrahan S., Santoro M., Ibáñez C. F., McDonald N. Q. (2006) J. Biol. Chem. 281, 33577–33587 [DOI] [PubMed] [Google Scholar]
- 17. Thomas J. W., Cooley M. A., Broome J. M., Salgia R., Griffin J. D., Lombardo C. R., Schaller M. D. (1999) J. Biol. Chem. 274, 36684–36692 [DOI] [PubMed] [Google Scholar]
- 18. Plaza-Menacho I., Morandi A., Robertson D., Pancholi S., Drury S., Dowsett M., Martin L. A., Isacke C. M. (2010) Oncogene 29, 4648–4657 [DOI] [PubMed] [Google Scholar]
- 19. Parsons J. T. (2003) J. Cell Sci. 116, 1409–1416 [DOI] [PubMed] [Google Scholar]
- 20. Parsons J. T., Schaller M. D., Hildebrand J., Leu T. H., Richardson A., Otey C. (1994) J. Cell Sci. Suppl. 18, 109–113 [DOI] [PubMed] [Google Scholar]
- 21. Ceccarelli D. F., Song H. K., Poy F., Schaller M. D., Eck M. J. (2006) J. Biol. Chem. 281, 252–259 [DOI] [PubMed] [Google Scholar]
- 22. Carlomagno F., Anaganti S., Guida T., Salvatore G., Troncone G., Wilhelm S. M., Santoro M. (2006) J. Natl. Cancer Inst. 98, 326–334 [DOI] [PubMed] [Google Scholar]
- 23. Plaza-Menacho I., Mologni L., Sala E., Gambacorti-Passerini C., Magee A. I., Links T. P., Hofstra R. M., Barford D., Isacke C. M. (2007) J. Biol. Chem. 282, 29230–29240 [DOI] [PubMed] [Google Scholar]
- 24. Liu T. J., LaFortune T., Honda T., Ohmori O., Hatakeyama S., Meyer T., Jackson D., de Groot J., Yung W. K. (2007) Mol. Cancer Ther. 6, 1357–1367 [DOI] [PubMed] [Google Scholar]
- 25. Ilić D., Furuta Y., Kanazawa S., Takeda N., Sobue K., Nakatsuji N., Nomura S., Fujimoto J., Okada M., Yamamoto T. (1995) Nature 377, 539–544 [DOI] [PubMed] [Google Scholar]
- 26. Pawson T., Nash P. (2003) Science 300, 445–452 [DOI] [PubMed] [Google Scholar]
- 27. Lietha D., Cai X., Ceccarelli D. F., Li Y., Schaller M. D., Eck M. J. (2007) Cell 129, 1177–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schaller M. D., Hildebrand J. D., Shannon J. D., Fox J. W., Vines R. R., Parsons J. T. (1994) Mol. Cell. Biol. 14, 1680–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lim S. T., Mikolon D., Stupack D. G., Schlaepfer D. D. (2008) Cell Cycle 7, 2306–2314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schutkowski M., Reineke U., Reimer U. (2005) Chembiochem 6, 513–521 [DOI] [PubMed] [Google Scholar]
- 31. Reineke U., Volkmer-Engert R., Schneider-Mergener J. (2001) Curr. Opin. Biotechnol. 12, 59–64 [DOI] [PubMed] [Google Scholar]
- 32. Murakami H., Iwashita T., Asai N., Iwata Y., Narumiya S., Takahashi M. (1999) Oncogene 18, 1975–1982 [DOI] [PubMed] [Google Scholar]
- 33. Panta G. R., Nwariaku F., Kim L. T. (2004) Surgery 136, 1212–1217 [DOI] [PubMed] [Google Scholar]
- 34. Panta G. R., Du L., Nwariaku F. E., Kim L. T. (2005) Surgery 138, 269–274 [DOI] [PubMed] [Google Scholar]
- 35. Ilic D., Furuta Y., Suda T., Atsumi T., Fujimoto J., Ikawa Y., Yamamoto T., Aizawa S. (1995) Biochem. Biophys. Res. Commun. 209, 300–309 [DOI] [PubMed] [Google Scholar]
- 36. Bonome T., Lee J. Y., Park D. C., Radonovich M., Pise-Masison C., Brady J., Gardner G. J., Hao K., Wong W. H., Barrett J. C., Lu K. H., Sood A. K., Gershenson D. M., Mok S. C., Birrer M. J. (2005) Cancer Res. 65, 10602–10612 [DOI] [PubMed] [Google Scholar]
- 37. Halder J., Lin Y. G., Merritt W. M., Spannuth W. A., Nick A. M., Honda T., Kamat A. A., Han L. Y., Kim T. J., Lu C., Tari A. M., Bornmann W., Fernandez A., Lopez-Berestein G., Sood A. K. (2007) Cancer Res. 67, 10976–10983 [DOI] [PubMed] [Google Scholar]
- 38. Cance W. G., Harris J. E., Iacocca M. V., Roche E., Yang X., Chang J., Simkins S., Xu L. (2000) Clin. Cancer Res. 6, 2417–2423 [PubMed] [Google Scholar]
- 39. Han N. M., Fleming R. Y., Curley S. A., Gallick G. E. (1997) Ann. Surg. Oncol. 4, 264–268 [DOI] [PubMed] [Google Scholar]
- 40. Natarajan M., Hecker T. P., Gladson C. L. (2003) Cancer J. 9, 126–133 [DOI] [PubMed] [Google Scholar]
- 41. Owens L. V., Xu L., Dent G. A., Yang X., Sturge G. C., Craven R. J., Cance W. G. (1996) Ann. Surg. Oncol. 3, 100–105 [DOI] [PubMed] [Google Scholar]
- 42. Tremblay L., Hauck W., Aprikian A. G., Begin L. R., Chapdelaine A., Chevalier S. (1996) Int. J. Cancer 68, 164–171 [DOI] [PubMed] [Google Scholar]
- 43. Shi Q., Hjelmeland A. B., Keir S. T., Song L., Wickman S., Jackson D., Ohmori O., Bigner D. D., Friedman H. S., Rich J. N. (2007) Mol. Carcinog. 46, 488–496 [DOI] [PubMed] [Google Scholar]
- 44. Golubovskaya V. M., Virnig C., Cance W. G. (2008) Mol. Carcinog. 47, 222–234 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.