

Abstract
JP4-039 is a novel nitroxide conjugate capable of crossing lipid bilayer membranes and scavenging reactive oxygen species (ROS). An efficient and scalable one-pot hydrozirconation-transmetalation-imine addition methodology has been developed for its asymmetric preparation. Furthermore, this versatile methodology allows for the synthesis of cyclopropyl and fluorinated analogs of the parent lead structure.
The 4-amino-Tempo (4-AT) derivative JP4-039 ((S)-6a, Scheme 2) is a lead structure among a new generation of peptidomimetic conjugates targeted to mitochondria and effective at scavenging reactive oxygen species (ROS) such as the superoxide radical anion.1,2 In particular, JP4-039 has been shown to protect cells from radiation damage2b and to prolong survival of mice subjected to high levels of ionizing irradiation.2c Moreover, the ameliorating effects of JP4-039 in irradiation-induced delay of bone wound healing were demonstrated in a murine model of combined bone wound/irradiation injury.2d JP4-039 and its larger congener, XJB-5-131, are found to enrich in mitochondria by a factor of 30–600 times over the cytosolic concentration, at least in part due to their affinity to the mitochondrial lipid, cardiolipin.1b,2a There, they serve to scavenge ROS and prevent hydroperoxidation of cardiolipin by cycling between nitroxide, hydroxylamine, and nitroxonium redox states.3 Their design was based on the structure of the antibiotic gramicidin S; the alkene peptide isostere replaces a polar internal amide bond and thus increases conformational regidity and membrane permeability.1a,g In order to further explore the therapeutic potential of JP4-039, a robust synthetic route was required to prepare multigram quantities of highly pure material.
Scheme 2.

Synthesis of the (E)-alkene isosteres (S)-6a-c
JP4-039 contains a single asymmetric carbon atom as part of an alkene isostere dipeptide moiety composed of leucine and glycine residues. Numerous methods have been developed to generate these α-chiral allylic amines.4 For example, Overman developed the rearrangement of allylic trichloroacetimidates,4b Berkowitz described an approach which relies on a Ni(0)-mediated allylic amination,4c and Krische reported a C-C bond-forming hydrogenation for this purpose.4k Overall, vinyl addition to imines remains the most commonly used strategy to prepare chiral allylic amines.4d-k Highly effective protocols for this route are based on the reductive coupling of alkynes,4d-f the diastereoselective addition of vinylorganometallic species,4g-i and the acylvinyl anion addition.4j
Our group has previously developed an efficient Zr-based method for the asymmetric synthesis of allylic amines.5 Hydrozirconation of alkyne 26 with Cp2ZrHCl,7 followed by transmetalation to trimethylaluminum and subsequent addition to chiral sulfinimine8 (R)-1 provided a diastereomerically pure (>20:1 dr) allylic amine according to 1H NMR analysis of the crude reaction mixture (Scheme 1). A four-membered chelate model has been proposed to account for this excellent diastereoselectivity.5a,c Treatment of the crude reaction mixture with HCl in diethyl ether yielded the amine hydrochloride (S)-3 on multigram scale and in 81% yield over 3 steps from 3-butyn-1-ol. The N-tert-butylsulfinimine (R)-1 could be easily obtained from isovaleraldehyde in 90% yield and proved remarkably stable to storage.
Scheme 1.

Synthesis of the common amine intermediate (S)-3
We were able to use the allylic sulfinylamine (S)-3 as a linchpin intermediate for the preparation of JP4-039 as well as a first series of analogs. Thus, acylation of (S)-3 with either a Boc, Cbz or diphenylphosphinoyl group, followed by TBAF-mediated deprotection of the silyl ether, afforded the alcohols (S)-5a-c (Scheme 2). Jones oxidation to the corresponding carboxylic acids and final coupling with 4-AT using the EDCI/HOBt/DMAP protocol provided the desired alkene isosteres (S)-6a-c in moderate to good yields.
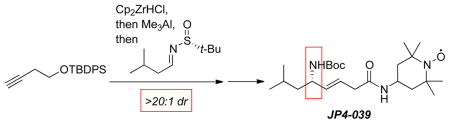
This methodology was further adapted for a >150 g scale preparation of JP4-039 ((S)-6a), obtained in 32% overall yield, 99% purity and >99% ee based on HPLC analysis. At this scale, the target compound was isolated from a 20:3 mixture of n-hexane/EtOAc as a crystalline solid. The X-ray structure of (S)-6a is in good agreement with a type II’ β-turn (Figure 1). The distance of 3.30 Å for the i to i+3 intramolecular hydrogen bond between the carbonyl oxygen of the Boc function and the nitrogen atom of 4-AT is indicative of a weak covalent component in this H-bond.
Figure 1.
X-ray structure of JP4-039.
The (R)-enantiomer of JP4-039 (R)-6a was obtained in 52% overall yield according to an analogous synthetic route starting from (S)-sulfinimine (S)-1 (Scheme 3). Chiral SFC on a Chiralpak-IC phase was used to determine an ee of 96.6% for alcohol (S)-5a and 98.0% for its enantiomer (R)-5a.
Scheme 3.

Synthesis of (R)-6a
An isosteric replacement of the (E)-alkene bond with a cyclopropyl moiety was also investigated as part of our medicinal chemistry program.9 Charette-modified Simmons-Smith cyclopropanation10 of the Cbz-protected allylic amine (S)-4b with Zn(CH2I)2•DME complex11 provided the cyclopropyl analog 7b on gram scale in 54% yield (65% based on recovered intermediate (S)-4b) over 2 steps (Scheme 4). Only one diastereomer was isolated after chromatography on SiO2 (>20:1 dr by 1H NMR). Since 7b was not crystalline and spectroscopic analysis did not allow an unambiguous assignment of its configuration, we resorted to an X-ray analysis of a suitable crystalline derivative. Hydrogenolysis, coupling with p-bromobenzoyl chloride, TBAF-desilylation and treatment with 1-naphthylisocyanate afforded urea 10, which gave fine colorless needles suitable for X-ray diffraction (Scheme 5). The X-ray analysis confirmed the anti-configuration of the cyclopropane vs the C-N bond (Figure 2). This diastereoselectivity had been previously noted by our group for the dimethylzinc-mediated addition of alkenylzirconocenes to N-diphenylphosphinoyl imines, which provided diastereomerically pure C-cyclopropylalkylamines.5b,9f The high level of anti-selectivity is also consistent with diastereoselectivities observed in the Simmons-Smith cyclopropanation of allylic ethers.12
Scheme 4.

Synthesis of the cyclopropyl isosteres 8a-b
Scheme 5.

Synthesis of 10
Figure 2.
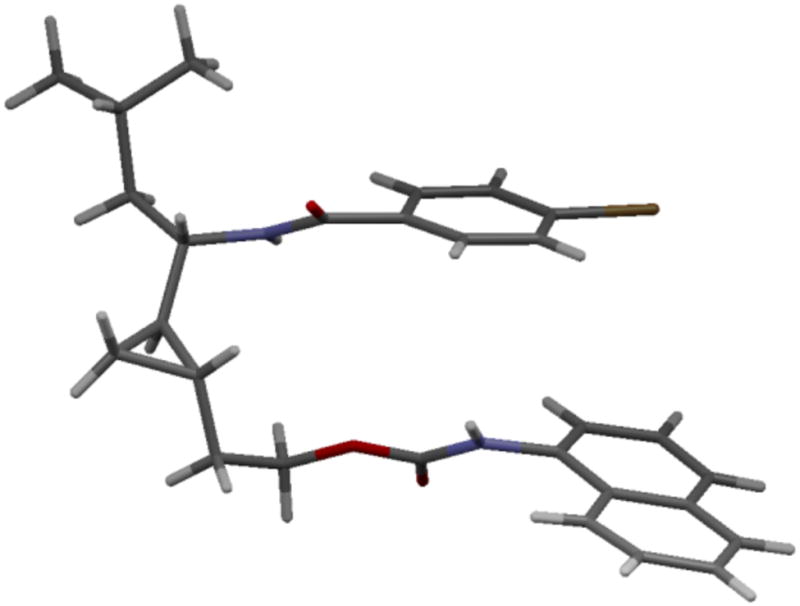
X-ray structure of 10
Somewhat surprisingly, the Boc group on (S)-4a was not compatible with the Simmons-Smith conditions. The desired intermediate 7a was therefore prepared by hydrogenolysis of 7b followed by Boc protection of the resulting amine (Scheme 4). Subsequent TBAF-desilylation of 7a and 7b, Jones oxidation, and coupling with 4-AT afforded the cyclopropyl isosteres 8a and 8b.
Finally, a difluorinated analog of JP4-039 was envisioned to enhance the bioavailability of the agent. The methyl ester (S)-11, prepared from alcohol (S)-5a by Jones oxidation and esterification of the acid with TMS-diazomethane, was treated with 3 equiv. of the fluorinating agent N-fluoro-N-(phenylsulfonyl)benzene-sulfonamide (accufluor, NFSi)13 and 2.3 equiv. of NaHMDS in THF at −78 °C to afford the desired α,α-difluoroester (S)-12 in 86% yield (Scheme 6).14 Saponification with tetra-butylammonium hydroxide (TBAH)15 and condensation with 4-AT provided the difluorinated JP4-039 analog (S)-13 in good yield. The biological activities of (S)-13 as well as (R)-6a are currently under investigation.
Scheme 6.

Synthesis of the difluoro analog (S)-13
In summary, the hydrozirconation-transmetalation-imine addition methodology was extended to the preparation of both enantiomers of a chiral allylic amine and the corresponding dipeptide alkene and cyclopropane isosteres. This straightforward approach enabled the synthesis of the bioprotective Tempo-conjugate JP4-039 on a 160 g scale. Noteworthy is also the ready access to diverse analogs of the parent compound, including the β,γ-cyclopropylamine isosteres and difluoromethylene derivatives. SAR studies of the JP4-039 scaffold are ongoing, and the synthesis of further derivatives will be reported in due course, along with their antioxidant and biological activities.
Supplementary Material
Acknowledgments
This project was supported by a BARDA contract (HHS0100200800062C), the NIH/NIAID CMCR program (AI068021) and the NIH/NIGMS CMLD program (GM067082). We thank the NMR and MS facilities at the University of Pittsburgh for their services, Dr. Steven J. Geib (University of Pittsburgh) for X-ray analyses, Mr. David M. Arnold (University of Pittsburgh) for SFC analyses, and Ms. Kayla R. Lloyd (University of Pittsburgh) for LC-MS analyses and for technical and administrative assistance. M.C.F. wishes to thank Mr. Chris J. Rosenker (University of Pittsburgh) and Dr. Gary C. Davis (University of Pittsburgh) for helpful comments. P.W. thanks Profs. Valerian Kagan, Laura Niedernhofer, Mike Epperly, Joel Greenberger, and Paul Robbins (all at University of Pittsburgh) for stimulating discussions and collaborative studies on the bioprotective properties of the JP4-series.
Footnotes
Supporting Information Available. Experimental procedures, x-ray and complete spectroscopic data for new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Wipf P, Xiao J, Jiang J, Belikova NA, Tyurin VA, Fink MP, Kagan VE. J Am Chem Soc. 2005;127:12460. doi: 10.1021/ja053679l.Jiang J, Kurnikov I, Belikova NA, Xiao J, Zhao Q, Amoscato AA, Braslau R, Studer A, Fink MP, Greenberger JS, Wipf P, Kagan VE. J Pharmacol Exp Ther. 2007;320:1050. doi: 10.1124/jpet.106.114769.Kanai A, Zabbarova I, Amoscato A, Epperly M, Xiao J, Wipf P. Org Biomol Chem. 2007;5:307. doi: 10.1039/b613334g.Macias CA, Chiao JW, Xiao J, Arora DS, Tyurina YY, Delude RL, Wipf P, Kagan VE, Fink MP. Ann Surg. 2007;245:305. doi: 10.1097/01.sla.0000236626.57752.8e.Jiang J, Belikova NA, Hoye AT, Zhao Q, Epperly MW, Greenberger JS, Wipf P, Kagan VE. Int J Radiat Oncol Biol Phys. 2008;70:816. doi: 10.1016/j.ijrobp.2007.10.047.For reviews on mitochondria targeting agents, see: Hoye AT, Davoren JE, Wipf P, Fink MP, Kagan VE. Acc Chem Res. 2008;41:87. doi: 10.1021/ar700135m.Frantz MC, Wipf P. Environ Mol Mutagen. 2010;51:462. doi: 10.1002/em.20554.
- 2.(a) Kagan VE, Wipf P, Stoyanovsky D, Greenberger JS, Borisenko G, Belikova NA, Yanamala N, Samhan Arias AK, Tungekar MA, Jiang J, Tyurina YY, Ji J, Klein-Seetharaman J, Pitt BR, Shvedova AA, Bayir H. Adv Drug Deliv Rev. 2009;61:1375. doi: 10.1016/j.addr.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rajagopalan MS, Gupta K, Epperly MW, Franicola D, Zhang X, Wang H, Zhao H, Tyurin VA, Pierce JG, Kagan VE, Wipf P, Kanai AJ, Greenberger JS. In Vivo. 2009;23:717. [PMC free article] [PubMed] [Google Scholar]; (c) Epperly MW, Goff JP, Li S, Gao X, Wipf P, Dixon T, Wang H, Franicola D, Shen H, Rwigema JCM, Kagan V, Bernard M, Greenberger JS. In Vivo. 2010;24:811. [PMC free article] [PubMed] [Google Scholar]; (d) Gokhale A, Rwigema JC, Epperly MW, Glowacki J, Wang H, Wipf P, Goff JP, Dixon T, Patrene K, Greenberger JS. In Vivo. 2010;24:377. [PMC free article] [PubMed] [Google Scholar]
- 3.Tyurina YY, Tyurin VA, Kaynar AM, Kapralova VI, Wasserloos K, Li J, Mosher M, Wright L, Wipf P, Watkins S, Pitt BR, Kagan VE. Am J Physiol Lung Cell Mol Physiol. 2010;299:L73. doi: 10.1152/ajplung.00035.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.For a review on allylic amination, see: Johannsen M, Jorgensen KA. Chem Rev. 1998;98:1689. doi: 10.1021/cr970343o.For selected examples, see: Anderson CE, Overman LE. J Am Chem Soc. 2003;125:12412. doi: 10.1021/ja037086r.Berkowitz DB, Maiti G. Org Lett. 2004;6:2661. doi: 10.1021/ol049159x.Grossman RB, Davis WM, Buchwald SL. J Am Chem Soc. 1991;113:2321.Patel SJ, Jamison TF. Angew Chem Int Ed. 2004;43:3941. doi: 10.1002/anie.200460044.Ngai MY, Barchuk A, Krische MJ. J Am Chem Soc. 2007;129:12644. doi: 10.1021/ja075438e.and references cited therein Denmark SE, Weber T, Piotrowski DW. J Am Chem Soc. 1987;109:2224.Cogan DA, Liu G, Ellman J. Tetrahedron. 1999;55:8883.Brak K, Ellman JA. J Am Chem Soc. 2009;131:3850. doi: 10.1021/ja9002603.Reynolds TE, Binkley MS, Scheidt KA. Org Lett. 2008;10:5227. doi: 10.1021/ol802227t.Skucas E, Ngai MY, Komanduri V, Krische MJ. Acc Chem Res. 2007;40:1394. doi: 10.1021/ar7001123.
- 5.Wipf P, Nunes RL, Ribe S. Helv Chim Acta. 2002;85:3478.Wipf P, Kendall C, Stephenson CRJ. J Am Chem Soc. 2003;125:761. doi: 10.1021/ja028092a.Wipf P, Pierce JG. Org Lett. 2006;8:3375. doi: 10.1021/ol0613057.For a review, see: Wipf P, Kendall C. Topics Organomet Chem. 2004;8:1.
- 6.Nicolaou KC, Lizos DE, Kim DW, Schlawe D, de Noronha RG, Longbottom DA, Rodriquez M, Bucci M, Cirino G. J Am Chem Soc. 2006;128:4460. doi: 10.1021/ja060064v. [DOI] [PubMed] [Google Scholar]
- 7.(a) Buchwald SL, LaMaire SJ, Nielsen RB, Watson BT, King SM. Org Synth. 1998;9:162. Coll. [Google Scholar]; (b) Wipf P, Jahn H. Tetrahedron. 1996;52:12853. and references cited therein. [Google Scholar]
- 8.Staas DD, Savage KL, Homnick CF, Tsou NN, Ball RG. J Org Chem. 2002;67:8276. doi: 10.1021/jo0259313.For a review on the use of sulfinimines, see: Robak MT, Herbage MA, Ellman JA. Chem Rev. 2010;110:3600. doi: 10.1021/cr900382t.
- 9.Wipf P, Henninger TC, Geib SJ. J Org Chem. 1998;63:6088. doi: 10.1021/jo981057v.Xiao J, Weisblum B, Wipf P. J Am Chem Soc. 2005;127:5742. doi: 10.1021/ja051002s.Xiao J, Weisblum B, Wipf P. Org Lett. 2006;8:4731. doi: 10.1021/ol0617704.Wipf P, Xiao J. Org Lett. 2005;7:103. doi: 10.1021/ol0477529.Wipf P, Werner S, Woo GHC, Stephenson CRJ, Walczak MAA, Coleman CM, Twining LA. Tetrahedron. 2005;61:11488.Wipf P, Stephenson CRJ. Org Lett. 2005;7:1137. doi: 10.1021/ol0501525.For a review, see: Wipf P, Xiao J, Stephenson CRJ. Chimia. 2009;63:764. doi: 10.2533/chimia.2009.764.
- 10.(a) Simmons HE, Smith RD. J Am Chem Soc. 1959;81:4256. [Google Scholar]; (b) Charette AB, Prescott S, Brochu C. J Org Chem. 1995;60:1081. [Google Scholar]
- 11.(a) Wittig G, Schwarzenbach K. Angew Chem. 1959;71:652. [Google Scholar]; (b) Furukawa J, Kawabata N, Nishimura J. Tetrahedron. 1968;24:53. [Google Scholar]
- 12.Charette A, Lebel H, Gagnon A. Tetrahedron. 1999;55:8845.For a review on the Simmons-Smith cyclopropanation, see: Charette AB, Beauchemin A. Org React. 2004;58:1.
- 13.Differding E, Ofner H. Synlett. 1991:187. [Google Scholar]
- 14.Liu S, Dockendorff C, Taylor SD. Org Lett. 2001;3:1571. doi: 10.1021/ol0158664. [DOI] [PubMed] [Google Scholar]
- 15.Abdel-Magid AF, Cohen JH, Maryanoff CA, Shah RD, Villani FJ, Zhang F. Tetrahedron Lett. 1998;39:3391. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.