

Abstract
Podocytes are dynamic polarized cells that lie on the surface of glomerular capillaries and comprise an essential component of the glomerular filitration barrier. Podocytes are affected in the earliest stages of diabetic nephropathy and insulin signaling to podocytes is essential for normal glomerular function. Large-conductance Ca2+-activated K+ channels (BKCa channels) encoded by the Slo1 gene are expressed in podocytes in a complex with multiple glomerular slit diaphragm proteins including nephrin, TRPC6 channels, and several different actin-binding proteins. Here we show that insulin increases cell surface expression of podocyte BKCa channels, which is accompanied by a corresponding increase in the density of current flowing through these channels. Insulin stimulation of BKCa channels was detectable in 15 minutes and required activation of both Erk and Akt signaling cascades. Exposure to high glucose (36.1 mM) for 24 hr caused a marked reduction in the steady-state surface expression of BKCa channels as well as of the slit diaphragm signaling molecule nephrin. High glucose treatment also abolished the stimulatory effects of insulin on BKCa current density, although insulin continued to increase phosphorylation of Erk and Akt under those conditions. Therefore, in contrast to most other cell types, high glucose abrogates the effects of insulin in podocytes at relatively distal steps in its signaling pathway. Insulin stimulation of BKCa channels in podocytes may prepare podocytes to adapt to changes in pressure gradients that occur during postprandial hyperfiltration.
Keywords: podocytes, diabetes, K channel, trafficking, nephrin
INTRODUCTION
Polarized cells known as podocytes are essential components of the glomerular filtration barrier in the kidney. These cells extend primary processes that ramify and terminate in foot processes that attach to the glomerular basement membrane. The specialized contacts between adjacent podocyte foot process form porous structures known as slit diaphragms that are 30–40 nm in width (Rodewald and Karnovsky, 1974). The glomerular filtration barrier is permeable to small solutes, but normally inhibits passage of plasma proteins such as albumin into the ultrafiltrate. Type 1 and type 2 diabetes can lead to changes in the structure of podocyte foot processes, loss of podocytes, reduction in slit diaphragm proteins, and failure of the glomerular filtration barrier (Pagtalunan et al., 1997; Steffes et al., 2001; Doublier et al., 2003). This in turn triggers a cascade of events that can ultimately lead to loss of the entire nephron and to end-stage renal failure (Kriz and Lehir, 2005; Shankland, 2006).
Podocytes are continually exposed to substantial mechanical forces owing to cyclical distension of the glomerular capillary wall and fluid shear stress (Endlich and Endlich, 2006; Peti-Peterdi and Sipos, 2010). The arrangement of F-actin, myosin, and α-actinin in podocyte foot processes has been proposed to facilitate adaptation of the glomerulus to changes in pressure gradients, or to modify the surface area for filtration (Drenckhahn and Franke, 1988; Kriz et al., 1994; Kriz et al., 1996). There is evidence that Ca2+ dynamics play a role in mediating adaptive responses in podocyte function (Peti-Peterdi, 2006; Saleem et al., 2008), although these cells are not excitable. Podocytes express large-conductance Ca2+-activated K+ channels (BKCa channels) encoded by the KCNMA1 gene (also known as Slo1 and KCa1.1) which are likely to play a role in regulating these dynamics (Morton et al., 2004; Kim et al., 2008; Dryer and Reiser, 2010). BKCa channels contain binding sites for Ca2+ on a large cytosolic domain in the COOH-terminus of the pore-forming subunits. Binding of Ca2+ to these sites allows for voltage-dependent gating of BKCa channels at physiological membrane potentials. Within glomeruli, BKCa channels are expressed in podocytes and mesangial cells (Morton et al., 2004; Kim et al., 2010). It is notable that in addition to Ca2+-dependent gating, the BKCa channels of cultured human podocytes also become active in response to membrane stretch (Morton et al., 2004). Moreover, they can be shown to interact directly with Ca2+-permeable TRPC6 channels in podocytes (Kim et al., 2009a). BKCa channels in podocytes may play a role to facilitate Ca2+ influx through non-selective cation channels such as TRPC6 (Dryer and Reiser, 2010), which require a large driving force in order to efficiently transport Ca2+ into cells (Estacion et al., 2006). The very large conductance of BKCa channels—greater than 100 pS with physiological ionic gradients—ensures that activation of a small number of them can dominate the electrical behavior of an electrically compact cell by effectively clamping the membrane to the potassium equilibrium potential.
Podocytes are direct targets for insulin, which has been shown to cause an acute increase in glucose uptake into cultured podocyte cell lines and in human podocytes ex vivo owing to translocation of the glucose transporters GLUT1 and GLUT4 to the plasma membrane (Coward et al., 2005, 2007). In podocytes this effect of insulin requires nephrin, a protein that is not expressed in skeletal muscle or adipocytes (Coward et al., 2007). Nephrin is a transmembrane cell adhesion molecule whose ectodomains form an essential component of glomerular slit diaphragms (Kestilä et al., 1998; Putaala et al., 2001). The cytoplasmic domains of nephrin form a platform for assembly of signaling complexes, including the p85 subunit of phosphoinositide-3’-OH-kinase (PI3K) and nephrin signaling is functionally connected to cytoskeletal dynamics in podocytes (Benzing et al., 2004). Importantly, insulin signaling specifically in podocytes has been recently shown to be essential for normal glomerular filtration (Welsh et al., 2010).
In several recent studies we have shown that the steady-state surface expression of BKCa channels in podocytes requires nephrin (Kim et al., 2008) as well as other proteins that are normally expressed in the slit diaphragm domains of foot processes (Kim et al., 2009a, 2009b, 2010; Ridgway et al., 2009). We have also shown that growth factors can stimulate surface expression of neuronal BKCa channels (Dryer et al., 2003; Chae et al., 2005). We now report that insulin increases macroscopic currents through endogenously expressed podocyte BKCa channels owing at least in part to an increase in their steady-state expression on the cell surface. Culturing podocytes in the presence of very high glucose (36.1 mM) markedly suppressed basal surface expression of both BKCa channels and nephrin, and prevented insulin from increasing BKCa current density in podocytes. Surprisingly, high glucose did not abrogate more proximal steps in insulin signaling in podocytes, as insulin-evoked phosphorylation of Erk and Akt was similar to that observed in cells maintained in normal glucose. These results suggest that metabolic changes in diabetes could affect glomerular filtration by altering ion channel expression in podocyte foot processes.
MATERIALS AND METHODS
Cell culture protocols
Mouse podocyte cell lines (obtained from Dr. Peter Mundel of the University of Miami Miller School of Medicine) were maintained in RPMI-1640 medium containing 11.1 mM glucose supplemented with 10% fetal bovine serum and 100U/ml penicillin-streptomycin, with or without recombinant mouse γ-interferon, in humidified 5% CO2 incubators. The cell line was allowed to propagate at 33°C. Removal of γ-interferon and temperature switch to 37°C induced podocyte differentiation and expression of podocyte markers in 14 days (Kim et al., 2009a, 2009b, 2010; Ridgway et al., 2009). Differentiated cells were transferred to serum-free media 24 hr before experiments with insulin. Cells were then treated with 100 nM recombinant human insulin for various lengths of time before analyses. High glucose media were prepared by adding 25 mM extra glucose to RPMI-1640 media (to make a total of 36.1 mM D-glucose). In some control experiments we used normal RPMI-1640, but in most of the experiments control cells were maintained in RPMI-1640 with 25 mM mannitol added to control for osmotic effects of high glucose.
Immunoblot analysis and cell-surface biotinylation assays
These were carried out as described in detail previously (Kim et al., 2008, 2010). Briefly, podocyte lysates were separated by SDS-PAGE on 10% gels and transferred to filters. Blots were blocked, incubated with a primary antibody overnight at 4 °C, washed again, and the membrane was incubated with horseradish peroxidase-conjugated secondary antibody for 1 h at room temperature. Proteins were visualized using a chemiluminescent substrate. The primary antibodies were anti-nephrin (Santa Cruz), anti-Slo1 (Chemicon), and anti-β-actin. In addition, immunoblot analyses of Akt and Erk1/2 phosphorylation were carried out using antibody pairs that recognize phosphorylated (active) and total forms of these enzymes (Cell Signaling Technology, Danvers, MA). For cell-surface biotinylation assays, intact podocytes were treated with a membrane impermeable biotinylation reagent, sulfo-N-hydroxy-succinimidobiotin (Pierce Biotechnology, Rockford, IL) (1 mg/ml) for 1 h. The reaction was stopped, cells were lysed, and biotinylated proteins from the cell surface were recovered by incubation with immobilized streptavidin-agarose beads. A sample of the initial cell lysate was retained for analysis of total proteins. These samples were quantified by immunoblot analysis using anti-Slo1 or anti-nephrin, followed by densitometric analysis using Image J software . All experiments were repeated at least three times.
Electrophysiology
Whole cell recordings were made as described in detail previously (Kim et al., 2008, 2009a, 2009b, 2010; Ridgway et al., 2009). Briefly, the bath solution contained 150 mM NaCl, 5.4 mM KCl, 0.8 mM MgCl2, 5.4 mM CaCl2, 10 mM HEPES, pH 7.4. Pipette solutions contained 10 mM NaCl, 125 mM KCl, 6.2 mM MgCl2, 10 mM HEPES, pH 7.2, and 5 μM free Ca2+. Currents were evoked by a series of depolarizing voltage steps from a holding potential of -60 mV. The recording electrodes had resistances of 3–4 MΩ and it was possible to compensate up to 85% of this without introducing oscillations into the current output of the patch clamp amplifier. Whole cell currents were not observed when recording pipettes contained solutions with no added Ca2+ and 10 mM EGTA (data not shown). PD98059 and LY294002 were dissolved in DMSO before adding to culture media.
Statistics
All quantitative data are presented as mean ± s.e.m. Data were analyzed by Student’s t-test, and by one-way ANOVA followed by post hoc analysis, when multiple comparisons were made. Throughout, p < 0.05 is regarded as significant.
RESULTS
In initial experiments we examined the effect of insulin (100 nM) on the steady-state surface expression of the pore-forming subunits of BKCa channels (Slo1 proteins) in differentiated cells of a mouse podocyte cell line. Cell-surface biotinylation assays showed that insulin exposure for 15 min could evoke an increase in the surface expression of Slo1 relative to the total amount of Slo1 expressed in podocytes (Fig. 1). Densitometric quantification of three repetitions of this experiment (Fig. 1B) showed a greater effect with longer insulin exposures of 3 hr and 24 hr. A stimulatory effect of insulin could also be seen in whole-cell recordings of BKCa channels from podocytes made using recording electrodes filled with a solution containing 5 μM free Ca2+ (Fig. 2). Including micromolar concentrations of free Ca2+ in the recording pipettes allowed measurement of BKCa current density in response to depolarizing voltage steps from a holding potential of −60 mV. In previous studies we have shown that the outward currents evoked using these protocols are entirely attributable to BKCa channels (Kim et al., 2008, 2010) and we observed here that the currents evoked in insulin-treated cells are completely blocked by the selective BKCa inhibitor paxilline (Fig. 2A, B). The results obtained by cell-surface biotinylation assays and whole cell recordings are not quantitatively identical. This may reflect differences intrinsic to the two methods, especially given the nonlinearity and high amplification of the chemiluminescent detection systems. However, we cannot exclude that insulin may also modulate activity of BKCa channels already in the plasma membrane as well as stimulating trafficking, especially given that insulin has been shown to regulate BKCa gating in other cell types (O’Malley and Harvey, 2004, 2007). However, we did not observe any effect of insulin on the voltage-dependence (Fig. 2C) or kinetics (Fig. 2D) of BKCa activation in podocyte cell lines, and insulin increased mean currents at all membrane potentials where they could be detected.
Figure 1.
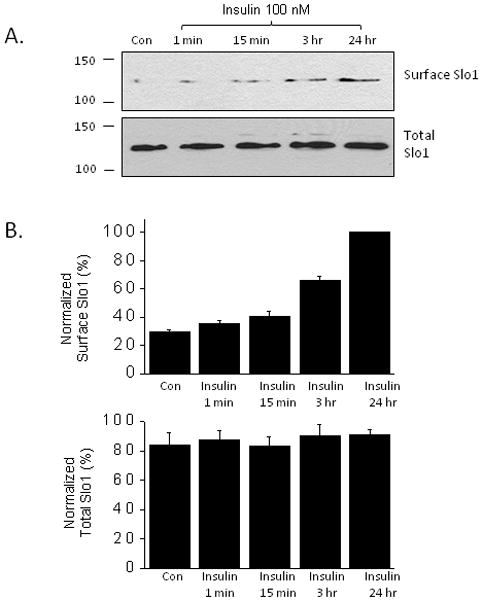
Exposure to insulin (100 nM) stimulates increases the steady-state expression of the pore-forming subunits of BKCa channels (Slo1 proteins) on the cell surface of podocytes. A, Example of typical cell-surface biotinylation assay with duration of insulin treatments as indicated. B, Densitometric quantification of three repititons of this experiment. In this and subsequent experiments, figures depict mean ± s.e.m.
Figure 2.

Insulin increases macroscopic currents carried by BKCa channels in cultured podocytes. A, Examples of families of currents evoked from a holding potential of -60 mV in a control cell and in a different cell exposed to insulin for 24 hr. The voltage-clamp protocol is denoted above traces. Recording pipettes contained 5 μM free Ca2+. Our previous studies (22) have shown that these protocols isolate macroscopic BKCa. Note complete inhibition of evoked outward currents in insulin-treated cells by bath application of the BKCa inhibitor paxilline (1 μM). B, Quantification of current densities. Numbers in parentheses indicate number of cells tested in each group. Asterisk indicates P < 0.05 by one-way ANOVA followed by Scheffe’s post hoc test. C, Mean currents (left) and conductance ratios (right) ± s.e.m. of control cells and cells treated for 24 hr with insulin. The ordinate G/G80 is the conductance at each voltage divided by the conductance evoked by a step depolarization to +80 mV. Note lack of effect of insulin on voltage-dependence of macroscopic BKCa. D, Typical currents from a control cell and from an insulin-treated cell (24 hr) with amplitudes normalized and overlaid so as to illustrate that activation kinetics are not altered by insulin.
The stimulatory effects of insulin on BKCa channels were associated with activation of mitogen-activated protein kinase (ERK1/2) and PI3K/Akt signaling cascades. Using immunoblot analysis we observed an increase in the phosphorylated (active) form of Erk1/2 (Fig. 3A) that could be seen as early as 1 min after insulin application and that was largely inhibited by pretreatment with the MEK1 inhibitor PD98059 (50 μM) (Fig. 3B, C). Moreover, insulin-evoked stimulation of BKCa channels was blocked by PD98059 as assessed by cell-surface biotinylation assays (Fig. 3D) and by whole-cell recordings (Fig. 3E). Insulin also evoked a rapid increase in the phosphorylated (active) form of Akt (Fig. 4A) and this effect was reduced by pretreatment with the PI3K inhibitor LY294002 (50 μM) (Fig. 4B, C). Moreover, insulin-evoked increases in Slo1 surface expression (Fig 4D) and macroscopic BKCa currents (Fig. 4E) were blocked by pretreatment of podocytes with LY294002 (Fig. 3F). These data indicate that both of these signaling pathways are necessary for insulin modulation of BKCa.
Figure 3.
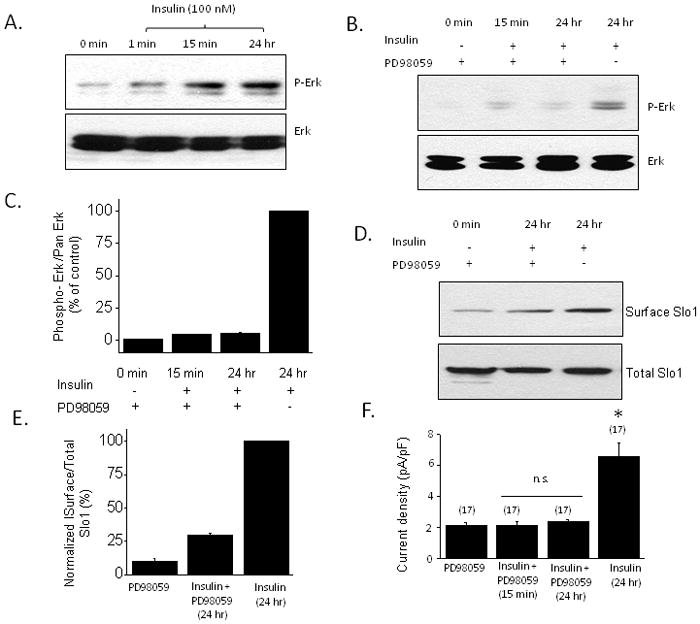
Insulin-evoked Erk is necessary for stimulation of current through podocyte BKCa channels. A, Typical immunoblot analysis showing rapid increase in Erk phosphorylation evoked by 100 nm insulin. B, This increase is markedly attenuated by pretreating cells with 50 μM PD98059, an inhibitor of MEK1. C, Densitometric analysis of three repetitions of the experiment shown in B. D, Cell-surface biotinylation assay showing that the effect insulin on BKCa trafficking is markedly attenuated by pretreatment with PD98059. E, Densitometric quantification of three repetitions of the experiment shown in D. F, Insulin stimulation of current through BKCa is blocked by pretreatment with PD98059. Asterisks indicate P < 0.05.
Figure 4.
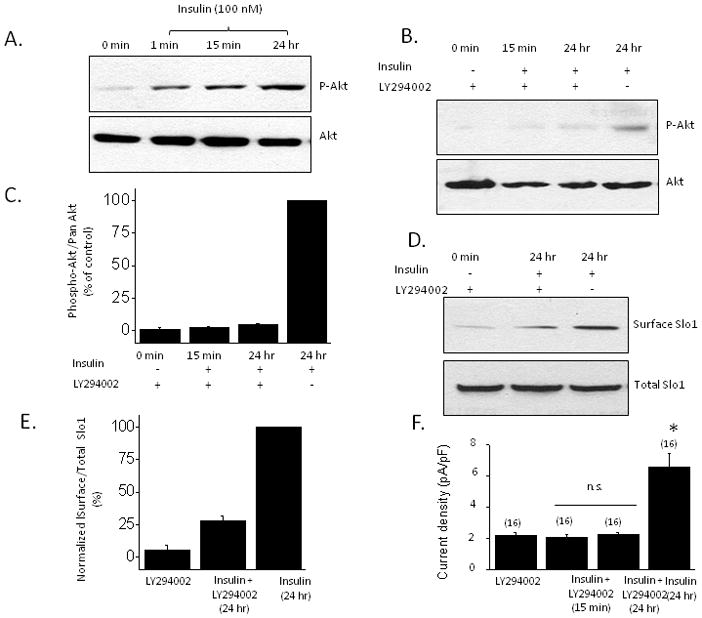
Insulin-evoked activation of PI3K/Akt signaling cascade is necessary for stimulation of current through podocyte BKCa channels. A, Typical immunoblot analysis showing rapid increase in Akt phosphorylation evoked by 100 nm insulin. B, This increase is markedly attenuated by pretreating cells with 50 μM LY294002, an inhibitor of PI3K. C, Densitometric analysis of three repetitions of the experiment shown in B. D, Cell-surface biotinylation assay showing that the effect insulin on BKCa trafficking is markedly attenuated by pretreatment with the PI3K inhibitor LY294002. E, Densitometric quantification of three repetitions of the experiment shown in D. F, Insulin stimulation of current through BKCa is blocked by pretreatment with LY294002. Asterisks indicate P < 0.05.
Elevated extracellular glucose is sufficient to cause insulin resistance in many different cells and tissues, and can act at several different steps along insulin signaling pathways (Pirola et al., 2004). We observed that culturing podocytes in high glucose (36.1 mM) for 24 hr or 72 hr caused a marked decrease in the steady-state expression of BKCa channels as assessed by cell-surface biotinylation assays (Fig. 5A, B). In these experiments, control cells were cultured in normal medium containing 11.1 mM glucose or in a medium containing 11.1 mM glucose and 25 mM mannitol in order to produce an osmotic effect similar to that of high glucose. A corresponding effect was also seen with whole-cell recordings, which showed a marked decrease in BKCa current density in cells cultured in high glucose (Fig. 5C). Given this, we were surprised to note that high glucose treatment did not completely abrogate insulin signaling in podocytes. Thus, treatment with insulin in the presence of 36.1 mM glucose evoked increases in Erk (Fig. 6A) and Akt (Fig. 6B) phosphorylation comparable to those observed in podocytes cultured in 11.1 mM glucose plus 25 mM mannitol. Insulin treatment also increased surface expression of Slo1 channels in cells cultured in 36.1 mM glucose (Fig. 7A, B) although it should be noted that Slo1 surface expression always remains low compared to what is observed in insulin-treated cells maintained in 11.1 mM glucose.
Figure 5.
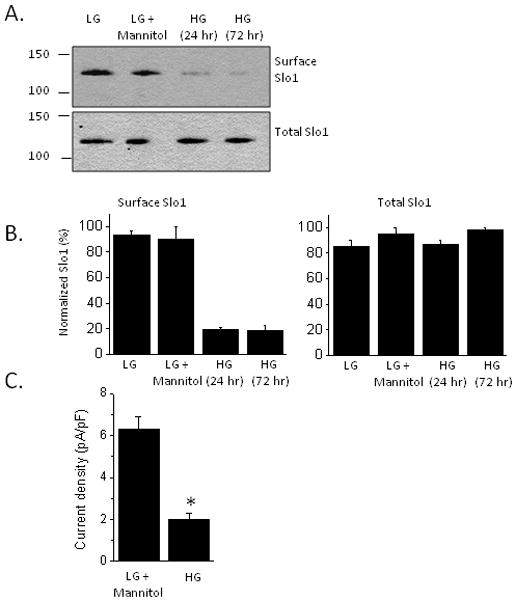
Exposure to high glucose markedly reduces basal expression of functional cell surface BKCa channels. A, Typical cell-surface biotinylation assay showing that exposing cells to 36.1 mM glucose (HG) for 24 hr or 72 hr reduces surface expression of pore-forming subunits of BKCa channels compared to controls. LG refers to cells cultured in normal medium containing 11.1 mM glucose. LG + mannitol refers to a separate set of controls in which 25 mM mannitol was used to control for osmotic effects of high glucose. B, Densitometric quantification of three repetitions of the experiment shown in A. C, High glucose for 24 hr decreases BKCa current density measured by whole-cell recording. Asterisk indicates P < 0.01 by Student’s unpaired t-test.
Figure 6.
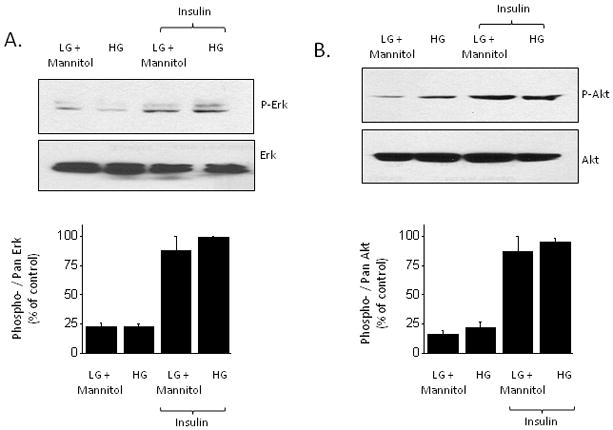
Insulin increases phosphorylation of Erk and Akt in cells cultured in high glucose. Typical immunoblots phosphorylated and total Erk (A) and Akt (B) are shown above densitometric analyses of three repetitions of these experiments.
Figure 7.
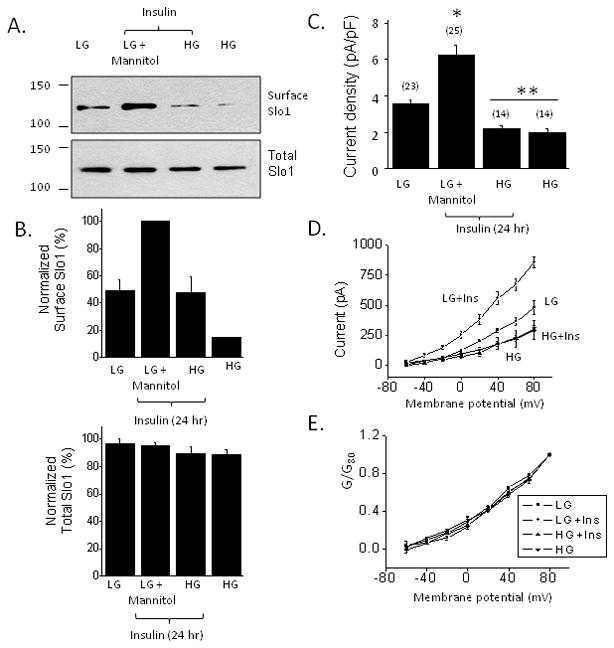
Effects of high glucose on insulin modulation of BKCa channels. Insulin treatment for 24 hr continues to increase surface expression of pore-forming subunits of BKCa channels in high glucose (36.1 mM), although this occurs from a lower baseline (A and B). However, insulin treatment for 24 hr does not increase current density through BKCa channels in cells grown in high glucose (C, D) indicating a functional insulin resistance. Voltage-dependence of BKCa activation is not changed by any of these treatments (E).
Using more quantitative electrophysiological methods, we observed podocytes were resistant to insulin when the measured endpoint was BKCa current density assayed by whole-cell recordings (Fig. 7C, D). With those methods we observed that culturing podocytes in 36.1 mM glucose resulted in markedly reduced basal levels of macroscopic current density compared to cells cultured in 11.1 mM glucose plus 25 mM mannitol, as expected from the results of cell-surface biotinylation assays. Moreover, insulin did not evoke any discernible increase in BKCa current density from the already low baseline in cells cultured in high glucose. Insulin and glucose concentration did not affect the voltage-dependence of BKCa activation and uniformly affected the amplitudes of the currents at all step voltages tested (Fig. 7D, E).
The slit diaphragm protein nephrin is required for normal trafficking of BKCa channels and glucose transporters to the cell surface (Coward et al., 2007; Kim et al., 2008) as well as for the normal function of the glomerular filtration slit (Kestilä et al., 1998; Putaala et al., 2001). Therefore, we examined the effects of high glucose on the surface expression of nephrin using cell-surface biotinylation assays. We observed that culturing cells in 36.1 mM glucose markedly reduced surface expression of nephrin compared to controls maintained in 11.1 mM glucose. Although insulin treatment could partially restore surface expression of nephrin in the presence of 36.1 mM glucose, it remained substantially less than what is seen in control media (Fig. 8).
Figure 8.
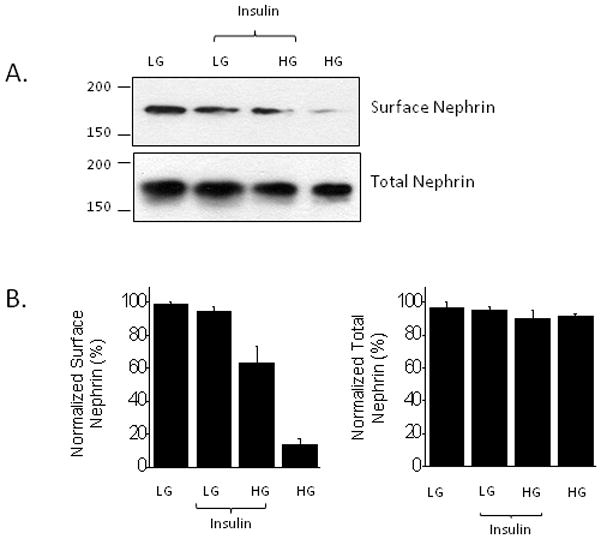
High glucose for 24 hr markedly reduces cell surface expression of nephrin, and insulin can partially restore normal expression. A, Typical cell-surface biotinylation assay. B, Densitometry of three repetitions of the experiment shown in A. Note that this experiment shows that some responses to insulin persist in podocytes cultured in high glucose.
DISCUSSION
In this study we have shown that insulin stimulates the surface expression of the pore-forming subunits (Slo1 proteins) of BKCa channels in mouse podocytes. This is accompanied by an increase in BKCa channel activity that can be readily observed in whole-cell recordings. Insulin effects on BKCa channels appear to be the result of a complex transduction cascade that includes activation of Erk MAP kinase and PI3K/Akt signaling cascades. Exposing podocytes to elevated extracellular glucose for 24 hr caused a marked reduction in the expression of BKCa channels and nephrin on the cell surface. High glucose also blocked insulin-evoked increases in BKCa currents measured by whole-cell recordings. However high glucose did not completely abrogate insulin signaling in podocytes, since insulin continued to evoke robust stimulation of Erk and Akt phosphorylation and could restore some of the glucose-evoked loss of nephrin expression on the cell surface.
Previous studies have shown that podocytes are direct targets for insulin action (Coward et al., 2005, 2007), and a more recent study has shown that insulin signaling in podocytes is required for normal glomerular function (Welsh et al., 2010). In podocytes, as in other cells, insulin increases glucose uptake owing to translocation of GLUT1 and GLUT4 transporters to the cell surface (Coward et al., 2005, 2007). However, those initial studies already identified unique features of insulin signaling in podocytes compared to what is seen in many other cell types, since nephrin was required for insulin stimulation of GLUT4 translocation (Coward et al., 2007). Nephrin is a single-pass transmembrane adhesion protein of the immunoglobulin superfamily. It has a large ectodomain comprised of multiple IgG-like domains and a single fibronectin-like motif, and a cytoplasmic domain that can interact with several other proteins (Kestilä et al., 1998). It is not generally expressed in skeletal muscle or adipocytes. We have previously shown that nephrin can interact with Slo1 proteins and β4-subunits of BKCa channels, and that nephrin and actin dynamics are necessary for the normal expression of functional BKCa channels on the podocyte cell surface (Kim et al., 2008, 2010).
The mechanisms by which insulin stimulates of BKCa activity in podocytes appear to be complex, and may have similarities to growth factor effects on BKCa channels of neurons. For example, certain growth factors such as TGFβ1 increase translocation of BKCa to the neuronal cell surface through a process that depends on activation of PIK3 and Erk (Dryer et al., 2003; Chae et al., 2005) and remodeling of cortical actin (Chae and Dryer, 2005; Zou et al., 2008). Insulin can modulate the gating of neuronal BKCa channels through an Erk-dependent signaling cascade that is also associated with actin remodeling (O’Malley and Harvey, 2004, 2007). Insulin also stimulates BKCa channels of mesangial cells via an Erk-dependent process that may entail increased expression on the cell surface (Foutz et al., 2008). It seems likely that insulin regulation of BKCa channels in podocytes is as complex as it is in neurons. However, effects of insulin on podocyte BKCa channels that are already in the plasma membrane, if they occur, must be subtle, as we did not observe any effects on the kinetics or voltage-dependence of the activation of macroscopic currents, which are usually sensitive indicators of changes in gating properties.
Exposure to high glucose for 24 hr caused a very marked reduction in the steady-state surface expression of podocyte BKCa channel subunits accompanied by a quantitatively similar reduction in the surface expression of nephrin. The reduction in the surface expression of nephrin is sufficient to explain the reduction in basal BKCa channels (Kim et al., 2008). Based on previous studies, one would predict that high glucose would also result in a reduction in insulin-evoked glucose uptake into podocytes, which also requires nephrin (Coward et al., 2007), although to our knowledge this has not been examined directly. This effect of high glucose may be pathophysiologically significant, as reductions in glomerular nephrin expression are a feature of both type 1 and type 2 diabetes (Doublier et al., 2003; , Benigni et al., 2004; Langham et al., 2002). They are also apparent in the earliest stages of animal models of diabetes (Aaltonen et al., 2001; Kim et al., 2007; Menne et al., 2006) and are mediated in part by increased endocytosis of nephrin (Tossidou et al., 2010). We should note that high glucose does not uniformly reduce surface expression of all membrane proteins, as we have observed that high glucose treatment causes a marked increase in the steady-state surface expression of TRPC6 cation channels (unpublished observations). Those observations will the subject of a later report.
In adipocytes and fibroblasts high glucose can induce insulin resistance by acting at the initial steps in the transduction cascade, resulting in inhibition of insulin receptor autophosphorylation and phosphorylation of insulin receptor substrate (IRS) proteins (Müller et al., 1991; Berti et al., 1994; Ide et al., 1994). Given that, we were surprised to observe that insulin-evoked phosphorylation of Erk or Akt persisted in podocytes cultured in high glucose. On the other hand, high glucose completely blocked insulin-evoked increases in density of current flowing through functional BKCa channels. This indicates that high glucose can impinge on insulin responses in podocytes at relatively distal stages in the transduction pathway. In any case, the effects of high glucose are likely to be complex, cell type-dependent, and may impact multiple cell physiological processes in podocytes.
Insulin modulation of BKCa channels may proceed through several different pathways that are not mutually exclusive. One potential pathway arises from insulin stimulation of PI3K activity, which leads to production of phosphatidylinositol–3,4,5-tris-phosphate [InsP(3,4,5)P3]. Phosphatidylinositol polyphosphates have been shown to stimulate activation of BKCa channels through a membrane delimited process that targets the channel or its proteolipid environment (Vaithianathan et al., 2008). Among the phosphoinositides, InsP(3,4,5)P3 is by far the most effective at causing activation of BKCa (Vaithianathan et al., 2008), and is therefore in a position to regulate both gating as well as cytoskeletal rearrangements that underlie trafficking to the cell surface. Disruption of nephrin expression could displace PI3K and therefore its catalytic product from the vicinity of BKCa channels (Benzing, 2004) thereby preventing their activation—even if total InsP(3,4,5)P3 and Akt activation persist at normal levels. It is also possible that insulin affects actin dynamics via PI3k and Erk mediated pathways to regulate trafficking or gating of BKCa channels (O’Malley and Harvey, 2007; Zou et al., 2008).
How might this response to insulin impact glomerular physiology? It has long been known that feeding (Corman et al., 1988; Uemasu et al., 1991; Finco and Cooper, 2000) or intravenous glucose (Christiansen et al., 1981) or amino acid (Castellino et al., 1986) infusion evoke an acute increase in the glomerular filtration rate (GFR). Increases in GFR are generally associated with an increase in the net pressure gradient across the glomerular filtration barrier, which can occur through multiple mechanisms (Persson et al., 2010). Podocytes are thought to play an active role in maintaining the permselectivity of the glomerular filtration barrier in the face of distending forces (Kriz et al., 1994, 1996) and there is evidence that the failure of podocytes to generate sufficient force to counteract filtration barrier pressure gradients can lead to glomerulosclerosis (Kriz et al., 2005). Insulin-evoked mobilization of BKCa channels may therefore be part of a signaling mechanism that allows the glomerular filtration apparatus to adapt to mechanical challenges associated with increases in GFR. In this regard, an elegant recent study has shown that deletion of insulin receptors selectively in podocytes leads over time to failure of the glomerular filtration barrier, even though the animals are normoglycemic (Welsh et al., 2010). The precise role of BKCa channels in podocyte function has not been established, but they interact directly with Ca2+-permeable TRPC6 channels in these cells (Kim et al. 2009) and their coordinated activation would be expected to increase the driving force for Ca2+ influx into podocytes through TRPC6 and related channels (Estacion et al., 2006; Dryer and Reiser , 2010), thereby contributing to active responses in these cells. BKCa channels may also be involved in Ca2+ oscillations and Ca2+ waves observed in podocytes within intact glomeruli during tubuloglomerular feedback (Peti-Peterdi, 2006).
In summary, we have shown that insulin modulates BKCa channels in podocytes. We have also shown that cell surface expression of BKCa channels and nephrin are down-regulated by high glucose, and that exposure to high glucose for as little as 24 hr blocks insulin responses at a downstream step in the overall signal-transduction pathway.
Acknowledgments
Supported by NIH Grant RO1-DK82529 to S. E. D.
References
- Aaltonen P, Luimula P, Aström E, Palmen T, Grönholm T, Palojoki E, Jaakkola I, Ahola H, Tikkanen I, Holthöfer H. Changes in the expression of nephrin gene and protein in experimental diabetic nephropathy. Lab Invest. 2001;81:1185–90. doi: 10.1038/labinvest.3780332. [DOI] [PubMed] [Google Scholar]
- Benigni A, Gagliardini E, Tomasoni S, Abbate M, Ruggenenti P, Kalluri R, Remuzzi G. Selective impairment of gene expression and assembly of nephrin in human diabetic nephropathy. Kidney Int. 2004;65:2193–200. doi: 10.1111/j.1523-1755.2004.00636.x. [DOI] [PubMed] [Google Scholar]
- Benzing T. Signaling at the slit diaphragm. J Am Soc Nephrol. 2004;15:1382–91. doi: 10.1097/01.asn.0000130167.30769.55. [DOI] [PubMed] [Google Scholar]
- Berti L, Mosthaf L, Kroder G, Kellerer M, Tippmer S, Mushack J, Seffer E, Seedorf K, Häring H. Glucose-induced translocation of protein kinase C isoforms in rat-1 fibroblasts is paralleled by inhibition of the insulin receptor tyrosine kinase. J Biol Chem. 1994;269:3381–6. [PubMed] [Google Scholar]
- Castellino P, Coda B, DeFronzo RA. Effect of amino acid infusion on renal hemodynamics in humans. Am J Physiol. 1986;251:F132–40. doi: 10.1152/ajprenal.1986.251.1.F132. [DOI] [PubMed] [Google Scholar]
- Chae KS, Dryer SE. The p38 mitogen-activated protein kinase pathway negatively regulates Ca2+-activated K+ channel trafficking in developing parasympathetic neurons. J Neurochem. 2005;94:367–79. doi: 10.1111/j.1471-4159.2005.03201.x. [DOI] [PubMed] [Google Scholar]
- Chae KS, Martin-Caraballo M, Anderson M, Dryer SE. Akt activation is necessary for growth factor-induced trafficking of functional KCa channels in developing parasympathetic neurons. J Neurophysiol. 2005;93:1174–82. doi: 10.1152/jn.00796.2004. [DOI] [PubMed] [Google Scholar]
- Christiansen JS, Frandsen M, Parving HH. Effect of intravenous glucose infusion on renal function in normal man and in insulin-dependent diabetics. Diabetologia. 1981;21:368–73. doi: 10.1007/BF00252683. [DOI] [PubMed] [Google Scholar]
- Corman B, Chami-Khazraji S, Schaeverbeke J, Michel JB. Effect of feeding on glomerular filtration rate and proteinuria in conscious aging rats. Am J Physiol. 1988;255:F250–6. doi: 10.1152/ajprenal.1988.255.2.F250. [DOI] [PubMed] [Google Scholar]
- Coward RJ, Welsh GI, Koziell A, Hussain S, Lennon R, Ni L, Tavaré JM, Mathieson PW, Saleem MA. Nephrin is critical for the action of insulin on human glomerular podocytes. Diabetes. 2007;56:1127–35. doi: 10.2337/db06-0693. [DOI] [PubMed] [Google Scholar]
- Coward RJ, Welsh GI, Yang J, Tasman C, Lennon R, Koziell A, Satchell S, Holman GD, Kerjaschki D, Tavaré JM, Mathieson PW, Saleem MA. The human glomerular podocyte is a novel target for insulin action. Diabetes. 2005;54:3095–102. doi: 10.2337/diabetes.54.11.3095. [DOI] [PubMed] [Google Scholar]
- Doublier S, Salvidio G, Lupia E, Ruotsalainen V, Verzola D, Deferrari G, Camussi G. Nephrin expression is reduced in human diabetic nephropathy: evidence for a distinct role for glycated albumin and angiotensin II. Diabetes. 2003;52:1023–30. doi: 10.2337/diabetes.52.4.1023. [DOI] [PubMed] [Google Scholar]
- Drenckhahn D, Franke RP. Ultrastructural organization of contractile and cytoskeletal proteins in glomerular podocytes of chicken, rat, and man. Lab Invest. 1988;59:673–82. [PubMed] [Google Scholar]
- Dryer SE, Lhuillier L, Cameron JS, Martin-Caraballo M. Expression of KCa channels in identified populations of developing vertebrate neurons: role of neurotrophic factors and activity. J Physiol Paris. 2003;97:49–58. doi: 10.1016/j.jphysparis.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Dryer SE, Reiser J. TRPC6 channels and their binding partners in podocytes: Role in glomerular filtration and pathophysiology. Am J Physiol Renal Physiol. 2010 doi: 10.1152/ajprenal.00298.2010. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endlich N, Endlich K. Stretch, tension and adhesion—adaptive mechanisms of the actin cytoskeleton in podocytes. Eur J Cell Biol. 2006;85:229–34. doi: 10.1016/j.ejcb.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Estacion M, Sinkins WG, Jones SW, Applegate MA, Schilling WP. Human TRPC6 expressed in HEK 293 cells forms non-selective cation channels with limited Ca2+ permeability. J Physiol. 2006;572:359–77. doi: 10.1113/jphysiol.2005.103143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finco DR, Cooper TL. Soy protein increases glomerular filtration rate in dogs with normal or reduced renal function. J Nutr. 2000;130:745–8. doi: 10.1093/jn/130.4.745. [DOI] [PubMed] [Google Scholar]
- Foutz RM, Grimm PR, Sansom SC. Insulin increases the activity of mesangial BK channels through MAPK signaling. Am J Physiol Renal Physiol. 2008;294:F1465–72. doi: 10.1152/ajprenal.00012.2008. [DOI] [PubMed] [Google Scholar]
- Frega NS, Weinberg JM, Ross BD, Leaf A. Stimulation of sodium transport by glucose in the perfused rat kidney. Am J Physiol. 1977;233:F235–40. doi: 10.1152/ajprenal.1977.233.3.F235. [DOI] [PubMed] [Google Scholar]
- Ide R, Maegawa H, Kikkawa R, Shigeta Y, Kashiwagi A. High glucose condition activates protein tyrosine phosphatases and deactivates insulin receptor function in insulin-sensitive rat 1 fibroblasts. Biochem Biophys Res Commun. 1994;201:71–7. doi: 10.1006/bbrc.1994.1670. [DOI] [PubMed] [Google Scholar]
- Kestilä M, Lenkkeri U, Männikkö M, Lamerdin J, McCready P, Putaala H, Ruotsalainen V, Morita T, Nissinen M, Herva R, Kashtan CE, Peltonen L, Holmberg C, Olsen A, Tryggvason K. Positionally cloned gene for a novel glomerular protein—nephrin—is mutated in congenital nephrotic syndrome. Mol Cell. 1998;1:575–82. doi: 10.1016/s1097-2765(00)80057-x. [DOI] [PubMed] [Google Scholar]
- Kim EY, Alvarez-Baron CP, Dryer SE. Canonical transient receptor potential channel (TRPC)3 and TRPC6 associate with large-conductance Ca2+-activated K+ (BKCa) channels: role in BKCa trafficking to the surface of cultured podocytes. Mol Pharmacol. 2009a;75:466–77. doi: 10.1124/mol.108.051912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EY, Chiu YH, Dryer SE. Neph1 regulates steady-state surface expression of Slo1 Ca2+-activated K+ channels: different effects in embryonic neurons and podocytes. Am J Physiol Cell Physiol. 2009b;297:C1379–88. doi: 10.1152/ajpcell.00354.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EY, Choi KJ, Dryer SE. Nephrin binds to the COOH terminus of a large-conductance Ca2+-activated K+ channel isoform and regulates its expression on the cell surface. Am J Physiol Renal Physiol. 2008;295:F235–46. doi: 10.1152/ajprenal.00140.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EY, Suh JM, Chiu YH, Dryer SE. Regulation of podocyte BKCa channels by synaptopodin, Rho, and actin microfilaments. Am J Physiol Renal Physiol. 2010 doi: 10.1152/ajprenal.00206.2010. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JJ, Li JJ, Jung DS, Kwak SJ, Ryu DR, Yoo TH, Han SH, Choi HY, Kim HJ, Han DS, Kang SW. Differential expression of nephrin according to glomerular size in early diabetic kidney disease. J Am Soc Nephrol. 2007;18:2303–10. doi: 10.1681/ASN.2006101145. [DOI] [PubMed] [Google Scholar]
- Kriz W, Hackenthal E, Nobiling R, Sakai T, Elger M, Hähnel B. A role for podocytes to counteract capillary wall distension. Kidney Int. 1994;45:369–76. doi: 10.1038/ki.1994.47. [DOI] [PubMed] [Google Scholar]
- Kriz W, Kretzler M, Provoost AP, Shirato I. Stability and leakiness: opposing challenges to the glomerulus. Kidney Int. 1996;49:1570–4. doi: 10.1038/ki.1996.227. [DOI] [PubMed] [Google Scholar]
- Kriz W, LeHir M. Pathways to nephron loss starting from glomerular diseases-insights from animal models. Kidney Int. 2005;67:404–19. doi: 10.1111/j.1523-1755.2005.67097.x. [DOI] [PubMed] [Google Scholar]
- Langham RG, Kelly DJ, Cox AJ, Thomson NM, Holthöfer H, Zaoui P, Pinel N, Cordonnier DJ, Gilbert RE. Proteinuria and the expression of the podocyte slit diaphragm protein, nephrin, in diabetic nephropathy: effects of angiotensin converting enzyme inhibition. Diabetologia. 2002;45:1572–6. doi: 10.1007/s00125-002-0946-y. [DOI] [PubMed] [Google Scholar]
- Menne J, Meier M, Park JK, Boehne M, Kirsch T, Lindschau C, Ociepka R, Leitges M, Rinta-Valkama J, Holthofer H, Haller H. Nephrin loss in experimental diabetic nephropathy is prevented by deletion of protein kinase C alpha signaling in-vivo. Kidney Int. 2006;70:1456–62. doi: 10.1038/sj.ki.5001830. [DOI] [PubMed] [Google Scholar]
- Morton MJ, Hutchinson K, Mathieson PW, Witherden IR, Saleem MA, Hunter M. Human podocytes possess a stretch-sensitive, Ca2+-activated K+ channel: potential implications for the control of glomerular filtration. J Am Soc Nephrol. 2004;15:2981–7. doi: 10.1097/01.ASN.0000145046.24268.0D. [DOI] [PubMed] [Google Scholar]
- Müller HK, Kellerer M, Ermel B, Mühlhöfer A, Obermaier-Kusser B, Vogt B, Häring HU. Prevention by protein kinase C inhibitors of glucose-induced insulin-receptor tyrosine kinase resistance in rat fat cells. Diabetes. 1991;40:1440–8. doi: 10.2337/diab.40.11.1440. [DOI] [PubMed] [Google Scholar]
- O'Malley D, Harvey J. Insulin activates native and recombinant large conductance Ca2+-activated potassium channels via a mitogen-activated protein kinase-dependent process. Mol Pharmacol. 2004;65:1352–63. doi: 10.1124/mol.65.6.1352. [DOI] [PubMed] [Google Scholar]
- O'Malley D, Harvey J. MAPK-dependent actin cytoskeletal reorganization underlies BK channel activation by insulin. Eur J Neurosci. 2007;25:673–82. doi: 10.1111/j.1460-9568.2007.05347.x. [DOI] [PubMed] [Google Scholar]
- Pagtalunan ME, Miller PL, Jumping-Eagle S, Nelson RG, Myers BD, Rennke HG, Coplon NS, Sun L, Meyer TW. Podocyte loss and progressive glomerular injury in type II diabetes. J Clin Invest. 1997;99:342–8. doi: 10.1172/JCI119163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson P, Hansell P, Palm F. Tubular reabsorption and diabetes-induced glomerular hyperfiltration. Acta Physiol (Oxf) 2010;200:3–10. doi: 10.1111/j.1748-1716.2010.02147.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peti-Peterdi J. Calcium wave of tubuloglomerular feedback. Am J Physiol Renal Physiol. 2006;291:F473–80. doi: 10.1152/ajprenal.00425.2005. [DOI] [PubMed] [Google Scholar]
- Peti-Peterdi J, Sipos A. A high-powered view of the filtration barrier. J Am Soc Nephrol. 2010 Jun 24; doi: 10.1681/ASN.2010040378. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirola L, Johnston AM, Van Obberghen E. Modulation of insulin action. Diabetologia. 2004;47:170–84. doi: 10.1007/s00125-003-1313-3. [DOI] [PubMed] [Google Scholar]
- Putaala H, Soininen R, Kilpeläinen P, Wartiovaara J, Tryggvason K. The murine nephrin gene is specifically expressed in kidney, brain and pancreas: inactivation of the gene leads to massive proteinuria and neonatal death. Hum Mol Genet. 2001;10:1–8. doi: 10.1093/hmg/10.1.1. [DOI] [PubMed] [Google Scholar]
- Ridgway LD, Kim EY, Dryer SE. MAGI-1 interacts with Slo1 channel proteins and suppresses Slo1 expression on the cell surface. Am J Physiol Cell Physiol. 2009;297:C55–65. doi: 10.1152/ajpcell.00073.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodewald R, Karnovsky MJ. Porous substructure of the glomerular slit diaphragm in the rat and mouse. J Cell Biol. 1974;60:423–33. doi: 10.1083/jcb.60.2.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleem MA, Zavadil J, Bailly M, McGee K, Witherden IR, Pavenstadt H, Hsu H, Sanday J, Satchell SC, Lennon R, Ni L, Bottinger EP, Mundel P, Mathieson PW. The molecular and functional phenotype of glomerular podocytes reveals key features of contractile smooth muscle cells. Am J Physiol Renal Physiol. 2008;295:F959–70. doi: 10.1152/ajprenal.00559.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankland SJ. The podocyte's response to injury: role in proteinuria and glomerulosclerosis. Kidney Int. 2006;69:2131–47. doi: 10.1038/sj.ki.5000410. [DOI] [PubMed] [Google Scholar]
- Steffes MW, Schmidt D, McCrery R, Basgen JM. Glomerular cell number in normal subjects and in type 1 diabetic patients. Kidney Int. 2001;59:2104–13. doi: 10.1046/j.1523-1755.2001.00725.x. [DOI] [PubMed] [Google Scholar]
- Tossidou I, Teng B, Menne J, Shushakova N, Park JK, Becker JU, Modde F, Leitges M, Haller H, Schiffer M, Tossidou I, Teng B, Menne J, Shushakova N, Park JK, Becker JU, Modde F, Leitges M, Haller H, Schiffer M. Podocytic PKC-alpha is regulated in murine and human diabetes and mediates nephrin endocytosis. PLoS One. 2010;5:e10185. doi: 10.1371/journal.pone.0010185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uemasu J, Hori T, Uemasu Y, Kawasaki H. Effects of a rice meal on renal hemodynamics and excretory functions in normal subjects. Nephron. 1991;57:187–91. doi: 10.1159/000186248. [DOI] [PubMed] [Google Scholar]
- Vaithianathan T, Bukiya A, Liu J, Liu P, Asuncion-Chin M, Fan Z, Dopico A. Direct regulation of BK channels by phosphatidylinositol 4,5-bisphosphate as a novel signaling pathway. J Gen Physiol. 2008;132:13–28. doi: 10.1085/jgp.200709913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh GI, Hale LJ, Eremina V, Jeansson M, Maezawa Y, Lennon R, Pons DA, Owen RJ, Satchell SC, Miles MJ, Caunt CJ, McArdle CA, Pavenstädt H, Tavaré JM, Herzenberg AM, Kahn CR, Mathieson PW, Quaggin SE, Saleem MA, Coward RJ. Insulin signaling to the glomerular podocyte is critical for normal kidney function. Cell Metab. 2010;12:329–40. doi: 10.1016/j.cmet.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou S, Jha S, Kim EY, Dryer SE. A novel actin-binding domain on Slo1 calcium-activated potassium channels is necessary for their expression in the plasma membrane. Mol Pharmacol. 2008;73:359–68. doi: 10.1124/mol.107.039743. [DOI] [PubMed] [Google Scholar]