

Abstract
A library composed of Nitazoxanide-based analogues was synthesized and assayed for increased antibacterial efficacy against the pyruvate:ferredoxin oxidoreductase (PFOR) utilizing microorganisms Helicobacter pylori, Campylobacter jejuni and Clostridium difficile. Derivatives were found to recapitulate and improve activity against these organisms and select analogues were tested for their ability to disrupt the PFOR enzyme directly. The library was also screened for activity against staphylococci and resulted in the identification of analogues capable of inhibiting both staphylococci and all PFOR organisms at low μM MIC concentrations with low toxicity to human foreskin cells.
Keywords: Antibiotics, Cytotoxicity, Drug design, Medicinal chemistry, Structure-activity relationships
Introduction
The identification and optimization of drugs is exceedingly expensive and complex and the introduction of new therapeutics to treat infectious diseases has declined significantly.[1, 2] Nitazoxanide (NTZ–1–Figure 1) is an FDA approved drug for treating infections caused by Giardia lamblia and Cryptosporidium parvum but its use is limited due to poor solubility and efficacy as nearly a gram per day is required for treatment.[3–6] NTZ also exhibits broad antimicrobial action against anaerobic intestinal pathogens and also has notable activity against microbial biofilms, rotavirus, influenza, hepatitis and Mycobacterium tuberculosis.[7–12]
Figure 1.

Representative Nitro Drugs.
NTZ is a unique member of the nitro-drug family (Figure 1). Unlike most nitro drugs,[3, 13, 14] the 5-nitro group of NTZ is metabolically stable and is not reduced as part of the mechanism of action (MoA).[15] Although nitro group containing drugs and analogues are seldom pursued in a drug discovery program due to mutagenic and potentially toxic side effects, NTZ’s stability and lack of nitro reduction make it an important exception.
NTZ’s activity against anaerobic pathogens strongly suggested a common target. Since NTZ does not rely on metabolic reduction of the nitro group for activity, a different mechanism was postulated to account for its efficacy. In previous mechanistic studies we determined that the amide anion of NTZ interfered with the vitamin co-factor thiamine pyrophosphate of the essential enzyme pyruvate:ferredoxin oxidoreductase (PFOR) (Figure 2).[15, 16] PFOR is utilized by all strictly anaerobic bacteria, anaerobic parasites and ε-proteobacteria (Figure 3).[15–20] Mammals and eubacteria oxidize pyruvate by the NTZ insensitive pyruvate dehydrogenase (PDH) enzyme complex.
Figure 2.

Binding of pyruvate to the activated vitamin co-factor thiamine pyrophosphate (TPP). Pyruvate is then converted to acetyl-CoA. Nitazoxanide is believed to bind to and abstract a proton from the activated TPP, essentially out competing pyruvate and inhibiting the enzymatic reaction. PP: Pyrophosphate.
Figure 3.

Pyruvate:ferredoxin oxidoreductase (PFOR) enzymatic reaction. Acetyl-CoA and CO2 are the oxidative by-products of the PFOR enzymatic reaction which requires ferredoxin (Fd) or flavodoxin (Fld) as electron acceptors. NADP oxidases or hydrogenases oxidize the reduced Fd/Fld to complete the cycle. Solid arrows indicate the forward reaction; hollow arrows indicate the reverse reaction.
In these studies, NTZ was shown to completely inhibit the production of both acetyl-coenzyme A and CO2 by PFOR with a Ki of ~5 × 10−6 M which is roughly two orders of magnitude lower than the Km for pyruvate (Km = ~3 × 10−4 M).[15] The anion of NTZ is the active form required for PFOR inhibition and NTZ is not chemically modified during the enzymatic reaction. NMR analysis of the NTZ anion revealed the expected four thiazolide resonance structures involving the 5-nitro, N2′ and N3 nitrogens and the amide oxygen.[15] The amide anion (pKa = 6.18) or other resonance forms are postulated to interact with the N4′ of the thiamine pyrophosphate pyrimidine ring and prevent pyruvate binding (Figure 2).
Aside from replacement of the nitro group of NTZ with halides, there has been little lead optimization. However, knowledge of target as well as of MoA should drive lead optimization of NTZ and produce next generation therapeutics to treat diseases that cause increased morbidity and mortality worldwide. Resistance to NTZ has not been observed clinically or induced experimentally in the laboratory, perhaps relating to drug interaction with the vitamin cofactor rather than with the enzyme directly.[21] Here we report the use of NTZ as a tool drug for probing structure-activity relationships (SAR) to direct lead optimization against the PFOR target essential in many intestinal human pathogens.
Results and Discussion
Synthesis and biological evaluation of the 2-amino-5-nitrothiazole library against PFOR organisms
In an effort to improve the biological activity of NTZ against PFOR utilizing organisms we envisioned the step-wise modification of benzene ring substitution patterns as well as replacement of the phenyl ring with aliphatic and heterocyclic moieties. NTZ and NTZ derivatives have been investigated previously but tend to retain the 2-acetoxy group with only minor changes to the benzene ring.[22, 23] We also sought to determine whether analogues without the 2-acetoxy group could recapitulate NTZ activity.
Helicobacter pylori and Campylobacter jejuni were chosen as PFOR utilizing organisms that could be quickly used to screen the NTZ library for biological activity. H. pylori is a microaerophilic Gram-negative bacterium that causes lifelong infections of the gastric mucosa which can lead to more severe diseases including duodenal and peptic ulcers and gastric cancer.[24] C. jejuni is also a microaerophilic Gram-negative bacterium that is the most common cause of severe lower gastrointestinal infections in mammals.[25] Analogues of interest possessing increased potency against H. pylori and C. jejuni were further assessed for their activity against Clostridium difficile, responsible for antibiotic associated enterocolitis in humans.[26] Secondary screens against Escherichia coli, Staphylococcus aureus (MRSA strain) and S. epidermidis were used to assess spectrum and as a means to explore any new targets that might arise.[27] Finally, NTZ-based derivatives were tested for cytotoxic properties in a human foreskin cell based assay.
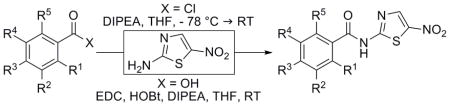
SAR studies began with the synthesis of derivatives primarily bearing systematic substitutions of halides, electron-withdrawing groups and some electron-donating groups (Table 1). Analogues in this sub-library were accessed by coupling 2-amino-5-nitrothiazole (2-ANT) to the respective benzoic acid through EDC coupling or through the corresponding benzoyl chloride derivative that was either commercially available or synthesized in situ from the benzoic acid.
Table 1.
Synthesis and biological evaluation of halide and mono-substituted NTZ analogues.
 | |||||
|---|---|---|---|---|---|
| NTZ Halide Analogues | NTZ Mono-subs Analogues | ||||
| Analogue[a] | MIC’s (μM) | Analogue[a] | MIC’s (μM) | ||
| H. pylori | C. jejuni | H. pylori | C. jejuni | ||
| Nitazoxanide (1) | 13.0 | 39.1 | Nitazoxanide (1) | 13.0 | 39.1 |
| R1 = F (6) | 0.5 | 5.6 | R2 = CN (20) | 4.1 | 36.5 |
| R2 = F (7) | 0.9 | 11.2 | R3 = CN (21) | 9.1 | 43.8 |
| R3 = F (8) | 0.9 | 2.8 | R1 = CF3 (22) | 1.6 | 18.9 |
| R1,3 = F (9) | 0.4 | 7.0 | R2 = CF3 (23) | 3.5 | 4.7 |
| R2,3 = F (10) | 1.8 | 5.3 | R3 = CF3 (24) | 1.6 | 4.7 |
| R1,5 = F (11) | 0.7 | 14.0 | R1 = NO2 (25) | 1.7 | 27.2 |
| R1,3,5 = F (12) | 4.9 | 9.9 | R2 = NO2 (26) | 1.3 | 27.2 |
| R2–4 = F (13) | 1.2 | 4.9 | R3 = NO2 (27) | 1.3 | 13.6 |
| R1,3,4 = F (14) | 0.8 | 9.9 | R1 = OCH3 (28) | 1.8 | 17.9 |
| R1–4 = F (15) | 1.2 | 4.7 | R2 = OCH3 (29) | 1.3 | 7.2 |
| R1–5 = F (16) | 7.4 | 23.6 | R3 = OCH3 (30) | 1.8 | 4.5 |
| R1 = Cl (17) | 0.3 | 7.8 | R2 = OCF3 (31) | 1.1 | 9.0 |
| R2 = Cl (18) | 1.0 | 6.5 | - | - | - |
| R3 = Cl (19) | 0.7 | 6.5 | - | - | - |
R = H unless otherwise noted.
These analogues were then assessed for biological activity through in vitro MIC testing against H. pylori and C. jejuni. The halide series proved useful in establishing that substitutions at any position of the ring appeared to be tolerated but yielded little effect when multiple substitutions of fluorine were investigated. Mono-substitutions of other functional groups resulted in the observation that the para-substituted analogues possessed more activity versus ortho or meta, with cyano analogue 21 being the exception (Table 1). Interestingly, the trifluoromethyl and methoxy analogues, which are nearly electronic opposites, possessed comparable antimicrobial potency. Similar activities for these derivatives strongly suggested that the electronic properties of the phenyl ring were not involved in the biological activity. The best library members in the series were undoubtedly the p-F (8), p-Cl (19), p-trifluoromethyl (24) and p-methoxy (30) analogues which displayed significant improvements in activity. Through the synthesis of halogen and mono-substituted derivatives, it became evident that the 2-acetoxy group was not necessary for NTZ analogue activity and that para-substitutions yielded improvements in activity.
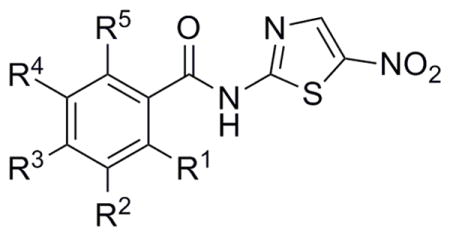
Following the synthesis of the halide and mono-substituted compounds, di-substituted analogues were envisioned that would couple some of the increased activity observed in Table 1 as well as determine if the ortho oxygen could further modulate activity. Derivative 32 was accessed through methylation of p-CF3 salicylic acid followed by saponification of the methyl ester and EDC coupling of 2-ANT with the resulting carboxylic acid.[28] Phenol 34 was synthesized through acylation of m-NO2 salicylic acid followed by EDC coupling of 2-ANT.[29] During the reaction the acetyl group was cleaved and the free phenol 34 was isolated and assayed for biological activity. The remainder of the di-substituted analogues were synthesized from commercially available starting materials through coupling of the acid chloride or EDC coupling of the carboxylic acid with 2-ANT.NTZ derivative activity did not increase with the appendage of ortho-oxygens to the ring system in a consistent manner and analogues 32 and 34 lost activity significantly against C. jejuni (Table 2). Additional di-substituted analogues bearing halogens and electron withdrawing groups were synthesized and assayed but activity for many of these analogues also did not correlate when compared to the activity of the mono-substituted parent derivatives (Table 2). Many of the para-substitutions that yielded increased activity in Table 1 failed to increase activity when further substituted at additional positions. Activity improvements brought about by di-substitution were finally discovered when derivatives bearing both chloro and trifluoromethyl groups were appended to the phenyl ring. The general trend of the additive effects of both the chloro and CF3 groups was noteworthy for analogues 40 and 41 and continued efforts to synthesize the fluoro derivatives may further increase biological activity.
Table 2.
Biological evaluation of di-substituted NTZ analogues.
 | ||
|---|---|---|
| Analogue[a] | MIC’s (μM) | |
| H. pylori | C. jejuni | |
| Nitazoxanide (1) | 13.0 | 39.1 |
| R1 = OCH3; R3 = CF3 (32) | 0.9 | 92.1 |
| R1 = OCH3; R3 = NO2 (33) | 0.6 | 9.3 |
| R1 = OH; R2 = NO2 (34) | 19.3 | 103.1 |
| R1 = CF3; R3 = F (35) | 3.4 | 23.9 |
| R1 = NO2; R3 = CF3 (36) | 8.3 | 88.3 |
| R2 = NO2; R3 = F (37) | 1.6 | 51.2 |
| R2,4 = CF3 (38) | 5.2 | 41.5 |
| R1 = Cl; R4 = CF3 (39) | 1.1 | 17.1 |
| R1 = Cl; R2 = CF3 (40) | 0.9 | 5.7 |
| R2 = CF3; R3 = Cl (41) | 2.8 | 2.8 |
R = H unless otherwise noted.
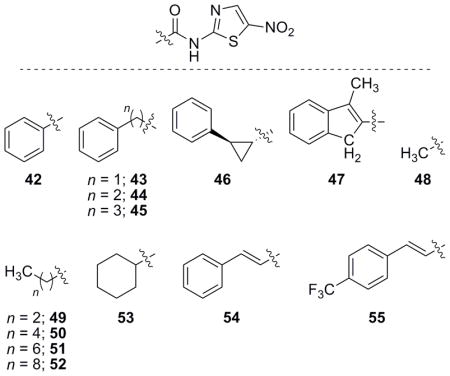
Aliphatic analogues of NTZ were also of interest as they have been previously investigated in relation to the 2-ANT head group,[30] but not in this biological context. Aliphatic derivatives were coupled to 2-ANT in a similar fashion to earlier library members via the carboxylic acid or acid chloride. In general, these derivatives possessed increased activity against H. pylori and C. jejuni in comparison to NTZ (Table 3). However, most analogues lacked significant activity trends to allow any hypotheses relating to their SAR. A small trend was apparent correlating increased chain length to increased activity of up to six carbons in length from the amide carbonyl carbon and that further increasing length (52 and 55) was deleterious to activity.
Table 3.
Biological evaluation of aliphatic NTZ analogues.
 | |||||
|---|---|---|---|---|---|
| Analogue | MIC’s (μM) | Analogue | MIC’s (μM) | ||
| H. pylori | C. jejuni | H. pylori | C. jejuni | ||
| NTZ (1) | 13.0 | 39.1 | NTZ (1) | 13.0 | 39.1 |
| 42 | 1.3 | 3.0 | 49 | 3.5 | 13.9 |
| 43 | 0.7 | 11.4 | 50 | 0.5 | 2.1 |
| 44 | 21.6 | 1.8 | 51 | 1.4 | 5.5 |
| 45 | 0.3 | 1.3 | 52 | 0.8 | 106.9 |
| 46 | 13.8 | 2.6 | 53 | 1.2 | 3.9 |
| 47 | 1.7 | 26.5 | 54 | 0.3 | 2.7 |
| 48 | 0.7 | 10.7 | 55 | 0.5 | 93.2 |
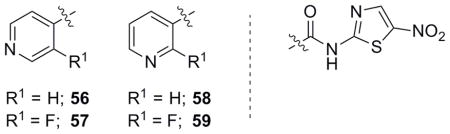
With both aromatic and aliphatic NTZ derivatives synthesized and assayed for biological activity, heteroaromatic derivatives were envisioned to further explore the SAR of NTZ. Pyridine analogues were accessed through both EDC/carboxylic acid couplings and acid chloride couplings with 2-ANT. The unsubstituted pyridine derivatives (56 and 58) displayed low activity compared to NTZ or phenyl derivative 42 (Table 4). Two fluorine substituted pyridine analogues were then synthesized and assayed to determine if the additive effects displayed in Table 1 may be applicable to heterocyclic systems. The ortho-fluoro-substituted pyridines (57 and 59) significantly improved activity over their non-substituted counterparts. Sensitivity to the substitution of ortho-fluorine on the pyridine ring system may allow for further derivatization of pyridine-related analogues with chlorines and trifluoromethyl group substitutions (as with 40 and 41) and is currently under investigation.
Table 4.
Biological evaluation of pyridine heterocyclic NTZ analogues.
 | ||
|---|---|---|
| Analogue | MIC’s (μM) | |
| H. pylori | C. jejuni | |
| Nitazoxanide (1) | 13.0 | 39.1 |
| 56 | 16.0 | 24.0 |
| 57 | 3.7 | 14.9 |
| 58 | 4.0 | 71.9 |
| 59 | 3.7 | 7.5 |
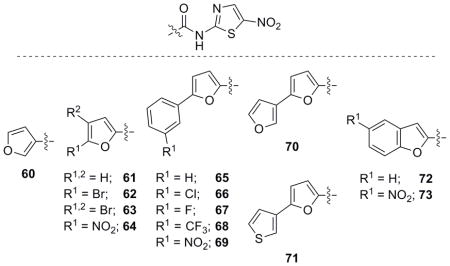
Furan derivatives were investigated next and many were synthesized following the established routes. Furan compounds displayed moderate activity similar to the aromatic and substituted aromatic congeners (Table 5) as several analogues were more active than NTZ. Aryl furan 2-ANT analogues that were accessed through commercially available carboxylic acids displayed moderate to good activity against H. pylori and C. jejuni (68 and 69) which necessitated the synthesis of additional derivatives.
Table 5.
Biological evaluation of furan and furan-related NTZ analogues.
 | |||||
|---|---|---|---|---|---|
| Analogue[a] | MIC’s (μM) | Analogue[a] | MIC’s (μM) | ||
| H. pylori | C. jejuni | H. pylori | C. jejuni | ||
| NTZ (1) | 13.0 | 39.1 | NTZ (1) | 13.0 | 39.1 |
| 60 | 0.8 | 12.5 | 67 | 1.5 | 24.0 |
| 61 | 1.0 | 8.4 | 68 | 1.1 | 8.5 |
| 62 | 1.6 | 12.6 | 69 | 6.9 | 16.7 |
| 63 | 2.8 | 20.2 | 70 | 1.6 | 13.1 |
| 64 | 21.1 | 7.0 | 71 | 6.2 | 99.6 |
| 65 | 2.4 | 101.5 | 72 | 0.6 | 110.6 |
| 66 | 1.1 | 22.9 | 73 | 4.5 | 4.5 |
R = H unless otherwise noted.
Utilizing the power and generality of the Suzuki-Miyaura Cross Coupling,[31] several meta-substituted furoic acid derivatives were synthesized to explore the activity around these phenyl-substituted furan analogues (Scheme 1). Analogues 65–67, 70 and 71 were synthesized using the Suzuki cross coupling and the derivatives produced an SAR that displayed increased activity with the introduction of electron withdrawing groups on the pendant phenyl ring. Analogue 68 was identified as being the best in the benzene ring series and the differential activity of analogues 70 and 71 was noteworthy as the pendant thiophene ring (71) appeared to completely abolish the efficacy of the compound.
Scheme 1.
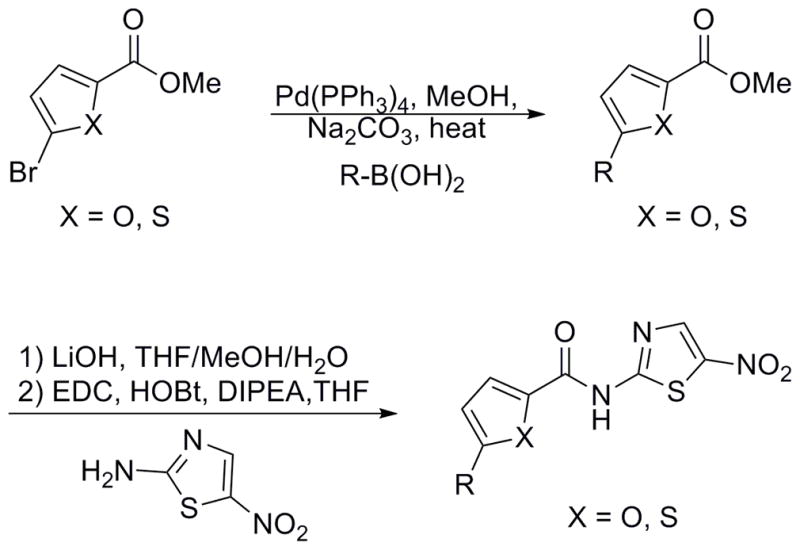
Synthesis of aryl furans and aryl thiophenes.
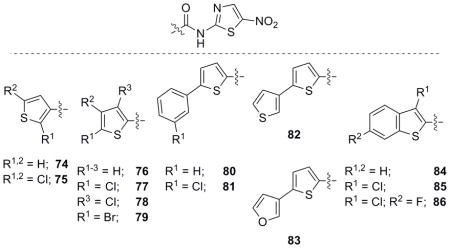
With the synthesis of a few simple pyridines and a more elaborate furan library, thiophenes were the final group to be assessed for activity. Thiophene analogues were synthesized and assayed following the established routes and several phenyl thiophene derivatives were also synthesized through Suzuki cross coupling reactions (Scheme 1).
Biological activity for the thiophene library members (Table 6) was comparable to the furan library with many of the analogues possessing potent activity against both bacteria. The Suzuki coupling analogues (80–83) displayed a similar trend compared to the furan derivatives and were slightly more potent in comparison (66 vs. 81). In contrast to the furan analogues, thiophenes and benzothiophenes were more potent with increased substitution of halogens. Bi-thiophene 82 vs. thiophene-furan 83 was also very interesting as the pendant furan analogue 83 was quite active compared to the latter. This is in direct correlation to the furan series (70 and 71) with the pendant furan derivative 70 possessing the more potent activity. The activity clearly shows the pendant 3-furanyl ring (70 and 83) retains activity and may allow for the appendage of this heterocycle on benzene ring systems as a means to explore the SAR. Further studies including derivatization of the pendant furan ring and modifications of the attachment position are currently in progress.
Table 6.
Biological evaluation of thiophene and related NTZ analogues.
 | |||||
|---|---|---|---|---|---|
| Analogue[a] | MIC’s (μM) | Analogue[a] | MIC’s (μM) | ||
| H. pylori | C. jejuni | H. pylori | C. jejuni | ||
| NTZ (1) | 13.0 | 39.1 | NTZ (1) | 13.0 | 39.1 |
| 74 | 0.7 | 5.9 | 81 | 0.7 | 5.5 |
| 75 | 1.5 | 2.3 | 82 | 3.0 | 94.8 |
| 76 | 2.9 | 2.9 | 83 | 7.8 | 12.4 |
| 77 | 5.2 | 6.9 | 84 | 2.5 | 9.8 |
| 78 | 5.2 | 20.7 | 85 | 2.2 | 5.9 |
| 79 | 3.0 | 9.9 | 86 | 1.0 | 5.6 |
| 80 | 1.5 | 72.4 | - | - | - |
R = H unless otherwise noted.
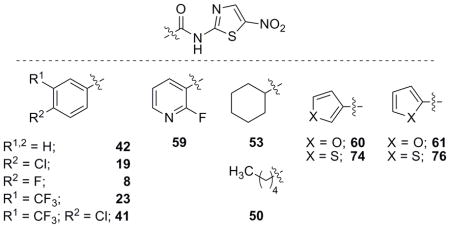
Evaluation of selected analogues against C. difficile and direct PFOR enzyme inhibition
We have synthesized a library of NTZ analogues and have shown the ability of various functional groups to outperform NTZ utilizing H. pylori and C. jejuni inhibition assays. From this library of compounds, several were chosen across the sub-libraries to be further screened for activity against the anaerobic PFOR-containing pathogen Clostridium difficile and in direct in vitro tests of inhibitory action against recombinant H. pylori PFOR purified from E. coli (Table 7).
Table 7.
Biological evaluation of selected analogues against C. difficile and direct PFOR inhibition.
 | ||
|---|---|---|
| Analogue[a] | MIC (μM) | PFOR Inhibition[c] |
| C. difficile | [Drug] = 40 μM (%) | |
| Nitazoxanide (1) | 1.2 | 54 ± 7 |
| 42 | 6.0 | 68 ± 5b |
| 19 | 0.8 | 55 |
| 8 | 3.3 | 85b |
| 23 | 0.5 | 41.5 ± 5.5 |
| 41 | 1.4 | 33 ± 1 |
| 59 | 2.8 | 61 ± 13 |
| 53 | 5.9 | 54 ± 1 |
| 50 | 8.2 | 64 ± 6b |
| 60 | 1.7 | 58 ± 2b |
| 74 | 2.9 | 58.5 ± 3.5b |
| 61 | 2.4 | 42b |
| 76 | 1.5 | 56 ± 6b |
R = H unless otherwise noted;
Complex pattern of inhibition with two different rates;
For PFOR inhibition assays, drug concentration was fixed at 40 μM which bench marks NTZ at ~50% inhibition.
C. difficile is a very common gut anaerobe that utilizes PFOR but unlike H. pylori and C. jejuni, is a Gram-positive bacterium.[32, 33] Many broad-spectrum antibiotics deplete natural gut floral which enables C. difficile to dominate the intestinal track causing severe enterocolitis. Due to recrudescence, C. difficile infections are much more challenging to eradicate.[32]
Upon examining the data in Table 7 much is left to be desired for the analogues as most are less than or equal to NTZ in potency against C. difficile. Analogues 19 and 23 are marginally more potent than NTZ against C. difficile and may represent the difficulties in designing new drugs to combat this bacterium. PFOR inhibition results are equally ambiguous as derivatives having efficacious effects against PFOR utilizing organisms displayed differing values in the direct enzyme inhibition assay. Activity evidenced from the PFOR enzyme assay and lack of effect in the C. difficile assay could be attributed to issues crossing the cell wall, slight differences in PFOR structure among these organisms or off-target effects.
Most of the library members selected and tested retained nearly equipotent inhibitory activity against the PFOR enzyme compared to NTZ but failed to significantly show increased efficacy at the enzymatic target to correlate the increase in activity against PFOR utilizing organisms. Of note are the relatively low PFOR inhibition values for analogues 23, 41 and 61 yet the potent values for inhibition against H. pylori, C. jejuni and to some extent, C. difficile. PFOR results were also complicated by a complex pattern of differing rates that occurred during the PFOR assay which indicated that some activity may be attributable to a nitroreduction mechanism. Experiments to delineate the differences in PFOR activity versus antibacterial assays and the possible role of nitroreduction in the MoA are currently being investigated.
Evaluation of library against non-PFOR utilizing organisms E. coli, S. epidermidis and S. aureus
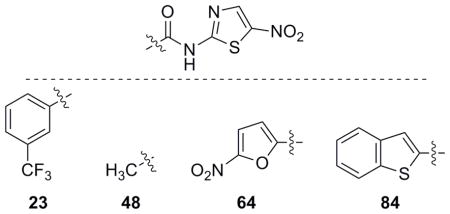
With the synthesis of an 81-member library we have shown that analogues based on the NTZ structure can recapitulate and improve activity against three strains of PFOR containing organisms. E. coli was chosen as a screening organism because NTZ has no antibacterial activity against E. coli (MIC > 32 μg/ml) as the putative target, PFOR, is not present. Ideally, NTZ-based analogues would also have little to no activity against E. coli. When tested, nearly the entire library had no antibacterial efficacy against E. coli. Only four derivatives (23, 48, 64 and 84) displayed activity lower than 50 μM indicating that the library had low off-target activity that may be related to PDH toxicity (Table 8). Low PDH toxicity was also used as a benchmark for low human toxicity as mammals also utilize PDH and this conclusion will be further supported using human foreskin cells (vide infra).
Table 8.
NTZ analogues with activity lower than 50 μM against E. coli.
 | |
|---|---|
| Analogue | E. coli MIC (μM) |
| Nitazoxanide (1) | > 104.1 |
| 23 | 37.8 |
| 48 | 42.7 |
| 64 | 14.1 |
| 84 | 39.8 |
Staphylococci also do not utilize PFOR for energy metabolism and antibacterial activity would represent a new MoA and different biological target. For staphylococcal activity assessment, the entire library was screened for activity against the Gram-positive bacteria S. aureus methicillin-resistant strain (MRSA) and S. epidermidis. MRSA has become especially problematic to treat because of enhanced resistance to standard antibiotic therapies while S. epidermidis has been linked to the increased rate of infection of indwelling medical devices.[34, 35]
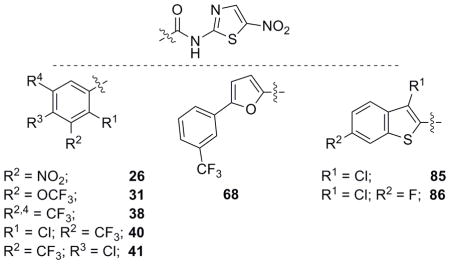
NTZ has moderate activity against MRSA and S. epidermidis but this action cannot be driven by the postulated PFOR MoA.[12] PDH is also not the likely target in staphylococci as the vast majority of the library was inactive at inhibiting E. coli. As a whole, the NTZ analogue library was relatively inactive against staphylococci with average MIC values near that of NTZ or higher (supporting info). Several derivatives displaying activity in the low μM range did stand out and analogues for Table 9 were selected with activity ≤ 12.0 μM against both strains of Staphylococcus.
Table 9.
Biological evaluation of selected analogues against Gram-positive bacteria S. aureus (MRSA) and S. epidermidis.
 | ||
|---|---|---|
| Analogue[a] | MIC’s (μM) | |
| S. aureus (MRSA) | S. epidermidis | |
| Nitazoxanide (1) | 39.1 | 52.1 |
| 26 | 10.2 | 10.2 |
| 31 | 6.0 | 12.0 |
| 38 | 2.6 | 1.9 |
| 40 | 11.4 | 11.4 |
| 41 | 2.8 | 2.8 |
| 68 | 11.4 | 5.7 |
| 85 | 2.9 | 5.9 |
| 86 | 2.1 | 5.6 |
R = H unless otherwise noted.
The first observation of the data strongly suggested the involvement of trifluoromethyl groups in the disruption of staphylococci versus any of the other pendant groups tested. Aromatic derivatives were also the only groups represented, with aliphatic moieties not displaying significant activity. Analogues 38 and 41 were of particular interest as they also displayed significant activity against PFOR utilizing organisms. Benzothiophenes 85 and 86 had moderate activity in the previous PFOR organisms and possessed potent activity against staphylococci. With the identification of several CF3-appended NTZ analogues and benzothiophenes, in particular, derivatives capable of inhibiting bacteria across genera have been identified in this library. As staphylococci do not utilize PFOR, analogues active against both PFOR organisms and staphylococci must be acting through unique MoAs with an unknown target. SAR studies utilizing several of the active compounds in Table 9 are underway to improve activity and aid in target identification.
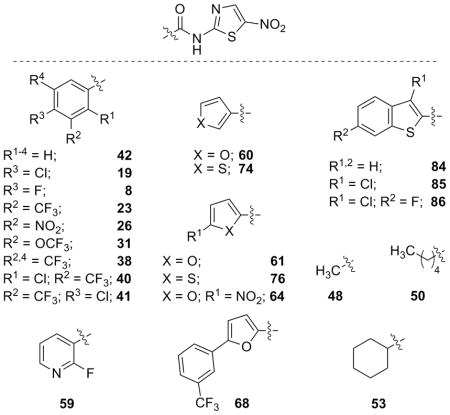
Evaluation of selected analogues for foreskin cell toxicity
As a final assessment of analogue activity and toxicity, a selection of the most active analogues was assayed against human foreskin cells. Table 10 summarizes the foreskin cell toxicity data and also the activity profiles of the selected NTZ derivatives. All of the NTZ analogues tested were relatively non-toxic compared to NTZ. Interestingly, all of the analogues that showed increased activity against E. coli were completely non-toxic to foreskin cells indicating that these analogues may, in fact, be efficacious candidates for non-toxic broad spectrum antibiotics. Only two analogues tested, a nitro aromatic (26) and di-meta trifluoromethyl analogue (38), stood out as being particularly toxic. Surprisingly, the mono-trifluoromethyl analogue 23 and nitrofuran 64 were not toxic compared to 26 or 38. Preliminary safety testing in mice suggests that these compounds (like NTZ) are relatively safe at 200 mg/Kg by oral administration (data not presented).
Table 10.
Evaluation of selected analogues against human foreskin cells and summation of biological data.
 | ||||||
|---|---|---|---|---|---|---|
| Analogue[a] | CC50 (μM) foreskin[b] | MIC’s (μM) | ||||
| H. P. | C. J. | C. D. | S. A. | S. E. | ||
| NTZ (1) | > 52.1 | 13.0 | 39.1 | 1.2 | 39.1 | 52.1 |
| 42 | 46.3 | 1.3 | 3.0 | 6.0 | 12.0 | 32.1 |
| 19 | 56.4 | 0.7 | 6.5 | 0.8 | 2.0 | 26.1 |
| 8 | 29.9 | 0.9 | 2.8 | 3.3 | 8.4 | 15.0 |
| 23 | > 50.4 | 3.5 | 4.7 | 0.5 | 3.2 | 12.6 |
| 26 | 27.2 | 1.3 | 27.2 | n.d. | 10.2 | 10.2 |
| 31 | > 48.0 | 1.1 | 9.0 | n.d. | 6.0 | 12.0 |
| 38 | 10.4 | 5.2 | 41.5 | n.d. | 2.6 | 1.9 |
| 40 | > 45.5 | 0.9 | 5.7 | n.d. | 11.4 | 11.4 |
| 41 | > 45.5 | 2.8 | 2.8 | 1.4 | 2.8 | 2.8 |
| 59 | > 59.7 | 3.7 | 7.5 | 2.8 | 119.3 | 119.3 |
| 60 | > 66.9 | 0.8 | 12.5 | 1.7 | 50.2 | 33.4 |
| 74 | > 62.7 | 0.7 | 5.9 | 2.9 | 23.5 | 15.7 |
| 61 | 66.9 | 1.0 | 8.4 | 2.4 | 50.2 | 66.9 |
| 76 | > 62.7 | 2.9 | 2.9 | 1.5 | 15.7 | 31.3 |
| 64 | > 56.3 | 21.1 | 7.0 | n.d. | 112.6 | 28.1 |
| 68 | > 41.7 | 1.1 | 8.5 | n.d. | 11.4 | 5.7 |
| 84 | > 52.6 | 2.5 | 9.8 | n.d. | 6.6 | 13.1 |
| 85 | > 47.1 | 2.2 | 5.9 | n.d. | 2.9 | 5.9 |
| 86 | 33.5 | 1.0 | 5.6 | n.d. | 2.1 | 5.6 |
| 48 | > 85.5 | 0.7 | 10.7 | n.d. | 171.0 | 21.4 |
| 50 | > 65.8 | 0.5 | 2.1 | 8.2 | 131.5 | 32.9 |
| 53 | > 62.7 | 1.2 | 3.9 | 5.9 | 125.2 | 31.3 |
R = H unless otherwise noted. n.d. = not determined.
48h time point.
Conclusion
From the synthesis and biological evaluation of NTZ-based analogues against three PFOR utilizing organisms, E. coli and two staphylococcal strains it is apparent that activity against these organisms can be improved beyond that of NTZ in both a broad and selective manner. Since NTZ targets PFOR, this was the logical first choice for investigation and numerous derivatives recapitulated and outperformed NTZ’s activity against both H. pylori and C. jejuni. We were able to determine that the 2-acetoxy group or simply an ortho-oxygen was not necessary for activity. The electronic properties of pendant benzene rings were also less important and we postulate that steric, ionic and hydrophobic interactions play major roles in the MoA.
Recapitulating gains in activity in the halogen and mono-substituted sub-libraries with di-substituted analogues was problematic and led to nearly every derivative being less potent. Activity against PFOR organisms in the di-substituted library was discovered for CF3/Cl combinations (40 and 41) (Table 10) and these will be further investigated with fluorine and additional substituents. These CF3/Cl di-substituted derivatives were also shown to be very active against both C. difficile and staphylococci.
Heterocycles displayed moderate to good activity against PFOR utilizing organisms and several had potent staphylococcal activity. Of particular note were the bi-aryl furan-furan (70) and thiophene-furan (83) analogues that displayed good activity against PFOR organisms but their pendant thiophene counterparts were completely inactive. Further studies modifying the connectivity and substitution profile of the pendant ring system of the bi-aryl groups and the substitution of furans on benzene rings are of great interest.
In general, C. difficile activity was moderate at best for the selected library members tested and may reflect the difficulty of treating this infection. As only selected analogues were tested, limited SAR can be drawn but it would be prudent to investigate halogen and CF3 substituted furans and thiophenes for activity improvements. PFOR enzyme results for the selected derivatives did provide evidence that PFOR is being targeted and should be responsible for the activity of the NTZ-based library of analogues. However, it was clear that factors independent of PFOR inhibition may account for the activity of some analogues.
Selective activity against PFOR utilizing organisms was observed for several library members that displayed moderate to good activity against all PFOR organisms and in the direct enzyme assay, but were not active against staphylococci. Of particular note were the heterocyclic analogues 59, 60, 61, 74 and 76 (Table 10) which represent the fluoro-substituted pyridine and unsubstituted furans and thiophenes. These analogues displayed good PFOR selectivity and moderate enzyme inhibition comparable to NTZ.
Broad spectrum activity against PFOR, PFOR utilizing organisms and staphylococci was discovered for several CF3-substituted benzene, aryl-furan and benzothiophene derivatives. Although compound 23 did indicate some possible activity against E. coli, di-substituted analogues bearing CF3 groups did not suffer from this effect (data not shown) and human foreskin toxicity data indicated that 23, 40 and 41 were not toxic (Table 10). In particular, derivative 41 was active against all of the PFOR and staphylococci organisms yet did not display a correlating increase in the direct PFOR enzymatic inhibition assay. Amide 41, therefore, must be acting through dual (multiple) pathways and this compound will require further investigation.
In conclusion, the exploration of NTZ-based analogues to improve activity yielded compounds capable of inhibiting across a broad spectrum and also selectively against PFOR organisms. This research has yielded much information regarding what can and can not improve activity and further studies and analogue derivatizations are needed to continue to improve activity. Efforts are underway to access and evaluate new derivatives based on the current SAR and to begin implementation of the PFOR crystal structure in analogue evaluation and design. Coupling the results presented herein with the recent identification of head groups able to recapitulate and replace the 2-ANT is expected to produce more efficacious analogues and results will be forth coming.[36]
Experimental Section
Determination of MIC values for H. pylori, C. jejuni and Staphylococci (liquid dilution)
H. pylori was grown overnight at 37 °C under microaerobic conditions in Bacto Brain Heart infusion (BHI) medium supplemented with 4% serum. C. jejuni was grown in BHI medium without supplementation. Staphylococci were grown in Bacto Trpytic soy medium without supplementation. For the microdilution assay, bacterial cultures were diluted to a final OD600 of 0.03 for H. pylori strain 26695, 0.01 for C. jejuni strain H840 and 0.01 for Staphylococci and 100 μL was dispensed into wells of a 96 well microplate. Analogues were diluted serially starting at 32 μg/mL in DMSO and the percent DMSO was always less than 4%. DMSO and NTZ served as controls. Plates were incubated with shaking at 37 °C in a microaerobic incubator (7% O2 and 10% CO2). The turbidity in the wells was read visually at 27 h or with a plate reader (Molecular Dynamics). MIC is defined as the concentration of drug that produced no detectable bacterial growth. All experiments were performed 3–6 times in triplicate.
Determination of MIC values for C. difficile (agar dilution)
C. difficile VPI 10463 was grown anaerobically overnight in chopped meat medium (Anaerobe system) from stock, and it was subcultured to a new chopped meat medium for 5 hours at 37 °C. It was standardized to an optical density of 0.1 at OD600. Analogues were then diluted into the agar media at concentrations ranging from 0.125–8 μg/ml. Ten-microliter volumes of the standardized inoculum were delivered to the surface of the agar plates. The number of viable bacteria contained in each inoculum was approximately 7 × 104 and 3.5 × 104 organisms. The plates were incubated for 18 hours in an anaerobic chamber and were read visually for growth or no growth. Anaerobic plates containing no compound were used as controls. All experiments were performed 3–6 times in triplicate.
Direct PFOR enzyme assay
H. pylori PFOR enzyme was overexpressed and purified from E. coli as described previously.[15] Enzymatic assays were carried out at 25 °C in 1-mL cuvettes in a modified Cary-14 spectrophotometer equipped with an OLIS data acquisition system (On Line Instrument Co., Bogart, Georgia). PFOR (EC 1.2.7.1) was assayed under anaerobic conditions with 100 mM potassium phosphate (pH 7.4), 10 mM sodium pyruvate, 5 mM benzyl viologen (BV; ε=9.2 mM−1 cm−1 at 546 nm), 0.18 mM CoA, and 1 mM MgCl2. The reaction was started by addition of enzyme, in the presence or absence of inhibitor (NTZ or its derivative in concentration of 40 μM) and the reduction of redox-active BV dye was monitored at 546 nm. Inhibition of PFOR was expressed in %.
Determination of Human Foreskin Cell Toxicity
Human foreskin cells were plated in 96-well plates at 1.6 × 103 cells per well using Medium 106® (Invitrogen). Plates were incubated overnight in a CO2 incubator at 37 °C to allow cells to adhere to the bottom of the wells. Test compounds were serially diluted in replicate sets of plates. After 24 hours, 0.02% resazurin sodium salt was added to each well of the first set of plates and placed back at 37 °C to incubate for 2 hours. At that time, the plates with resazurin were read on a plate reader at OD570. The second set of plates was treated in the same manner at the 48 hour time point. All assays were performed in triplicate and in two independent assays. The CC50 was recorded as the drug concentration that inhibited 50% of the resazurin reduction by the untreated controls. All experiments were performed 2–3 times in triplicate. DMSO concentration did not exceed 0.6%.
Chemistry
All reagents were purchased from commercially available sources and used as is without further purification. All reactions were run under a nitrogen or argon atmosphere unless otherwise noted. Flash silica gel chromatography was performed with 60 Å mesh standard grade silica gel (Sorbtech). 1H and 13C NMR spectra were obtained using Varian 300 MHz or 500 MHz spectrometers and recorded at 23 °C. Chemical shifts (s = singlet, bs = broad singlet, d = doublet, t = triplet, dd = doublet of doublets, dt = doublet of triplets, td = triplet of doublets, ddd = doublet of doublet of doublets, m = multiplet) are given in parts per million relative to DMSO-d6 (δ 2.50) and CDCl3 (δ 7.27) for proton spectra and relative to DMSO-d6 (δ 39.51) for carbon spectra. Mass spectra were obtained at the NCSU Department of Chemistry Mass Spectrometry Facility which is funded by the North Carolina Biotechnology Center and the NCSU Department of Chemistry.
Method A - Acid Chloride Coupling
Acid chloride (~100 mg or ~0.1 mL, 1 eq) was dissolved in THF (0.1 M) and cooled to −78 °C then 2-amino-5-nitrothiazole (1 eq) was added in one portion. DIPEA (1.1 eq) was added to the resulting slurry at −78 °C and the solution was held at this temperature for 10 mins then allowed to warm to room temperature overnight. The solution was judged complete by TLC analysis (~24 h) and was diluted with EtOAc (30 mL) and washed with sat. NaHCO3 (3 × 20 mL), 1 M HCl (3 × 20 mL) and brine (2 × 20 mL) then dried (MgSO4) followed by filtration and evaporation to dryness. The resulting residue was purified by gradient flash column chromatography (10–60% EtOAc/hexanes or 1–2% MeOH/CH2Cl2) to obtain the product.
Method B - Carboxylic Acid EDC Coupling
Carboxylic acid (~100 mg, 1 eq), EDC (2 eq), HOBT (2 eq) and DIPEA (3 eq) were dissolved in THF (0.1 M) and stirred for 15 mins. 2-Amino-5-nitrothiazole (1 eq) was then added in one portion and the reaction was stirred at ambient temperature. Once judged complete by TLC analysis (~24 h), the resulting suspension was diluted with EtOAc (30 mL) and washed with sat. NaHCO3 (3 × 20 mL), 1 M HCl (3 × 20 mL) and brine (2 × 20 mL) then dried (MgSO4) followed by filtration and evaporation to dryness. The resulting residue was purified by gradient flash column chromatography (10–60% EtOAc/hexanes or 1–2% MeOH/CH2Cl2) to obtain the product.
Method C - Acid Chloride Formation
Carboxylic acid (100 mg, 1 eq) was dissolved in CH2Cl2 (0.3 M) with a drop of DMF (catalytic) and cooled to 0 °C then 2 M (COCl)2 in CH2Cl2 (1.0 mL, 3 eq) was added dropwise to the stirring solution. The slurry was allowed to warm to room temperature for 2 h then concentrated to dryness using hexanes to remove the excess (COCl)2. The crude acid chloride obtained was used in the next step without further purification.
Method D - Suzuki Coupling of Furans and Thiophenes
Methyl 5-bromofuran-2-carboxylate (100–150 mg, 1 eq), Pd(PPh3)4 (5 mol%) or PdCl2(PPh3)2 (5 mol%), 2 M Na2CO3 (2 eq) and the respective boronic acid (1.3 eq) in 1,4-dioxanes (0.1 M) was warmed to 90 °C. The solution was then held at this temperature for 5–24 h and judged complete by TLC then cooled and washed with 1 M HCl (2 × 20 mL), brine (2 × 20 mL) then dried (MgSO4) followed by filtration and evaporation to dryness. The resulting residue was then purified by flash column chromatography (5–15% EtOAc/hexanes) to obtain the product.
Method E - Alkyl Ester Saponification
Ester (1 eq) was dissolved in a mixture of MeOH:THF:H2O (1 M: 1 M: 1 M) then LiOH·H2O (3 eq) was added. The solution was stirred for 24 h then was quenched with 1 M HCl (20 mL) and extracted with EtOAc (4 × 15 mL). The combined organic layers were then washed with brine (2 × 20 mL) then dried (MgSO4) followed by filtration and evaporation to dryness to obtain the product.
2-Fluoro-N-(5-nitrothiazol-2-yl)benzamide (6)
Method A yielded the title compound 6 (138 mg, 61%) as an orange solid. 1H NMR (300 MHz, DMSO-d6) δ 13.58 (s, 1H), 8.70 (s, 1H), 7.80 (t, J = 7.5 Hz, 1H), 7.69 (q, J = 7.5 Hz, 1H), 7.48–7.30 (m, 1H); 13C NMR (75 MHz, DMSO-d6) δ 164.1, 161.7, 159.5 (d, JCF = 253 Hz), 142.6, 134.6 (d, JCF = 8.5 Hz), 130.6, 124.8 (d, JCF = 3.4 Hz), 120.9 (d, JCF = 13.0 Hz), 116.5 (d, JCF = 21.2 Hz); HRMS (ESI) calcd for [C10H6FN3O3S + H]+ 268.0187, found 268.0193.
3-Fluoro-N-(5-nitrothiazol-2-yl)benzamide (7)
Method A yielded the title compound 7 (164 mg, 73%) as a light yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 13.69 (s, 1H), 8.74 (s, 1H), 8.04–7.88 (m, 2H), 7.70–7.49 (m, 2H); 13C NMR (75 MHz, DMSO-d6) δ 165.3, 162.4, 161.9 (d, JCF = 244 Hz), 142.5, 142.2, 133.1 (d, JCF = 7.5 Hz), 131.1 (d, JCF = 8.1 Hz), 124.9 (d, JCF = 2.8 Hz), 120.4 (d, JCF = 21.2 Hz, 1H), 115.3 (d, JCF = 23.6 Hz); HRMS (ESI) calcd for [C10H6FN3O3S + H]+ 268.0187, found 268.0195.
4-Fluoro-N-(5-nitrothiazol-2-yl)benzamide (8)
Method A yielded the title compound 8 (96 mg, 43%) as a tan solid. 1H NMR (300 MHz, DMSO-d6) δ 13.62 (s, 1H), 8.72 (s, 1H), 8.22 (dd, J = 8.8, 5.4 Hz, 2H), 7.43 (t, J = 8.9 Hz, 2H); 13C NMR (125 MHz, DMSO-d6) δ 165.3, 165.2 (d, JCF =150 Hz), 162.6, 142.6, 142.1, 131.6 (d, JCF = 9.5 Hz), 127.4, 115.9 (d, JCF = 22.1 Hz); HRMS (ESI) calcd for [C10H6FN3O3S + H]+ 268.0187, found 268.0196.
2,4-Difluoro-N-(5-nitrothiazol-2-yl)benzamide (9)
Method A yielded the title compound 9 (158 mg, 68%) as a pale yellow solid. 1H NMR (500 MHz, DMSO-d6) δ 13.60 (bs, 1H), 8.70 (s, 1H), 7.91 (dd, J = 14.9, 8.4 Hz, 1H), 7.59–7.40 (m, 1H), 7.29 (td, J = 8.6, 2.4 Hz, 1H); 13C NMR (125 MHz, DMSO-d6) δ 165.7 (d, JCF = 12.5 Hz), 163.7 (d, JCF = 12.2 Hz), 162.4 (d, JCF = 198 Hz), 161.4, 142.5, 142.2, 132.6 (d, JCF = 10.3 Hz), 117.7 (d, JCF = 15.5 Hz), 112.2 (d, JCF = 21.7 Hz), 105.1 (t, JCF = 26.0 Hz); HRMS (ESI) calcd for [C10H5F2N3O3S + H]+ 286.0092, found 286.0103.
3,4-Difluoro-N-(5-nitrothiazol-2-yl)benzamide (10)
Method B yielded the title compound 10 (104 mg, 58%) as a pale yellow solid. 1H NMR (500 MHz, DMSO-d6) δ 13.62 (bs, 1H), 8.67 (s, 1H), 8.17 (ddd, J = 11.2, 7.7, 2.1 Hz, 1H), 8.14–7.95 (m, 1H), 7.64 (dt, J = 10.2, 8.4 Hz, 1H); 13C NMR (125 MHz, DMSO-d6) δ 164.3, 162.4, 152.6 (dd, JCF = 254, 12.6 Hz), 149.2 (dd, JCF = 248, 13.1 Hz), 142.3, 142.1, 128.2 (d, JCF = 3.8 Hz), 126.5 (dd, JCF = 7.5, 3.0 Hz), 118.1 (dd, JCF = 18.4, 6.1 Hz); HRMS (ESI) calcd for [C10H5F2N3O3S + H]+ 286.0092, found 286.0098.
2,6-Difluoro-N-(5-nitrothiazol-2-yl)benzamide (11)
Method A yielded the title compound 11 (160 mg, 70%) as a dark yellow solid. 1H NMR (500 MHz, DMSO-d6) δ 13.97 (s, 1H), 8.69 (s, 1H), 7.74–7.68 (m, 1H), 7.31 (t, J = 8.4 Hz, 2H); 13C NMR (125 MHz, DMSO-d6) δ 160.8, 160.1, 159.0 (dd, JCF = 247, 5.0 Hz), 142.6, 142.4, 134.2 (t, JCF = 10.2 Hz), 112.4 (dd, JCF = 20.3, 3.7 Hz), 111.8 (t, JCF = 20.5 Hz); HRMS (ESI) calcd for [C10H5F2N3O3S + H]+ 286.0092, found 286.0095.
2,4,6-Trifluoro-N-(5-nitrothiazol-2-yl)benzamide (12)
Method B with DMAP (cat.) yielded the title compound 12 (89 mg, 65%) as a light yellow solid.. 1H NMR (500 MHz, DMSO-d6) δ 13.97 (bs, 1H), 8.69 (s, 1H), 7.45 (t, J = 8.9 Hz, 2H); 13C NMR (125 MHz, DMSO-d6) δ 163.9 (dt, JCF = 250, 15.7 Hz), 160.8, 159.7 (dt, JCF = 245, 13.3 Hz), 159.2, 142.5, 142.4, 108.8 (t, JCF = 22.8 Hz), 101.7 (t, JCF = 26.7 Hz); HRMS (ESI) calcd for [C10H4F3N3O3S + H]+ 303.9998, found 304.0010.
3,4,5-Trifluoro-N-(5-nitrothiazol-2-yl)benzamide (13)
Method B yielded the title compound 13 (68 mg, 40%) as a beige solid. After reaction completion, as judged by TLC analysis, solution was concentrated to dryness and purified directly from the residue without dilutions or washings. 1H NMR (500 MHz, DMSO-d6) δ 13.78 (bs, 1H), 8.73 (s, 1H), 8.13–8.07 (m, 2H); 13C NMR (125 MHz, DMSO-d6) δ 163.7, 162.4, 150.2 (dd, JCF = 249, 6.8 Hz), 142.3, 142.2, 142.1 (dt, JCF = 255, 15.2 Hz), 127.4, 114.0 (d, JCF = 22.9 Hz); HRMS (ESI) calcd for [C10H4F3N3O3S + H]+ 303.9998, found 304.0007.
2,4,5-Trifluoro-N-(5-nitrothiazol-2-yl)benzamide (14)
Method A yielded the title compound 14 (161 mg, 68%) as a beige solid. 1H NMR (500MHz, DMSO-d6) δ 13.67 (bs, 1H), 8.68 (s, 1H), 7.98 (ddd, J = 10.3, 9.0, 6.5 Hz, 1H), 7.79 (td, J = 10.4, 6.5 Hz, 1H); 13C NMR (125 MHz, DMSO-d6) δ 162.2, 161.5, 155.7 (dd, JCF = 254, 9.2 Hz), 151.8 (dt, JCF = 253, 13.8 Hz), 145.8 (dd, JCF = 246, 9.6 Hz), 142.3, 118.9 (d, JCF = 21.2 Hz), 117.6 (d, JCF = 15.7 Hz), 107.4 (dd, JCF = 28.3, 22.0 Hz); HRMS (ESI) calcd for [C10H4F3N3O3S + H]+ 303.9998, found 304.0007.
2,3,4,5-Tetrafluoro-N-(5-nitrothiazol-2-yl)benzamide (15)
Method A yielded the title compound 15 (158 mg, 66%) as a light orange solid. 1H NMR (300 MHz, DMSO-d6) δ 13.74 (bs, 1H), 8.73 (s, 1H), 8.14–7.75 (m, 1H); 13C NMR (125 MHz, DMSO-d6) δ 161.5, 146.0 (dd, JCF = 248, 8.4 Hz), 145.7 (dd, JCF = 254, 9.8 Hz), 142.3, 142.2 (dt, JCF = 256, 13.6 Hz), 142.1, 139.1, 117.4, 112.7 (d, JCF = 20.9 Hz); HRMS (ESI) calcd for [C10H3F4N3O3S + H]+ 321.9904, found 321.9911.
2,3,4,5,6-Pentafluoro-N-(5-nitrothiazol-2-yl)benzamide (16)
Method A yielded the title compound 16 (95 mg, 65%) as a light yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 8.74 (s, 1H); 13C NMR (125 MHz, DMSO-d6) δ 160.8, 157.3, 144.9, 143.7, 142.7, 142.2, 141.7, 138.2, 136.2; HRMS (ESI) calcd for [C10H2F5N3O3S + H]+ 339.9810, found 339.9820.
2-Chloro-N-(5-nitrothiazol-2-yl)benzamide (17)
Method A yielded the title compound 17 (94 mg, 42%) as a yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 13.74 (s, 1H), 8.70 (s, 1H), 7.75–7.70 (m, 1H), 7.66–7.45 (m, 3H); 13C NMR (125 MHz, DMSO-d6) δ 166.2, 161.4, 142.6, 142.4, 132.9, 132.7, 130.4, 130.0, 129.8, 127.4; HRMS (ESI) calcd for [C10H6ClN3O3S + H]+ 283.9891, found 283.9900.
3-Chloro-N-(5-nitrothiazol-2-yl)benzamide (18)
Method A yielded the title compound 18 (128 mg, 58%) as a light yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 8.68 (s, 1H), 8.17 (s, 1H), 8.05 (d, J = 7.8 Hz, 1H), 7.73 (d, J = 7.0 Hz, 1H), 7.59 (t, J = 7.9 Hz, 1H); 13C NMR (75 MHz, DMSO-d6) δ 166.0, 163.8, 143.0, 142.7, 133.7, 133.5, 132.9, 130.7, 128.3, 127.3; HRMS (ESI) calcd for [C10H6ClN3O3S + H]+ 283.9891, found 283.9902.
4-Chloro-N-(5-nitrothiazol-2-yl)benzamide (19)
Method A yielded the title compound 19 (125 mg, 57%) as a light yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 13.67 (s, 1H), 8.72 (s, 1H), 8.13 (d, J = 8.5 Hz, 2H), 7.66 (d, J = 8.7 Hz, 2H); 13C NMR (125 MHz, DMSO-d6) δ 165.5, 162.5, 142.5, 142.1, 138.4, 130.5, 129.6, 128.9; HRMS (ESI) calcd for [C10H6ClN3O3S + H]+ 283.9891, found 283.9900.
3-Cyano-N-(5-nitrothiazol-2-yl)benzamide (20)
Method A yielded the title compound 20 (145 mg, 88%) as a bright yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 13.68 (s, 1H), 8.68 (s, 1H), 8.51 (s, 1H), 8.34 (d, J = 8.0 Hz, 1H), 8.11 (d, J = 7.7 Hz, 1H), 7.76 (t, J = 7.9 Hz, 1H); 13C NMR (75 MHz, DMSO-d6) δ 164.7, 162.1, 141.9, 141.6, 136.0, 132.7, 132.0, 131.9, 129.7, 117.5, 111.7; HRMS (ESI) calcd for [C11H6N4O3S + H]+ 275.0233, found 275.0243.
4-Cyano-N-(5-nitrothiazol-2-yl)benzamide (21)
Method C followed by Method A yielded the title compound 21 (134 mg, 72%) as a yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 13.74 (s, 1H), 8.70 (s, 1H), 8.21 (d, J = 8.4 Hz, 2H), 8.03 (d, J = 8.4 Hz, 2H); 13C NMR (75 MHz, DMSO-d6) δ 165.4, 162.3, 142.4, 142.2, 134.9, 132.7, 129.3, 118.0, 115.4; HRMS (ESI) calcd for [C11H6N4O3S + H]+ 275.0233, found 275.0243.
N-(5-Nitrothiazol-2-yl)-2-(trifluoromethyl)benzamide (22)
Method A yielded the title compound 22 (55 mg, 25%) as a yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 13.84 (bs, 1H), 8.71 (s, 1H), 8.07–7.71 (m, 4H); 13C NMR (75 MHz, DMSO-d6) δ 166.9, 161.3, 132.7, 132.0 (q, JCF = 2.1 Hz), 131.6, 129.5 (q, JCF = 265 Hz), 129.2, 127.9 (q, JCF = 268 Hz), 126.7 (q, JCF = 4.7 Hz), 123.5 (d, JCF = 274 Hz); HRMS (ESI) calcd for [C11H6F3N3O3S + H]+ 318.0155, found 318.0165.
N-(5-Nitrothiazol-2-yl)-3-(trifluoromethyl)benzamide (23)
Method A yielded the title compound 23 (157 mg, 75%) as a light yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 13.85 (bs, 1H), 8.74 (s, 1H), 8.51 (s, 1H), 8.39 (d, J = 8.2 Hz, 1H), 8.06 (d, J = 7.9 Hz, 1H), 7.83 (t, J = 7.9 Hz, 1H); 13C NMR (125 MHz, DMSO-d6) δ 165.2, 162.4, 142.5, 142.2, 132.7, 131.9, 130.1, 129.8 (q, JCF = 3.4 Hz), 129.4 (q, JCF = 32.6 Hz), 125.2 (q, JCF = 3.9 Hz), 123.8 (q, JCF = 273 Hz); HRMS (ESI) calcd for [C11H6F3N3O3S + H]+ 318.0155, found 318.0164.
N-(5-Nitrothiazol-2-yl)-4-(trifluoromethyl)benzamide (24)
Method A yielded the title compound 24 (159 mg, 75%) as a light yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 13.86 (bs, 1H), 8.74 (s, 1H), 8.30 (d, J = 8.2 Hz, 2H), 7.97 (d, J = 8.4 Hz, 2H); 13C NMR (75 MHz, DMSO-d6) δ 165.6, 162.4, 142.5, 142.2, 134.8, 132.7 (q, JCF = 32.2 Hz), 129.5, 125.7 (q, JCF = 3.4 Hz), 123.7 (q, JCF = 273 Hz); HRMS (ESI) calcd for [C11H6F3N3O3S + H]+ 318.0155, found 318.0162.
2-Nitro-N-(5-nitrothiazol-2-yl)benzamide (25)
Method A yielded the title compound 25 (83 mg, 37%) as a tan solid. 1H NMR (300 MHz, DMSO-d6) δ 13.80 (s, 1H), 8.71 (s, 1H), 8.25 (d, J = 8.2 Hz, 1H), 8.04 – 7.72 (m, 3H); 13C NMR (75 MHz, DMSO-d6) δ 165.9, 161.4, 146.1, 142.5, 142.4, 134.6, 132.4, 129.8, 129.2, 124.6; HRMS (ESI) calcd for [C10H6N4O5S + H]+ 295.0132, found 295.0135.
3-Nitro-N-(5-nitrothiazol-2-yl)benzamide (26)
Method A yielded the title compound 26 (131 mg, 82%) as a bright yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 13.92 (s, 1H), 8.94 (t, J = 1.8 Hz, 1H), 8.69 (s, 1H), 8.48 (dd, J = 8.0, 1.8 Hz, 2H), 7.85 (t, J = 8.0 Hz, 1H); 13C NMR (75 MHz, DMSO-d6) δ 164.7, 162.4, 147.8, 142.3, 134.8, 132.4, 130.5, 127.7, 123.3; HRMS (ESI) calcd for [C10H6N4O5S + H]+ 295.0132, found 295.0137.
4-Nitro-N-(5-nitrothiazol-2-yl)benzamide (27)
Method A yielded the title compound 27 (133 mg, 84%) as a yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 8.76 (s, 1H), 8.40 (d, J = 8.5 Hz, 2H), 8.33 (d, J = 9.0 Hz, 2H); 13C NMR (125 MHz, DMSO-d6) δ 165.2, 162.4, 150.0, 142.4, 142.2, 136.5, 130.2, 123.7; HRMS (ESI) calcd for [C10H6N4O5S + H]+ 295.0132, found 295.0139.
2-Methoxy-N-(5-nitrothiazol-2-yl)benzamide (28)
Method A with DMAP (cat.) yielded the title compound 28 (29 mg, 16%) as a tan solid. 1H NMR (300 MHz, DMSO-d6) δ 12.90 (s, 1H), 8.69 (s, 1H), 7.68 (dd, J = 7.6, 1.7 Hz, 1H), 7.65 – 7.57 (m, 1H), 7.23 (d, J = 8.4 Hz, 1H), 7.11 (t, J = 7.5 Hz, 1H), 3.90 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 165.9, 161.5, 157.2, 142.7, 134.0, 130.2, 121.0, 120.6, 112.3, 56.1; HRMS (ESI) calcd for [C11H9N3O4S + H]+ 280.0387, found 280.0396.
3-Methoxy-N-(5-nitrothiazol-2-yl)benzamide (29)
Method A yielded the title compound 29 (31 mg, 16%) as a tan solid. 1H NMR (300 MHz, DMSO-d6) δ 13.61 (s, 1H), 8.73 (s, 1H), 7.71 (t, J = 4.1 Hz, 2H), 7.50 (t, J = 7.8 Hz, 1H), 7.31 – 7.21 (m, 1H), 3.86 (s, 3H); 13C NMR (75 MHz, DMSO-d6) δ 166.2, 162.6, 159.3, 142.7, 142.1, 132.0, 130.0, 120.9, 119.9, 113.0, 55.5; HRMS (ESI) calcd for [C11H9N3O4S + H]+ 280.0387, found 280.0395.
4-Methoxy-N-(5-nitrothiazol-2-yl)benzamide (30)
Method A yielded the title compound 30 (78 mg, 38%) as a tan solid. 1H NMR (300 MHz, DMSO-d6) δ 13.37 (s, 1H), 8.70 (s, 1H), 8.14 (d, J = 8.7 Hz, 2H), 7.11 (d, J = 8.7 Hz, 2H), 3.86 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 165.7, 163.4, 163.1, 142.8, 141.7, 130.8, 122.9, 114.1, 55.6; HRMS (ESI) calcd for [C11H9N3O4S + H]+ 280.0387, found 280.0388.
N-(5-Nitrothiazol-2-yl)-3-(trifluoromethoxy)benzamide (31)
Method B yielded the title compound 31 (91 mg, 56%) as an orange solid. 1H NMR (300 MHz, DMSO-d6) δ 13.77 (bs, 1H), 8.74 (s, 1H), 8.20 – 8.14 (m, 1H), 8.12 (s, 1H), 7.78 – 7.67 (m, 2H); 13C NMR (75 MHz, DMSO-d6) δ 165.0, 162.4, 148.4, 142.4, 142.2, 133.0, 131.0, 127.7, 125.9, 120.9, 120.0 (d, JCF = 256 Hz); HRMS (ESI) calcd for [C11H6F3N3O4S + H]+ 334.0104, found 334.0115.
2-Methoxy-N-(5-nitrothiazol-2-yl)-4-(trifluoromethyl)benzamide (32)
Method B employing known 2-methoxy-4-(trifluoromethyl)benzoic acid[28] yielded the title compound 32 (61 mg, 77%) as an orange solid. 1H NMR (300 MHz, DMSO-d6) δ 13.32 (s, 1H), 8.70 (s, 1H), 7.82 (d, J = 7.9 Hz, 1H), 7.51 (s, 1H), 7.46 (d, J = 7.9 Hz, 1H), 3.96 (s, 3H); 13C NMR (75 MHz, DMSO-d6) δ 165.3, 161.3, 157.3, 142.7, 142.2, 133.1 (q, JCF = 32.0 Hz), 130.9, 125.80, 123.6 (q, JCF = 273 Hz), 117.2, 109.1, 56.6; HRMS (ESI) calcd for [C12H8F3N3O4S + H]+ 348.0260, found 348.0276.
2-Methoxy-4-nitro-N-(5-nitrothiazol-2-yl)benzamide (33)
Method C followed by Method A yielded the title compound 33 (96 mg, 59%) as a light yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 13.48 (s, 1H), 8.70 (s, 1H), 7.97 – 7.89 (m, 2H), 7.88 – 7.82 (m, 1H), 3.99 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 165.0, 161.3, 157.5, 150.4, 142.6, 131.0, 127.9, 115.4, 107.1, 56.9; HRMS (ESI) calcd for [C11H8N4O6S + H]+ 325.0237, found 325.0246.
2-Hydroxy-3-nitro-N-(5-nitrothiazol-2-yl)benzamide (34)
Method C employing known 2-acetoxy-3-nitrobenzoic acid[29] followed by Method A yielded the title compound 34 (60 mg, 44%) as a yellow solid. Note: 2-O-acetyl group cleaved under the reaction conditions to afford the 2-hydroxy derivative. 1H NMR (300 MHz, DMSO-d6) δ 8.61 (s, 1H), 8.10 (dd, J = 7.7, 1.9 Hz, 1H), 7.91 (dd, J = 8.0, 1.9 Hz, 1H), 6.64 (t, J = 7.8 Hz, 1H); 13C NMR (125 MHz, DMSO-d6) δ 167.5, 165.7, 160.8, 144.1, 140.1, 135.0, 129.8, 120.6, 112.6; HRMS (ESI) calcd for [C10H6N4O6S + H]+ 311.0081, found 311.0092.
4-Fluoro-N-(5-nitrothiazol-2-yl)-2-(trifluoromethyl)benzamide (35)
Method A yielded the title compound 35 (89 mg, 40%) as a light yellow solid. 1H NMR (500 MHz, DMSO-d6) δ 8.70 (s, 1H), 7.98 (dd, J = 8.6, 5.3 Hz, 1H), 7.88 (dd, J = 9.2, 2.5 Hz, 1H), 7.75 (td, J = 8.4, 2.5 Hz, 1H); 13C NMR (125 MHz, DMSO-d6) δ 166.0, 162.9 (d, JCF = 251 Hz), 161.4, 142.5, 132.4 (d, JCF = 9.0 Hz), 129.0 (dd, JCF = 32.8, 8.5 Hz), 128.6, 122.6 (q, JCF = 272 Hz), 119.7 (d, JCF = 21.4 Hz), 114.8 (dd, JCF = 25.9, 4.9 Hz); HRMS (ESI) calcd for [C11H5F4N3O3S + H]+ 336.0061, found 336.0065.
2-Nitro-N-(5-nitrothiazol-2-yl)-4-(trifluoromethyl)benzamide (36)
Method B yielded the title compound 36 (84 mg, 58%) as a light yellow solid. 1H NMR (500 MHz, DMSO-d6) δ 13.96 (bs, 1H), 8.72 (s, 1H), 8.58 (s, 1H), 8.38 (d, J = 7.9 Hz, 1H), 8.16 (d, J = 8.0 Hz, 1H); 13C NMR (125 MHz, DMSO-d6) δ 164.7, 161.2, 146.6, 142.5, 142.4, 132.6, 132.0 (q, JCF = 34.2 Hz), 131.4, 131.3 (d, JCF = 3.4 Hz), 122.6 (d, JCF = 272 Hz), 121.9 (q, JCF = 7.5 Hz, 1H); HRMS (ESI) calcd for [C11H5F3N4O5S + H]+ 363.0006, found 363.0016.
4-Fluoro-3-nitro-N-(5-nitrothiazol-2-yl)benzamide (37)
Method B yielded the title compound 37 (25 mg, 16%) as a pale yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 8.95 (d, J = 7.2 Hz, 1H), 8.76 (s, 1H), 8.60 – 8.38 (m, 1H), 7.90 – 7.78 (m, 1H); 13C NMR (75 MHz, DMSO-d6) δ 162.7, 157.3 (d, JCF = 268 Hz), 142.4, 136.6 (d, JCF = 10.4 Hz), 128.3, 126.9, 119.3 (d, JCF = 21.4 Hz); HRMS (ESI) calcd for [C10H5FN4O5S + H]+ 313.0037, found 313.0044.
N-(5-Nitrothiazol-2-yl)-3,5-bis(trifluoromethyl)benzamide (38)
Method B yielded the title compound 38 (59 mg, 40%) as a light orange solid. 1H NMR (300 MHz, DMSO-d6) δ 8.76 (s, 1H), 8.76 (s, 2H), 8.48 (s, 1H); 13C NMR (75 MHz, DMSO-d6) δ 164.3, 162.6, 142.4, 133.7, 130.7 (q, JCF = 33.5 Hz, 129.5 (d, JCF = 3.2 Hz), 126.6 (d, JCF = 3.3 Hz), 123.0 (d, JCF = 273 Hz); HRMS (ESI) calcd for [C12H5F6N3O3S + H]+ 386.0029, found 386.0040.
2-Chloro-N-(5-nitrothiazol-2-yl)-5-(trifluoromethyl)benzamide (39)
Method B yielded the title compound 39 (81 mg, 52%) as a light orange solid. 1H NMR (300 MHz, DMSO-d6) δ 13.85 (bs, 1H), 8.71 (s, 1H), 8.22 (d, J = 1.6 Hz, 1H), 7.97 (dd, J = 8.5, 1.8 Hz, 1H), 7.87 (d, J = 8.5 Hz, 1H); 13C NMR (75 MHz, DMSO-d6) δ 164.8, 161.3, 142.5, 135.0, 133.7, 131.2, 129.3 (d, JCF = 2.6 Hz), 127.9 (q, JCF = 33.1 Hz), 127.1 (d, JCF = 3.0 Hz), 123.4 (d, JCF = 273 Hz), 119.4; HRMS (ESI) calcd for [C11H5ClF3N3O3S + H]+ 351.9765, found 351.9767.
2-Chloro-N-(5-nitrothiazol-2-yl)-3-(trifluoromethyl)benzamide (40)
Method B yielded the title compound 40 (97 mg, 62%) as a beige solid. 1H NMR (300 MHz, DMSO-d6) δ 13.88 (bs, 1H), 8.72 (s, 1H), 8.13 – 7.97 (m, 2H), 7.73 (t, J = 7.8 Hz, 1H); 13C NMR (75 MHz, DMSO-d6) δ 165.3, 161.1, 142.6, 142.5, 136.0, 133.5, 130.0 (d, JCF = 4.7 Hz), 128.3, 128.2, 127.5 (q, JCF = 31.0 Hz), 122.6 (q, JCF = 274 Hz); HRMS (ESI) calcd for [C11H5ClF3N3O3S + H]+ 351.9765, found 351.9775.
4-Chloro-N-(5-nitrothiazol-2-yl)-3-(trifluoromethyl)benzamide (41)
Method B yielded the title compound 41 (126 mg, 80%) as an orange solid. 1H NMR (300 MHz, DMSO-d6) δ 13.89 (bs, 1H), 8.71 (d, J = 0.6 Hz, 1H), 8.58 (d, J = 1.9 Hz, 1H), 8.35 (dd, J = 8.4, 2.1 Hz, 1H), 7.94 (d, J = 8.4 Hz, 1H); 13C NMR (75 MHz, DMSO-d6) δ 164.4, 162.4, 142.3, 135.7, 134.2, 132.3, 130.4, 127.9 (d, JCF = 4.8 Hz), 126.9 (q, JCF = 31.6 Hz), 122.5 (q, JCF = 274 Hz); HRMS (ESI) calcd for [C11H5ClF3N3O3S + H]+ 351.9765, found 351.9775.
N-(5-Nitrothiazol-2-yl)benzamide (42)
Method A yielded the title compound 42 (90 mg, 42%) as a beige solid. 1H NMR (300 MHz, DMSO-d6) δ 13.61 (s, 1H), 8.73 (s, 1H), 8.13 (d, J = 7.3 Hz, 2H), 7.70 (t, J = 7.4 Hz, 1H), 7.59 (t, J = 7.6 Hz, 2H); 13C NMR (125 MHz, DMSO-d6) δ 166.4, 162.6, 142.6, 142.1, 133.5, 130.8, 128.8, 128.6; HRMS (ESI) calcd for [C10H7N3O3S + H]+ 250.0281, found 250.0287.
N-(5-Nitrothiazol-2-yl)-2-phenylacetamide (43)
Method B yielded the title compound 43 (70 mg, 36%) as a beige solid. 1H NMR (300 MHz, DMSO-d6) δ 13.33 (s, 1H), 8.63 (s, 1H), 7.80 – 7.02 (m, 5H), 3.87 (s, 2H); 13C NMR (75 MHz, DMSO-d6) δ 171.1, 161.7, 142.7, 141.9, 134.0, 129.4, 128.5, 127.1, 41.5; HRMS (ESI) calcd for [C11H9N3O3S + H]+ 264.0437, found 264.0447.
N-(5-Nitrothiazol-2-yl)-3-phenylpropanamide (44)
Method B yielded the title compound 44 (122 mg, 66%) as a pale yellow solid. 1H NMR (500 MHz, DMSO-d6) δ 13.10 (bs, 1H), 8.60 (s, 1H), 7.32 – 7.26 (m, 1H), 7.23 (d, J = 7.5 Hz, 1H), 7.19 (t, J = 6.8 Hz, 1H), 2.94 (t, J = 7.4 Hz, 2H), 2.88 – 2.81 (m, 2H); 13C NMR (125 MHz, DMSO-d6) δ 172.8, 162.1, 143.1, 142.1, 140.8, 128.8, 128.7, 126.6, 37.0, 30.4; HRMS (ESI) calcd for [C12H11N3O3S + H]+ 278.0594, found 278.0604.
N-(5-Nitrothiazol-2-yl)-4-phenylbutanamide (45)
Method B yielded the title compound 45 (133 mg, 84%) as an orange solid. 1H NMR (300 MHz, DMSO-d6) δ 13.07 (bs, 1H), 8.60 (s, 1H), 7.35 – 7.05 (m, 5H), 2.64 (t, J = 7.6 Hz, 2H), 2.56 (t, J = 7.4 Hz, 2H), 1.94 (quint., J = 7.6 Hz, 2H); 13C NMR (75 MHz, DMSO-d6) δ 172.8, 161.7, 142.6, 141.6, 141.3, 128.3, 125.9, 34.4, 25.8; HRMS (ESI) calcd for [C13H13N3O3S + H]+ 292.0750, found 292.0753.
(1R,2R)-N-(5-Nitrothiazol-2-yl)-2-phenylcyclopropanecarboxamide (46)
Method B yielded the title compound 46 (43 mg, 24%) as a white solid. 1H NMR (500 MHz, DMSO-d6) δ 13.35 (bs, 1H), 8.62 (s, 1H), 7.30 (t, J = 7.5 Hz, 2H), 7.22 (t, J = 7.6 Hz, 3H), 2.64 – 2.52 (m, 1H), 2.31 – 2.22 (m, 1H), 1.68 – 1.54 (m, 2H); 13C NMR (125 MHz, DMSO-d6) δ 171.8, 161.7, 142.8, 141.8, 139.7, 128.5, 126.6, 126.2, 27.3, 25.5, 17.1; HRMS (ESI) calcd for [C13H11N3O3S + H]+ 290.0594, found 290.0599.
3-Methyl-N-(5-nitrothiazol-2-yl)indene-2-carboxamide (47)
Method B yielded the title compound 47 (25 mg, 17%) as an orange solid. 1H NMR (500 MHz, DMSO-d6) δ 12.66 (bs, 1H), 8.61 (s, 1H), 7.66 – 7.55 (m, 2H), 7.48 – 7.39 (m, 2H), 3.98 (d, J = 2.3 Hz, 2H), 2.57 (t, J = 2.3 Hz, 3H); 13C NMR (125 MHz, DMSO-d6) δ 162.3, 152.8, 144.3, 143.2, 142.7, 129.8, 128.3, 126.8, 124.1, 121.5, 37.8, 12.5; HRMS (ESI) calcd for [C14H11N3O3S + H]+ 302.0594, found 302.0601.
N-(5-Nitrothiazol-2-yl)acetamide (48)
Method A yielded the title compound 48 (132 mg, 50%) as a beige solid. 1H NMR (300 MHz, DMSO-d6) δ 13.02 (s, 1H), 8.56 (s, 1H), 2.21 (s, 3H); 13C NMR (75 MHz, DMSO-d6) δ 170.3, 161.8, 142.6, 141.7, 22.5; HRMS (ESI) calcd for [C5H5N3O3S + H]+ 188.0124, found 188.0124.
N-(5-Nitrothiazol-2-yl)butyramide (49)
Method A yielded the title compound 49 (112 mg, 55%) as an orange solid. 1H NMR (500 MHz, DMSO-d6) δ 13.04 (bs, 1H), 8.60 (s, 1H), 2.50 (t, J = 7.3 Hz, 2H), 1.96 – 1.40 (m, 2H), 0.91 (t, J = 7.4 Hz, 2H); 13C NMR (125 MHz, DMSO-d6) δ 173.0, 161.7, 142.7, 141.6, 36.8, 17.8, 13.4; HRMS (ESI) calcd for [C7H9N3O3S + H]+ 216.0437, found 216.0439.
N-(5-Nitrothiazol-2-yl)hexanamide (50)
Method A yielded the title compound 50 (126 mg, 71%) as a light orange solid. 1H NMR (500 MHz, DMSO-d6) δ 13.02 (s, 1H), 8.57 (s, 1H), 2.50 (t, J = 7.5 Hz, 2H), 1.67 – 1.44 (m, 2H), 1.44 – 1.07 (m, 4H), 0.85 (t, J = 7.0 Hz, 3H); 13C NMR (125 MHz, DMSO-d6) δ 173.1, 161.7, 142.6, 141.6, 34.9, 30.7, 24.0, 21.8, 13.8; HRMS (ESI) calcd for [C9H13N3O3S + H]+ 244.0750, found 244.0757.
N-(5-Nitrothiazol-2-yl)octanamide (51)
Method A yielded the title compound 51 (61 mg, 38%) as a beige solid. 1H NMR (500 MHz, DMSO-d6) δ 13.02 (bs, 1H), 8.59 (s, 1H), 2.50 (t, J = 7.4 Hz, 2H), 1.75 – 1.43 (m, 2H), 1.31 – 1.19 (m, 8H), 0.84 (t, J = 6.8 Hz, 3H); 13C NMR (125 MHz, DMSO-d6) δ 173.1, 161.7, 142.7, 141.6, 34.9, 31.1, 28.43, 28.37, 24.3, 22.1, 13.9; HRMS (ESI) calcd for [C11H17N3O3S + H]+ 272.1063, found 272.1071.
N-(5-Nitrothiazol-2-yl)decanamide (52)
Method A yielded the title compound 52 (53 mg, 36%) as a white solid. 1H NMR (500 MHz, DMSO-d6) δ 13.02 (bs, 1H), 8.59 (s, 1H), 2.50 (t, J = 7.4 Hz, 2H), 1.59 (quint., J = 7.0 Hz, 2H), 1.24 (d, J = 13.0 Hz, 12H), 0.84 (t, J = 6.8 Hz, 3H); 13C NMR (125 MHz, DMSO-d6) δ 173.1, 161.7, 142.6, 141.6, 34.9, 31.3, 28.8, 28.67, 28.66, 28.4, 24.3, 22.1, 13.9; HRMS (ESI) calcd for [C13H21N3O3S + H]+ 300.1376, found 300.1383.
N-(5-Nitrothiazol-2-yl)cyclohexanecarboxamide (53)
Method A yielded the title compound 53 (128 mg, 68%) as a beige solid. 1H NMR (500 MHz, DMSO-d6) δ 12.98 (s, 1H), 8.57 (s, 1H), 2.58 – 2.46 (m, 1H), 1.83 (d, J = 12.9 Hz, 2H), 1.80 – 1.64 (m, 2H), 1.62 (d, J = 11.5 Hz, 1H), 1.38 (qd, J = 12.3, 2.8 Hz, 2H), 1.30 – 1.13 (m, 3H); 13C NMR (125 MHz, DMSO-d6) δ 175.8, 161.9, 142.6, 141.7, 43.4, 28.5, 25.2, 24.9; HRMS (ESI) calcd for [C10H13N3O3S + H]+ 256.0750, found 256.0755.
N-(5-Nitrothiazol-2-yl)cinnamamide (54)
Method B yielded the title compound 54 (78 mg, 42%) as a beige solid. 1H NMR (300 MHz, DMSO-d6) δ 13.31 (s, 1H), 8.66 (s, 1H), 7.85 (d, J = 15.9 Hz, 1H), 7.72 – 7.59 (m, 2H), 7.53 – 7.40 (m, 3H), 6.92 (d, J = 15.9 Hz, 1H); 13C NMR (125 MHz, DMSO-d6) δ 164.6, 161.9, 144.6, 142.9, 142.1, 133.9, 130.9, 129.2, 128.3, 118.1; HRMS (ESI) calcd for [C12H9N3O3S + H]+ 276.0437, found 276.0445.
(E)-N-(5-Nitrothiazol-2-yl)-3-(4-(trifluoromethyl)phenyl)acrylamide (55)
Method B with DMAP (cat.) yielded the title compound 55 (51 mg, 32%) as a tan solid. 1H NMR (300 MHz, DMSO-d6) δ 13.42 (s, 1H), 8.69 (s, 1H), 7.97 – 7.80 (m, 5H), 7.03 (d, J = 16.0 Hz, 1H); 13C NMR (75 MHz, DMSO-d6) δ 164.2, 161.8, 142.8, 142.6, 142.2, 137.8, 129.9 (q, JCF = 26.5 Hz), 128.9, 126.0 (q, JCF = 3.7 Hz), 124.0 (d, JCF = 269 Hz), 120.9; HRMS (ESI) calcd for [C13H8F3N3O3S + H]+ 344.0311, found 344.0319.
N-(5-Nitrothiazol-2-yl)isonicotinamide (56)
Method A without an aqueous workup yielded the title compound 56 (90 mg, 56%) as a yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 8.85 (d, J = 4.1 Hz, 2H), 8.75 (s, 1H), 8.01 (d, J = 6.0 Hz, 2H); 13C NMR (125 MHz, DMSO-d6) δ 165.5, 162.4, 150.5, 142.5, 142.2, 138.4, 122.0; HRMS (ESI) calcd for [C9H6N4O3S + H]+ 251.0233, found 251.0239.
3-Fluoro-N-(5-nitrothiazol-2-yl)isonicotinamide (57)
Method C followed by Method A without an aqueous workup and quenching with 2 M HCl in Et2O yielded the title compound 57 (112 mg, 59%) as a yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 8.82 (s, 1H), 8.72 (s, 1H), 8.65 (d, J = 4.7 Hz, 1H), 7.82 (t, J = 5.4 Hz, 1H); 13C NMR (75 MHz, DMSO-d6) δ 162.6, 161.3, 155.2 (d, JCF = 262 Hz), 146.4 (d, JCF = 4.8 Hz), 142.4, 139.4 (d, JCF = 23.5 Hz), 127.9 (d, JCF = 11.1 Hz), 123.5; HRMS (ESI) calcd for [C9H5FN4O3S + H]+ 269.0139, found 269.0142.
N-(5-Nitrothiazol-2-yl)nicotinamide (58)
Method A without an aqueous workup and quenching with 2 M HCl in Et2O yielded the title compound 58 (16 mg, 10%) as a yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 9.24 (s, 1H), 8.83 (d, J = 4.7 Hz, 1H), 8.74 (s, 1H), 8.45 (d, J = 8.0 Hz, 1H), 7.62 (dd, J = 8.0, 4.9 Hz, 1H); 13C NMR (75 MHz, DMSO-d6) δ 174.5, 171.7, 162.3, 158.3, 151.5, 145.1, 136.2, 132.6; HRMS (ESI) calcd for [C9H6N4O3S + H]+ 251.0233, found 251.0238.
2-Fluoro-N-(5-nitrothiazol-2-yl)nicotinamide (59)
Method C followed by Method A without an aqueous workup and quenching with 2 M HCl in Et2O yielded the title compound 59 (118 mg, 62%) as a yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 13.77 (bs, 1H), 8.70 (s, 1H), 8.54 – 8.44 (m, 1H), 8.41 – 8.35 (m, 1H), 7.63 – 7.50 (m, 1H); 13C NMR (75 MHz, DMSO-d6) δ 163.0 (d, JCF = 5.9 Hz), 161.5, 159.4 (d, JCF = 242 Hz), 151.3 (d, JCF = 15.2 Hz), 142.4, 142.3, 122.3 (d, JCF = 4.1 Hz), 115.9 (d, JCF = 28.5 Hz); HRMS (ESI) calcd for [C9H5FN4O3S + H]+ 269.0139, found 269.0143.
N-(5-Nitrothiazol-2-yl)furan-3-carboxamide (60)
Method B yielded the title compound 60 (105 mg, 49%) as a yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 13.44 (bs, 1H), 8.70 (s, 1H), 8.68 (dd, J = 1.5, 0.8 Hz, 1H), 7.93 – 7.85 (m, 1H), 7.17 – 7.09 (m, 1H); 13C NMR (75 MHz, DMSO-d6) δ 162.2, 161.1, 148.4, 145.1, 144.7, 142.6, 120.0, 109.1; HRMS (ESI) calcd for [C8H5N3O4S + H]+ 240.0074, found 240.0082.
N-(5-Nitrothiazol-2-yl)furan-2-carboxamide (61)
Method A yielded the title compound 61 (39 mg, 16%) as a beige solid. 1H NMR (300 MHz, DMSO-d6) δ 8.68 (s, 1H), 8.12 – 8.05 (m, 1H), 7.76 (d, J = 3.5 Hz, 1H), 6.78 (dd, J = 3.5, 1.4 Hz, 1H); 13C NMR (125 MHz, DMSO-d6) δ 162.5, 157.0, 149.0, 145.1, 143.1, 142.5, 118.9, 113.2; HRMS (ESI) calcd for [C8H5N3O4S + H]+ 240.0074, found 240.0077.
5-Bromo-N-(5-nitrothiazol-2-yl)furan-2-carboxamide (62)
Method B yielded the title compound 62 (72 mg, 44%) as a red solid. 1H NMR (300 MHz, DMSO-d6) δ 13.57 (bs, 1H), 8.63 (s, 1H), 7.73 (d, J = 3.7 Hz, 1H), 6.89 (d, J = 3.7 Hz, 1H); 13C NMR (75 MHz, DMSO-d6) δ 161.9, 155.5, 146.6, 142.4, 142.1, 128.9, 120.5, 114.9; HRMS (ESI) calcd for [C8H4BrN3O4S + H]+ 317.9179, found 317.9180.
4,5-Dibromo-N-(5-nitrothiazol-2-yl)furan-2-carboxamide (63)
Method B yielded the title compound 63 (53 mg, 18%) as a tan solid. 1H NMR (300 MHz, DMSO-d6) δ 8.72 (s, 1H), 7.90 (s, 1H); 13C NMR (75 MHz, DMSO-d6) δ 161.9, 155.2, 146.7, 142.3, 142.1, 130.0, 121.5, 104.0; HRMS (ESI) calcd for [C8H3Br2N3O4S + H]+ 395.8284, found 395.8295.
5-Nitro-N-(5-nitrothiazol-2-yl)furan-2-carboxamide (64)
Method A yielded the title compound 64 (128 mg, 79%) as a tan solid. 1H NMR (500 MHz, DMSO-d6) δ 8.71 (s, 1H), 7.87 (d, J = 3.8 Hz, 1H), 7.81 (d, J = 3.9 Hz, 1H); 13C NMR (125 MHz, DMSO-d6) δ 162.1, 156.3, 152.5, 145.6, 141.8, 141.5, 118.9, 113.1; HRMS (ESI) calcd for [C8H4N4O6S + H]+ 284.9924, found 284.9928.
N-(5-Nitrothiazol-2-yl)-5-phenylfuran-2-carboxamide (65)
Method D yielded the intermediate Suzuki-coupling product (94 mg, 95%) as a white solid.[37] 1H NMR (300 MHz, CDCl3) δ 7.86 – 7.76 (m, 2H), 7.50 – 7.34 (m, 3H), 7.28 (d, J = 3.8 Hz, 1H), 6.77 (d, J = 3.6 Hz, 1H), 3.94 (s, 3H). Method E yielded the intermediate carboxylic acid (92 mg, 100%) as a white solid. 1H NMR (500 MHz, DMSO-d6) δ 7.84 – 7.75 (m, 2H), 7.52 – 7.43 (m, 2H), 7.44 – 7.36 (m, 1H), 7.32 (d, J = 3.6 Hz, 1H), 7.15 (d, J = 3.6 Hz, 1H). Method B yielded the title compound 65 (29 mg, 24%) as a yellow solid. 1H NMR (500 MHz, DMSO-d6) δ 13.79 (bs, 1H), 8.72 (s, 1H), 8.06 – 7.99 (m, 2H), 7.77 (d, J = 3.8 Hz, 1H), 7.55 – 7.47 (m, 2H), 7.46 – 7.40 (m, 1H), 7.29 (d, J = 3.7 Hz, 1H). 13C NMR (125 MHz, DMSO-d6) δ 162.2, 157.5, 156.3, 143.9, 142.7, 142.0, 129.4, 129.0, 128.8, 125.0, 120.9, 108.6. HRMS (ESI) calcd for [C14H9N3O4S + H]+ 316.0387, found 316.0388.
5-(3-Chlorophenyl)-N-(5-nitrothiazol-2-yl)furan-2-carboxamide (66)
Method D yielded the intermediate Suzuki-coupling product (154 mg, 88%) as a pale yellow solid. 1H NMR (500 MHz, DMSO-d6) δ 7.82 (s, 1H), 7.73 (d, J = 7.7 Hz, 1H), 7.48 (t, J = 7.9 Hz, 1H), 7.44 – 7.40 (m, 1H), 7.39 (d, J = 3.7 Hz, 1H), 7.26 (d, J = 3.7 Hz, 1H), 3.83 (s, 3H). Method E yielded the intermediate carboxylic acid (85 mg, 89%) as a white solid.[38] 1H NMR (500 MHz, DMSO-d6) δ 13.24 (bs, 1H), 7.87 (s, 1H), 7.77 (dd, J = 7.7, 1.1 Hz, 1H), 7.52 (t, J = 7.9 Hz, 1H), 7.50 – 7.42 (m, 1H), 7.33 (d, J = 3.6 Hz, 1H), 7.28 (d, J = 3.6 Hz, 1H). Method B yielded the title compound 66 (53 mg, 45%) as a yellow solid. 1H NMR (500 MHz, DMSO-d6) δ 13.70 (bs, 1H), 8.67 (s, 1H), 8.12 (t, J = 1.8 Hz, 1H), 8.00 – 7.92 (m, 1H), 7.71 (d, J = 3.8 Hz, 1H), 7.51 (t, J = 7.9 Hz, 1H), 7.47 – 7.43 (m, 1H), 7.35 (d, J = 3.8 Hz, 1H); 13C NMR (125 MHz, DMSO-d6) δ 161.9, 156.1, 155.5, 144.2, 142.5, 142.0, 133.8, 130.8, 130.6, 128.9, 124.3, 123.4, 120.7, 109.8; HRMS (ESI) calcd for [C14H8ClN3O4S + H]+ 349.9997, found 350.0010.
5-(3-Fluorophenyl)-N-(5-nitrothiazol-2-yl)furan-2-carboxamide (67)
Method D yielded the intermediate Suzuki-coupling product (133 mg, 83%) as a white solid. 1H NMR (500 MHz, DMSO-d6) δ 7.70 – 7.62 (m, 2H), 7.54 (td, J = 8.1, 6.2 Hz, 1H), 7.45 (dd, J = 3.7, 0.4 Hz, 1H), 7.30 (d, J = 3.7 Hz, 1H), 7.26 (td, J = 8.7, 2.5 Hz, 1H), 3.85 (s, 3H). Method E yielded the intermediate carboxylic acid (93 mg, 99%) as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 13.22 (bs, 1H), 7.71 – 7.59 (m, 2H), 7.53 (td, J = 8.0, 6.0 Hz, 1H), 7.34 (d, J = 3.6 Hz, 1H), 7.28 – 7.16 (m, 2H). Method B yielded the title compound 67 (28 mg, 29%) as a yellow solid. 1H NMR (500 MHz, DMSO-d6) δ 13.71 (bs, 1H), 8.68 (s, 1H), 7.94 (d, J = 10.2 Hz, 1H), 7.84 (d, J = 7.8 Hz, 1H), 7.71 (d, J = 3.7 Hz, 1H), 7.54 (dd, J = 14.1, 7.9 Hz, 1H), 7.34 (d, J = 3.0 Hz, 1H), 7.24 (td, J = 8.5, 1.8 Hz, 1H); 13C NMR (125 MHz, DMSO-d6) δ 162.8 (d, JCF = 180 Hz), 161.5, 156.2, 155.8, 144.2, 142.4 (d, JCF = 15.3 Hz), 142.0, 130.9, 121.0, 120.6 (d, JCF = 8.6 Hz), 116.0, 115.9, 111.6 (d, JCF = 24.0 Hz), 109.6 (d, JCF = 19.8 Hz); HRMS (ESI) calcd for [C14H8FN3O4S + H]+ 334.0292, found 334.0303.
N-(5-Nitrothiazol-2-yl)-5-(3-(trifluoromethyl)phenyl)furan-2-carboxamide (68)
Method B with DMAP (cat.) yielded the title compound 68 (79 mg, 46%) as a pale yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 13.82 (s, 1H), 8.72 (s, 1H), 8.35 (s, 1H), 8.32 (s, 1H), 7.82 – 7.67 (m, 3H), 7.50 (d, J = 3.8 Hz, 1H); 13C NMR (75 MHz, DMSO-d6) δ 162.1, 156.3, 155.5, 144.5, 142.6, 142.1, 130.2, 129.7, 128.8, 125.6 (q, JCF = 3.9 Hz), 124.0 (q, JCF = 271 Hz), 121.3 (q, JCF = 3.8 Hz), 120.8, 110.2; HRMS (ESI) calcd for [C15H8F3N3O4S + H]+ 384.0260, found 384.0275.
5-(3-Nitrophenyl)-N-(5-nitrothiazol-2-yl)furan-2-carboxamide (69)
Method B yielded the title compound 69 (31 mg, 20%) as a bright yellow solid. 1H NMR (500 MHz, DMSO-d6) δ 13.83 (bs, 1H), 8.75 (s, 1H), 8.67 (s, 1H), 8.41 (d, J = 7.8 Hz, 1H), 8.23 – 8.19 (m, 1H), 7.76 (t, J = 8.0 Hz, 1H), 7.72 (d, J = 3.8 Hz, 1H), 7.51 (d, J = 3.7 Hz, 1H); 13C NMR (125 MHz, DMSO-d6) δ 162.1, 156.2, 154.7, 148.4, 144.7, 142.5, 142.1, 131.0, 130.6, 130.2, 123.6, 120.7, 119.1, 110.7; HRMS (ESI) calcd for [C14H8N4O6S + H]+ 361.0237, found 361.0241.
N-(5-Nitrothiazol-2-yl)-2,3′-bifuran-5-carboxamide (70)
Method D yielded the intermediate Suzuki-coupling product (133 mg, 95%) as a pale yellow solid. 1H NMR (500 MHz, DMSO-d6) δ 8.22 (s, 1H), 7.84 – 7.77 (m, 1H), 7.36 (d, J = 3.6 Hz, 1H), 6.92 – 6.89 (m, 1H), 6.85 (d, J = 3.6 Hz, 1H), 3.81 (s, 3H). Method E yielded the intermediate carboxylic acid (92 mg, 99%) as a white solid. 1H NMR (500 MHz, DMSO-d6) δ 13.08 (bs, 1H), 8.22 – 8.16 (m, 1H), 7.77 (t, J = 1.7 Hz, 1H), 7.27 (d, J = 3.5 Hz, 1H), 6.88 (dd, J = 1.9, 0.8 Hz, 1H), 6.80 (d, J = 3.5 Hz, 1H). Method B yielded the title compound 70 (42 mg, 32%) as a yellow solid. 1H NMR (500 MHz, DMSO-d6) δ 13.55 (s, 1H), 8.66 (s, 1H), 8.33 – 8.28 (m, 1H), 7.82 (d, J = 1.5 Hz, 1H), 7.73 (d, J = 3.7 Hz, 1H), 6.99 (d, J = 0.6 Hz, 1H), 6.92 (d, J = 3.7 Hz, 1H); 13C NMR (125 MHz, DMSO-d6) δ 162.2, 156.1, 152.1, 144.8, 143.0, 142.6, 142.0, 141.1, 120.5, 116.5, 108.4, 108.0; HRMS (ESI) calcd for [C12H7N3O5S + H]+ 306.0179, found 306.0189.
N-(5-Nitrothiazol-2-yl)-5-(thiophen-3-yl)furan-2-carboxamide (71)
Method D yielded the intermediate Suzuki-coupling product (128 mg, 84%) as a yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 7.97 (dd, J = 2.9, 1.2 Hz, 1H), 7.70 (dd, J = 5.1, 2.9 Hz, 1H), 7.54 (dd, J = 5.1, 1.2 Hz, 1H), 7.40 (d, J = 3.6 Hz, 1H), 6.99 (d, J = 3.6 Hz, 1H), 3.83 (s, 3H). Method E yielded the intermediate carboxylic acid (92 mg, 99%) as a pink solid. 1H NMR (500 MHz, DMSO-d6) δ 13.08 (bs, 1H), 7.93 (dd, J = 2.9, 1.3 Hz, 1H), 7.69 (dd, J = 5.1, 2.9 Hz, 1H), 7.52 (dd, J = 5.1, 1.3 Hz, 1H), 7.29 (d, J = 3.6 Hz, 1H), 6.94 (d, J = 3.6 Hz, 1H). Method B yielded the title compound 71 (23 mg, 19%) as a yellow solid. 1H NMR (500 MHz, DMSO-d6) δ 13.66 (bs, 1H), 8.71 (s, 1H), 8.17 (dd, J = 2.9, 1.2 Hz, 1H), 7.74 (d, J = 3.7 Hz, 1H), 7.72 (dd, J = 5.0, 2.9 Hz, 1H), 7.67 (dd, J = 5.0, 1.2 Hz, 1H), 7.07 (d, J = 3.7 Hz, 1H); 13C NMR (125 MHz, DMSO-d6) δ 162.3, 156.4, 154.5, 143.1, 142.8, 142.0, 130.7, 128.0, 125.3, 123.6, 120.8, 108.2; HRMS (ESI) calcd for [C12H7N3O4S2 + H]+ 321.9951, found 321.9953.
N-(5-Nitrothiazol-2-yl)benzofuran-2-carboxamide (72)
Method C followed by Method A yielded the title compound 72 (57 mg, 32%) as a light orange solid. 1H NMR (300 MHz, DMSO-d6) δ 8.71 (s, 1H), 8.16 (s, 1H), 7.87 (d, J = 7.7 Hz, 1H), 7.74 (d, J = 8.4 Hz, 1H), 7.61 – 7.49 (m, 1H), 7.39 (t, J = 7.6 Hz, 1H); 13C NMR (75 MHz, DMSO-d6) δ 162.0, 157.7, 155.1, 145.9, 142.5, 142.1, 128.4, 126.7, 124.3, 123.6, 113.9, 112.2; HRMS (ESI) calcd for [C12H7N3O4S + H]+ 290.0230, found 290.0241.
5-Nitro-N-(5-nitrothiazol-2-yl)benzofuran-2-carboxamide (73)
Method B yielded the title compound 73 (16 mg, 10%) as a light orange solid. 1H NMR (300 MHz, DMSO-d6) δ 8.91 (d, J = 2.4 Hz, 1H), 8.75 (s, 1H), 8.39 (dd, J = 9.2, 2.4 Hz, 1H), 8.32 (s, 1H), 8.01 (d, J = 9.2 Hz, 1H); 13C NMR (75 MHz, DMSO-d6) δ 162.6, 157.5, 144.4, 142.4, 127.4, 123.3, 120.5, 114.1, 113.3, 95.8, 91.6; HRMS (ESI) calcd for [C12H6N4O6S + H]+ 335.0081, found 335.0088.
N-(5-Nitrothiazol-2-yl)thiophene-3-carboxamide (74)
Method B yielded the title compound 74 (76 mg, 38%) as a yellow solid. 1H NMR (500 MHz, DMSO-d6) δ 13.43 (bs, 1H), 8.75 – 8.68 (m, 1H), 8.65 (s, 1H), 7.75 – 7.72 (m, 1H), 7.72 – 7.67 (m, 1H); 13C NMR (125 MHz, DMSO-d6) δ 162.4, 161.2, 142.6, 142.0, 133.7, 133.4, 127.9, 127.2; HRMS (ESI) calcd for [C8H5N3O3S2 + H]+ 255.9845, found 255.9848.
2,5-Dichloro-N-(5-nitrothiazol-2-yl)thiophene-3-carboxamide (75)
Method B yielded the title compound 75 (109 mg, 66%) as a tan solid. 1H NMR (300 MHz, DMSO-d6) δ 13.47 (bs, 1H), 8.69 (s, 1H), 7.72 (d, J = 0.7 Hz, 1H); 13C NMR (75 MHz, DMSO-d6) δ 161.7, 159.8, 142.4, 132.9, 129.4, 126.9, 125.6; HRMS (ESI) calcd for [C8H3Cl2N3O3S2 + H]+ 323.9066, found 323.9074.
N-(5-Nitrothiazol-2-yl)thiophene-2-carboxamide (76)
Method A yielded the title compound 76 (85 mg, 36%) as a light yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 13.70 (bs, 1H), 8.72 (s, 1H), 8.32 (dd, J = 3.8, 1.1 Hz, 1H), 8.09 (dd, J = 5.0, 1.1 Hz, 1H), 7.30 (dd, J = 5.0, 3.9 Hz, 1H); 13C NMR (125 MHz, DMSO-d6) δ 162.5, 160.8, 142.6, 142.0, 135.7, 135.3, 132.4, 128.9; HRMS (ESI) calcd for [C8H5N3O3S2 + H]+ 255.9845, found 255.9846.
5-Chloro-N-(5-nitrothiazol-2-yl)thiophene-2-carboxamide (77)
Method B yielded the title compound 77 (122 mg, 69%) as a yellow solid. 1H NMR (500 MHz, DMSO-d6) δ 13.79 (s, 1H), 8.71 (s, 1H), 8.17 (d, J = 4.2 Hz, 1H), 7.35 (d, J = 4.2 Hz, 1H); 13C NMR (125 MHz, DMSO-d6) δ 162.4, 160.0, 142.3, 142.0, 137.1, 134.9, 132.4, 129.1; HRMS (ESI) calcd for [C8H4ClN3O3S2 + H]+ 289.9455, found 289.9465.
3-Chloro-N-(5-nitrothiazol-2-yl)thiophene-2-carboxamide (78)
Method B yielded the title compound 78 (133 mg, 75%) as a yellow solid. 1H NMR (500 MHz, DMSO-d6) δ 8.74 (s, 1H), 8.02 (d, J = 5.2 Hz, 1H), 7.25 (d, J = 5.2 Hz, 1H); 13C NMR (125 MHz, DMSO-d6) δ 163.0, 161.9, 140.5, 139.9, 132.3, 129.9, 129.0, 128.0; HRMS (ESI) calcd for [C8H4ClN3O3S2 + H]+ 289.9455, found 289.9467.
5-Bromo-N-(5-nitrothiazol-2-yl)thiophene-2-carboxamide (79)
Method B yielded the title compound 79 (128 mg, 75%) as a tan solid. 1H NMR (300 MHz, DMSO-d6) δ 13.77 (bs, 1H), 8.71 (s, 1H), 8.11 (d, J = 4.1 Hz, 1H), 7.44 (d, J = 4.1 Hz, 1H); 13C NMR (75 MHz, DMSO-d6) δ 162.3, 159.9, 142.3, 142.0, 137.5, 133.1, 132.5, 121.4; HRMS (ESI) calcd for [C8H4BrN3O3S2 + H]+ 333.8950, found 333.8959.
N-(5-Nitrothiazol-2-yl)-5-phenylthiophene-2-carboxamide (80)
Method D yielded the intermediate Suzuki-coupling product (92 mg, 94%) as a white solid.[39] 1H NMR (300 MHz, DMSO-d6) δ 7.81 (d, J = 3.9 Hz, 1H), 7.78 – 7.69 (m, 2H), 7.62 (d, J = 4.0 Hz, 1H), 7.52 – 7.36 (m, 3H), 3.84 (s, 3H). Method E yielded the intermediate carboxylic acid (74 mg, 95%) as a white solid. 1H NMR (500 MHz, DMSO-d6) δ 13.17 (bs, 1H), 7.78 – 7.69 (m, 3H), 7.58 (d, J = 3.9 Hz, 1H), 7.49 – 7.43 (m, 2H), 7.42 – 7.37 (m, 1H). Method B yielded the title compound 80 (68 mg, 57%) as a yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 13.73 (bs, 1H), 8.72 (s, 1H), 8.32 (d, J = 4.1 Hz, 1H), 7.84 – 7.76 (m, 2H), 7.71 (d, J = 4.1 Hz, 1H), 7.61 – 7.29 (m, 3H); 13C NMR (75 MHz, DMSO-d6) δ 162.6, 160.7, 151.6, 142.7, 142.0, 134.5, 133.5, 132.5, 129.4, 126.11, 125.3; HRMS (ESI) calcd for [C14H9N3O3S2+ H]+ 332.0158, found 332.0161.
5-(3-Chlorophenyl)-N-(5-nitrothiazol-2-yl)thiophene-2-carboxamide (81)
Method D yielded the intermediate Suzuki-coupling product (165 mg, 96%) as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 7.86 (t, J = 1.8 Hz, 1H), 7.82 (d, J = 4.0 Hz, 1H), 7.74 – 7.67 (m, 2H), 7.50 – 7.44 (m, 2H), 3.84 (s, 3H). Method E yielded the intermediate carboxylic acid (88 mg, 93%) as a white solid. 1H NMR (500 MHz, DMSO-d6) δ 13.26 (bs, 1H), 7.83 (t, J = 1.8 Hz, 1H), 7.73 (d, J = 3.9 Hz, 1H), 7.70 – 7.65 (m, 2H), 7.51 – 7.41 (m, 2H). Method B yielded the title compound 81 (28 mg, 33%) as a bright yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 13.73 (bs, 1H), 8.69 (s, 1H), 8.28 (d, J = 4.0 Hz, 1H), 7.85 (s, 1H), 7.77 (d, J = 4.0 Hz, 1H), 7.75 – 7.63 (m, 1H), 7.54 – 7.41 (m, 2H); 13C NMR (75 MHz, DMSO-d6) δ 162.7, 160.8, 149.4, 142.7, 141.8, 135.6, 134.5, 134.1,133.3, 131.2, 128.9, 126.3, 125.5, 124.8; HRMS (ESI) calcd for [C14H8ClN3O3S2 + H]+ 365.9768, found 365.9764.
N-(5-Nitrothiazol-2-yl)-2,3′-bithiophene-5-carboxamide (82)
Method D yielded the intermediate Suzuki-coupling product (145 mg, 95%) as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 7.99 (dd, J = 2.9, 1.3 Hz, 1H), 7.77 (d, J = 3.9 Hz, 1H), 7.70 (dd, J = 5.0, 2.9 Hz, 1H), 7.53 (dd, J = 5.0, 1.3 Hz, 1H), 7.50 (d, J = 3.9 Hz, 1H), 3.83 (s, 3H). Method E yielded the intermediate carboxylic acid (90 mg, 96%) as a white solid. 1H NMR (500 MHz, DMSO-d6) δ 13.10 (bs, 1H), 7.95 (dd, J = 2.9, 1.3 Hz, 1H), 7.70 – 7.67 (m, 2H), 7.51 (dd, J = 5.0, 1.3 Hz, 1H), 7.45 (d, J = 3.8 Hz, 1H). Method B yielded the title compound 82 (44 mg, 37%) as a yellow solid. 1H NMR (500 MHz, DMSO-d6) δ 13.65 (bs, 1H), 8.68 (s, 1H), 8.25 (d, J = 4.1 Hz, 1H), 8.01 (dd, J = 2.9, 1.3 Hz, 1H), 7.70 (dd, J = 5.0, 2.9 Hz, 1H), 7.55 (d, J = 4.0 Hz, 1H), 7.53 (dd, J = 5.0, 1.3 Hz, 1H); 13C NMR (125 MHz, DMSO-d6) δ 162.7, 160.7, 146.6, 142.7, 141.8, 133.9, 133.4, 133.3, 128.2, 126.0, 125.2, 123.3; HRMS (ESI) calcd for [C12H7N3O3S3 + H]+ 337.9722, found 337.9729.
5-(Furan-3-yl)-N-(5-nitrothiazol-2-yl)thiophene-2-carboxamide (83)
Method D yielded the intermediate Suzuki-coupling product (123 mg, 87%) as a white solid. 1H NMR (500 MHz, DMSO-d6) δ 8.30 – 8.26 (m, 1H), 7.80 (t, J = 1.7 Hz, 1H), 7.76 (d, J = 3.9 Hz, 1H), 7.39 (d, J = 3.9 Hz, 1H), 6.93 (dd, J = 1.9, 0.9 Hz, 1H), 3.82 (s, 3H). Method E yielded the intermediate carboxylic acid (91 mg, 98%) as a white solid. 1H NMR (500 MHz, DMSO-d6) δ 13.08 (bs, 1H), 8.25 (s, 1H), 7.81 – 7.76 (m, 1H), 7.69 – 7.65 (m, 1H), 7.35 (d, J = 3.8 Hz, 1H), 6.94 – 6.90 (m, 1H). Method B yielded the title compound 83 (26 mg, 21%) as a yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 13.66 (bs, 1H), 8.71 (s, 1H), 8.33 (s, 1H), 8.26 (d, J = 4.1 Hz, 1H), 7.82 (s, 1H), 7.47 (d, J = 4.0 Hz, 1H), 6.96 (s, 1H); 13C NMR (125 MHz, DMSO-d6) δ 162.5, 160.6, 145.0, 143.1, 142.6, 141.9, 140.7, 133.2, 133.0, 125.3, 119.5, 109.0; HRMS (ESI) calcd for [C12H7N3O4S2 + H]+ 321.9951, found 321.9960.
N-(5-Nitrothiazol-2-yl)benzo[b]thiophene-2-carboxamide (84)
Method A yielded the title compound 84 (39 mg, 25%) as a light yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 13.97 (s, 1H), 8.75 (s, 1H), 8.67 (s, 1H), 8.12 (d, J = 7.9 Hz, 1H), 8.06 (d, J = 8.0 Hz, 1H), 7.62 – 7.54 (m, 1H), 7.54 – 7.45 (m, 1H); 13C NMR (125 MHz, DMSO-d6) δ 162.3, 161.7, 142.5, 142.0, 141.4, 138.8, 135.7, 129.5, 127.6, 126.3, 125.5, 123.1; HRMS (ESI) calcd for [C12H7N3O3S2 + H]+ 306.0002, found 306.0011.
3-Chloro-N-(5-nitrothiazol-2-yl)benzo[b]thiophene-2-carboxamide (85)
Method A yielded the title compound 85 (79 mg, 54%) as a bright yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 8.84 (s, 1H), 8.17 (dd, J = 6.6, 2.0 Hz, 1H), 7.98 (dd, J = 6.1, 1.9 Hz, 1H), 7.69 – 7.60 (m, 2H); 13C NMR (75 MHz, DMSO-d6) δ 163.8, 163.6, 139.0, 137.7, 136.1, 128.4, 126.2, 125.9, 123.8, 123.6, 123.0; HRMS (ESI) calcd for [C12H6ClN3O3S2 + H]+ 339.9612, found 339.9616.
3-Chloro-6-fluoro-N-(5-nitrothiazol-2-yl)benzo[b]thiophene-2-carboxamide (86)
Method A yielded the title compound 86 (69 mg, 48%) as a light yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 8.83 (s, 1H), 8.08 (dd, J = 9.1, 2.2 Hz, 1H), 7.97 (dd, J = 9.0, 5.1 Hz, 1H), 7.48 (td, J = 9.0, 2.3 Hz, 1H); 13C NMR (75 MHz, DMSO-d6) δ 163.9, 163.5, 160.3, 139.6, 139.1, 138.4, 133.1, 129.9, 125.1, 123.5, 115.5, 109.8; HRMS (ESI) calcd for [C12H5ClFN3O3S2 + H]+ 357.9518, found 357.9529.
Supplementary Material
Acknowledgments
This work was supported by U01 grant AI075520 from the National Institute of Allergy and Infectious Diseases to PSH.
Footnotes
Supporting information for this article is available on the WWW under http://www.chemmedchem.org or from the author.
References
- 1.Brown D, Superti-Furga G. Drug Discov Today. 2003;8:1067. doi: 10.1016/s1359-6446(03)02902-7. [DOI] [PubMed] [Google Scholar]
- 2.Carney S. Drug Discov Today. 2005;10:1011. doi: 10.1016/S1359-6446(05)03533-6. [DOI] [PubMed] [Google Scholar]
- 3.Anderson VR, Curran MP. Drugs. 2007;67:1947. doi: 10.2165/00003495-200767130-00015. [DOI] [PubMed] [Google Scholar]
- 4.Gilles HM, Hoffman PS. Trends Parasitol. 2002;18:95. doi: 10.1016/s1471-4922(01)02205-x. [DOI] [PubMed] [Google Scholar]
- 5.Ortiz JJ, Chegne NL, Gargala G, Favennec L. Trans R Soc Trop Med Hyg. 2002;96:193. doi: 10.1016/s0035-9203(02)90301-9. [DOI] [PubMed] [Google Scholar]
- 6.Pankuch GA, Appelbaum PC. Antimicrob Agents Chemother. 2006;50:1112. doi: 10.1128/AAC.50.3.1112-1117.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Carvalho LPS, Lin G, Jiang X, Nathan C. J Med Chem. 2009;52:5789. doi: 10.1021/jm9010719. [DOI] [PubMed] [Google Scholar]
- 8.Rossignol JF, Abu-Zekry M, Hussein A, Santoro MG. Lancet. 2006;368:124. doi: 10.1016/S0140-6736(06)68852-1. [DOI] [PubMed] [Google Scholar]
- 9.Rossignol JF, Elfert A, El-Gohary Y, Keeffe EB. Gastroenterology. 2009;136:856. doi: 10.1053/j.gastro.2008.11.037. [DOI] [PubMed] [Google Scholar]
- 10.Rossignol JF, La Frazia S, Chiappa L, Ciucci A, Santoro MG. J Biol Chem. 2009;284:29798. doi: 10.1074/jbc.M109.029470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shamir ER, Warthan M, Brown SP, Nataro JP, Guerrant RL, Hoffman PS. Antimicrob Agents Chemother. 2010;54:1526. doi: 10.1128/AAC.01279-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tchouaffi-Nana F, Ballard TE, Cary CH, Macdonald TL, Sifri CD, Hoffman PS. Antimicrob Agents Chemother. 2010;54 doi: 10.1128/AAC.00901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Broekhuysen J, Stockis A, Lins RL, De Graeve J, Rossignol JF. Int J Clin Pharmacol Ther. 2000;38:387. doi: 10.5414/cpp38387. [DOI] [PubMed] [Google Scholar]
- 14.Olekhnovich IN, Goodwin A, Hoffman PS. FEBS J. 2009;276:3354. doi: 10.1111/j.1742-4658.2009.07060.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoffman PS, Sisson G, Croxen MA, Welch K, Harman WD, Cremades N, Morash MG. Antimicrob Agents Chemother. 2007;51:868. doi: 10.1128/AAC.01159-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sisson G, Goodwin A, Raudonikiene A, Hughes NJ, Mukhopadhyay AK, Berg DE, Hoffman PS. Antimicrob Agents Chemother. 2002;46:2116. doi: 10.1128/AAC.46.7.2116-2123.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cremades N, Bueno M, Toja M, Sancho J. Biophys Chem. 2005;115:267. doi: 10.1016/j.bpc.2004.12.045. [DOI] [PubMed] [Google Scholar]
- 18.Evans MC, Buchanan BB, Arnon DI. Proc Natl Acad Sci U S A. 1966;55:928. doi: 10.1073/pnas.55.4.928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoffman PS, Goodman TG. J Bacteriol. 1982;150:319. doi: 10.1128/jb.150.1.319-326.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kletzin A, Adams MW. J Bacteriol. 1996;178:248. doi: 10.1128/jb.178.1.248-257.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Megraud F, Occhialini A, Rossignol JF. Antimicrob Agents Chemother. 1998;42:2836. doi: 10.1128/aac.42.11.2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rossignol JF. 20060194853. US. 2006
- 23.Rossignol JF, Cavier R. 19763950351. US. 1976
- 24.Fuccio L, Zagari RM, Minardi ME, Bazzoli F. Aliment Pharmacol Ther. 2007;25:133. doi: 10.1111/j.1365-2036.2006.03183.x. [DOI] [PubMed] [Google Scholar]
- 25.Young KT, Davis LM, DiRita VJ. Nat Rev Micro. 2007;5:665. doi: 10.1038/nrmicro1718. [DOI] [PubMed] [Google Scholar]
- 26.Lyerly DM, Krivan HC, Wilkins TD. Clin Microbiol Rev. 1988;1:1. doi: 10.1128/cmr.1.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kimon JA, Gordon GH. Proc Natl Acad Sci U S A. 1978;75:4877. [Google Scholar]
- 28.Lynch JK, Freeman JC, Judd AS, Iyengar R, Mulhern M, Zhao G, Napier JJ, Wodka D, Brodjian S, Dayton BD, Falls D, Ogiela C, Reilly RM, Campbell TJ, Polakowski JS, Hernandez L, Marsh KC, Shapiro R, Knourek-Segel V, Droz B, Bush E, Brune M, Preusser LC, Fryer RM, Reinhart GA, Houseman K, Diaz G, Mikhail A, Limberis JT, Sham HL, Collins CA, Kym PR. J Med Chem. 2006;49:6569. doi: 10.1021/jm060683e. [DOI] [PubMed] [Google Scholar]
- 29.Kanamori D, Okamura TA, Yamamoto H, Ueyama N. Angew Chem Int Ed Engl. 2005;44:969. doi: 10.1002/anie.200461842. [DOI] [PubMed] [Google Scholar]
- 30.Bushby SRM, Copp FC. J Pharm Pharmacol. 1955;7:112. doi: 10.1111/j.2042-7158.1955.tb12014.x. [DOI] [PubMed] [Google Scholar]
- 31.Miyaura N, Suzuki A. Chem Rev. 1995;95:2457. [Google Scholar]
- 32.Kelly CP, LaMont JT. N Engl J Med. 2008;359:1932. doi: 10.1056/NEJMra0707500. [DOI] [PubMed] [Google Scholar]
- 33.Mylonakis E, Ryan ET, Calderwood SB. Arch Intern Med. 2001;161:525. doi: 10.1001/archinte.161.4.525. [DOI] [PubMed] [Google Scholar]
- 34.Menichetti F. Clin Microbiol Infec. 2005;11:22. doi: 10.1111/j.1469-0691.2005.01138.x. [DOI] [PubMed] [Google Scholar]
- 35.Vuong C, Otto M. Microb Infect. 2002;4:481. doi: 10.1016/s1286-4579(02)01563-0. [DOI] [PubMed] [Google Scholar]
- 36.Ballard TE, Wang X, Olekhnovich I, Koerner T, Seymour C, Hoffman PS, Macdonald TL. Bioorg Med Chem Lett. 2010;20:3537. doi: 10.1016/j.bmcl.2010.04.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee S, Yi KY, Hwang SK, Lee BH, Yoo S-e, Lee K. J Med Chem. 2005;48:2882. doi: 10.1021/jm0492305. [DOI] [PubMed] [Google Scholar]
- 38.Ye QZ, Xie SX, Huang M, Huang WJ, Lu JP, Ma ZQ. J Am Chem Soc. 2004;126:13940. doi: 10.1021/ja045864p. [DOI] [PubMed] [Google Scholar]
- 39.McAllister LA, Hixon MS, Kennedy JP, Dickerson TJ, Janda KD. J Am Chem Soc. 2006;128:4176. doi: 10.1021/ja057699z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.