

Abstract
Purpose of review
Investigation into the underlying mechanisms of salt sensitivity has made important advances in recent years. This review examines in particular the effects of sodium and potassium on vascular function.
Recent findings
Sodium chloride (salt) intake promotes cutaneous lymphangiogenesis mediated through tissue macrophages and directly alters endothelial cell function, promoting increased production of transforming growth factor-β (TGF-β) and nitric oxide (NO). In the setting of endothelial dysfunction, such as occurs with aging, diminished NO production exacerbates the vascular effects of TGF-β, promoting decreased arterial compliance and hypertension. Dietary potassium intake may serve as an important countervailing influence on the effects of salt in the vasculature.
Summary
There is growing appreciation that, independently of alterations in blood pressure, dietary intake of sodium and potassium promote functional changes in the vasculature and lymphatic system. These changes may serve as compensatory changes that protect against development of salt-sensitive hypertension. While salt sensitivity cannot be ascribed exclusively to these factors, perturbation of these processes promote hypertension during high-salt intake. These studies add to the list of genetic and environmental factors that are associated with salt sensitivity, but in particular provide insight into adaptive mechanisms during high salt intake.
Keywords: dietary sodium, dietary potassium, nitric oxide, TGF-β, arterial compliance
Introduction
Hypertension is one of the most common diseases in modern society. Twenty-four percent of American adults are estimated to have hypertension [1]. The importance of hypertension derives not just from the high prevalence but also from the close relationship with cardiovascular morbidity and mortality [2]. In considering the pathogenesis of hypertension, dietary NaCl (termed “salt” in this review) has long been appreciated as a major contributing environmental factor. However, despite intense interest and research in the blood pressure response to salt intake, it is remarkable that a consistent definition of salt-sensitivity has not emerged. It is clear that blood pressure responses to dietary salt intake vary among individuals, and the degree of these responses can be grouped into categories of salt sensitivity and salt resistance, but exact definitions and protocols used to determine the effect of salt differ among investigators. Some investigators, for example, define salt sensitivity by a shift in the pressure-natriuresis curve, an approach that is focused on renal mechanisms of salt handling. Other approaches have been used to examine the cardiovascular effects of salt sensitivity. In one protocol championed by Weinberger [3], intravenous infusion of two liters of saline is administered over four hours in the morning and blood pressure is determined at the end of the infusion. On the following day, dietary sodium is decreased to 10 mmol and three doses of furosemide, 40 mg, are administered orally. Blood pressure is determined again the following morning. Following this two-day protocol, salt-sensitive subjects demonstrated a decrease in mean arterial pressure of > 10 mm Hg, while mean arterial pressures of salt resistant subjects decreased by < 5 mm Hg. A third (intermediate) group consisted of those whose decrease in blood pressure ranged between 6 and 9 mm Hg. Using these criteria and extrapolating to the total population, among the 43,186,000 hypertensive patients in the US, 51% are salt-sensitive and 33% are salt-resistant. That is, blood pressure control would improve in half of the hypertensive population with dietary salt restriction. Among normotensive subjects, 26% are salt-sensitive and 58% are salt-resistant. Thus, about a quarter of otherwise healthy adults are salt sensitive and likely are unaware that they respond to increases in dietary salt intake with an increase in blood pressure. This latter observation is important, because a subsequent follow-up study documented that these normotensive salt-sensitive subjects had a cumulative mortality that was similar to that of hypertensive subjects [4]. Risk factors that promote salt sensitivity include race (African Americans in particular), intrinsic kidney disease and aging per se [5]. Based largely on blood pressure effects of salt intake and demographics, epidemiologists recently estimated that a reduction in sodium intake to about 1.2 g/day (3 g/day NaCl) would reduce the annual number of new cases of coronary heart disease by 60,000, stroke by 32,000, and MI by 54,000; while all would benefit, relative benefits would be greater for blacks, (and presumably all patients with salt-sensitivity), across all age groups [6].
The magnitude of the problem demands an understanding of the pathomechanisms of salt sensitivity, but conclusive evidence of the cause(s) remains elusive. Recent studies have suggested some new insights into the fundamental pathophysiological aspects of salt-induced hypertension and findings from these publications are the major focus of this review.
Is salt sensitivity a disorder of the immune system?
The traditional paradigm that increased salt intake promotes water retention and expands extracellular fluid volume has been recently challenged by Machnik, et al. [7]. Using elegant techniques, these investigators demonstrated that increased salt intake (8% NaCl plus 1% saline as the drinking solution) in rats led to accumulation of Na+ in the skin, promoting an increase in interstitial tonicity along with hyperplasia of the lymphocapillary network. Exploring the mechanism further, they observed that the increase in interstitial tonicity in rats on the high salt diet promoted infiltrating macrophages to express TonEBP, a transcription factor that increased the production of vascular endothelial growth factor-C (VEGF-C). Interestingly, depleting macrophages using liposomal clodronate blocked the local production of VEGF-C and prevented the lymphatic hyperplasia. As a consequence of preventing this compensatory response, blood pressure increased. Years ago, our laboratory demonstrated an increase in the endothelial isoform of nitric oxide synthase (NOS3) in the vasculature of rats on a high salt diet. This increase was mediated in part through transforming growth factor-beta (TGF-β) [8, 9]. The study by Machnik confirmed an increase in NOS3 in the capillary endothelium, but the investigators further observed involvement of macrophages and VEGF-C in this process [7] Figure 1.
Figure 1.
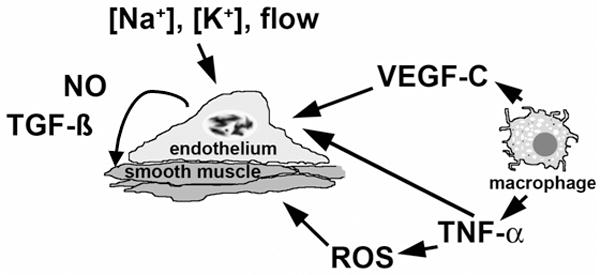
Cartoon summarizing data suggesting the endothelium as a “salt sensor” that responds to changes in [Na+], [K+] and flow by altering production of TGF-β, a fibrogenic growth factor that modifies endothelial and smooth muscle function and promotes vascular stiffness, and NO, a vasodilator that also serves as a potential countervailing influence on TGF-β. Macrophage-derived cytokines, such as VEGF-C and TNF-α, and reactive oxygen species may also directly affect endothelia and vascular smooth muscle and contribute to salt sensitivity. Please see text for details.
This novel compensatory response provides a reservoir for Na+ ions to defend against salt-induced volume expansion and likely increases NOS3-generated nitric oxide (NO) to prevent an increase in blood pressure. Undoubtedly, this system plays a role in the regulation of blood pressure, since interruption of this protective mechanism promotes salt-sensitive hypertension, at least in small animals. The findings support a role for lymphangiogenesis mediated through the macrophage phagocyte system in the regulation of the blood pressure response to salt intake.
Macrophages may also participate directly in end-organ damage associated with salt-sensitive hypertension. Elmarakby, et al. [10], used etanercept in the desoxycorticosterone acetate (DOCA)-salt rodent model of hypertension to inhibit tumor necrosis factor-alpha (TNF-α), a cytokine produced primarily by monocytes and macrophages. Etanercept reduced renal inflammation and proteinuria without altering the blood pressure effect of DOCA-salt treatment. Interestingly, plasma TNF-α levels are increased in Dahl salt-sensitive rats maintained on a high salt diet for five weeks [11] and in Sabra salt-sensitive rats prior to development of hypertension [12]. TNF-α levels increase further during salt-loading and development of hypertension in the Sabra strain. This cytokine might play a role in activating NADPH oxidase in polymorphonuclear leukocytes and thereby promote systemic oxidative stress and hypertension in that model of salt-sensitive hypertension [12]. Additional studies to determine the effect of TNF-α inhibitors, such as etanercept, in these rodent strains may prove fruitful.
Recent research also suggested that T cells might participate in the pathogenesis of salt-sensitive hypertension. Guzik et al. [13], found that RAG-1−/− mice, which lack both T and B cells, demonstrated a blunted increase in blood pressure and reduced vascular oxidative stress in response to DOCA-salt. The potential role of adaptive immunity in vascular pathology associated with salt-sensitive hypertension has also been examined using normotensive (Brown Norway), hypertensive (Dahl salt-sensitive), and consomic rats (SSBN2; in which chromosome 2 has been transferred from Brown Norway to Dahl rats). Tail-cuff systolic blood pressures were elevated in Dahl rats compared with Brown Norway rats and were reduced in SSBN2 rats compared with Dahl rats. Compared with Brown Norway and SSBN2 rats, Dahl rats exhibited increased inflammatory markers in the aorta, including activation of NF-κB and increased infiltration of CD4+ T cells. Also observed in aortic tissue of Dahl rats was decreased infiltration of cells of the regulatory T cell lineage, relative to the SSBN2 strain. The authors concluded that this genetic rodent model of salt-sensitive hypertension exhibited increased vascular inflammatory responses that are reduced by transfer of chromosome 2 from a normotensive strain, which also displayed enhanced production of immunosuppressive mediators [14]. Although incompletely characterized, the combined findings support a role for adaptive immunity and T cell function in the development of salt-sensitive hypertension and end organ damage.
Dietary Salt, TGF-β and NO
TGF-β consists of a family of three pleiotropic growth factors that have complex effects on organ development and cell growth and differentiation, but they are particularly important in the expression of extracellular matrix proteins and promotion of vascular and renal fibrosis in a variety of disease states [15]. While evidence supports similar functions among these three TGF-βs, TGF-β1 is considered the most important mammalian TGF-β family member. TGF-β1 is synthesized by many cell types including endothelium and is secreted as a latent dimeric protein complex [16]. A latency-associated peptide (LAP) is cleaved from the molecule during intracellular processing, but remains noncovalently complexed to the mature peptide after secretion. Binding of this complex to latent TGF-β binding proteins (LTBP) directs it to the adjacent interstitium. Removal of latent TGF-β permits the mature, biologically active form of TGF-β to interact with the receptor [17]. Thus, TGF-β1 secreted by endothelium typically acts on adjacent vascular smooth muscle or endothelium. TGF-β appears to be involved in blood pressure regulation. EMILIN1 knockout mice display increased TGF-β1 signaling in the vessel wall. These animals develop peripheral vasoconstriction and arterial hypertension, which was prevented by inactivation of one TGFB1 allele [18]. Parenteral administration of an anti-TGF-β antibody to Dahl salt-sensitive rats significantly reduced blood pressure and the associated proteinuria, glomerulosclerosis and interstitial and vascular fibrosis [19].
Studies in normotensive rats demonstrated excess salt intake rapidly increased endothelial production of active TGF-β [9, 20]. As described, dietary salt intake modulates NO production in normotensive, Dahl/Rapp salt-resistant (R) rats [21–24] and healthy humans [25]. In addition to hemodynamic effects, NO also modulates salt-induced TGF-β1 production [26]. The intracellular signaling events involved in salt-induced increase in NO production relate to Akt-mediated phosphorylation of NOS3 at amino acid residue 1176 in rats. Activation of Akt occurs with the formation of a Pyk2/c-src/phosphatidylinositol 3-kinase complex [27]. Excess salt intake therefore promotes production of TGF-β and simultaneously NO, which serves as a factor that mitigates the effects of TGF-β. Ultimately, however, it appears that the pro-fibrotic effect of TGF-β cannot be inhibited over time. Yu, et al. [28], demonstrated collagen deposition in the arteries, arterioles, glomeruli, and interstitium of the hearts and kidneys of normotensive (WKY) and hypertensive (SHR) rats fed 8.0% salt diet for eight weeks. Deposition of collagen promotes arterial stiffness, which may also stimulate cardiac hypertrophy and remodeling of the microcirculation. The effect of reduced vascular compliance on the microcirculation is dramatic in the brain and kidney, two organs that are perfused at high flow rates and pulsatile pressures [29].
Recent studies support a deleterious effect of increased dietary NaCl on vascular structure and function in humans. In a group of 34 hypertensive patients who were otherwise healthy, an increase in sodium intake from 60 mmol/d to 150 and 200 mmol/d for four weeks resulted in a significant increase (0.35–0.39 m/s) in pulse wave velocity (PWV), an indicator of arterial stiffness. Systolic and diastolic blood pressures also increased, but the poor correlation of PWV with blood pressure suggested a blood pressure-independent effect on the vasculature [30]. Conversely, in a double-blind, placebo-controlled, crossover study of older adults with prehypertension, low salt intake increased carotid arterial compliance by 27% by the end of the first week and the improvement stabilized at 46% by the end of the study at week 2 [31]. Dickinson, et al. [32], examined the effect of reduction in sodium intake from 150 mmol/d to 50 mmol/d for two weeks in overweight (BMI 31.6±2.6 kg/m2) normotensive men and women and observed an improvement in endothelial function, as demonstrated by increased flow mediated vasodilation in patients while on the low salt diet. The authors observed no improvement in PWV but the study was of short duration and was underpowered to detect a < 2 m/s difference. The effect of salt was also examined in a heterogeneous population of adults with uncomplicated hypertension by administering dietary sodium either at 165 or 110 mmol/d (9.7 to 6.5 gms of salt/day) for six weeks. With this modest reduction in salt intake, blood pressure, urinary albumin excretion rates and PWV fell [33]. The combined evidence supports a role for dietary salt intake in the regulation of arterial stiffness and perhaps endothelial function.
Evidence in vitro demonstrates a direct effect of sodium concentration present in the medium on endothelial stiffness [34, 35]. Endothelial cells in culture stiffened and produced less flow-induced NO when sodium concentration in the medium increased from 135 to 145 mEq/L. Altered endothelial cell function from increased medium sodium concentration was observed, however, only in the presence of aldosterone [34]. This finding may have clinical relevance, since reduction of salt intake to very low levels (10 mmol/d) produced a small but significant reduction in plasma sodium concentration (about 3 mEq/L), compared to values obtained from the same patients while on 350 mmol/d salt diet [36].
Endothelial dysfunction with diminished NO production, such as occurs with aging or in conditions that produce oxidative stress, might accelerate the effects of TGF-β in the vasculature. Mice lacking NOS3 demonstrate higher mean PWV than wild-type mice [37]. Aging decreases flow-mediated activation of Akt and NO production and an associated increase in PWV, compared to vascular tissue from young animals [37]. Unlike Dahl/Rapp salt-resistant (R) rats, Dahl/Rapp salt-sensitive (S) rats manifest impaired NO production that is exacerbated by increased salt intake [21, 22]. Vascular and glomerular production of active TGF-β1 and NO correlate directly in both R and S rats, but production of TGF-β1 is increased in prehypertensive S rats and differences in endothelial production of TGF-β1 and NO between S and R rats are further exaggerated with institution of the increased salt diet [26]. Inhibition of NO promotes salt retention and salt-sensitive hypertension [38] and, if protracted, results in renal injury if the animals are maintained on a high-salt diet [39].
To explore the potential for oxidative stress to deplete NO and promote salt-sensitive hypertension, Ma et al. [40], examined the effects of salt intake on mice that lacked the mitochondrial uncoupling protein 2 (UCP2). Homozygous mice lacking UCP2 demonstrated increased blood pressure and superoxide production and vascular dysfunction with diminished NO production, while wild-type mice showed no effects of the high salt intake. The findings implicate ROS in the pathogenesis of salt-sensitive hypertension through depletion of NO. However, this concept was recently challenged by Kopkan, et al. [41], who demonstrated that administration of a high salt diet to mice that lack NOS3 promoted hypertension and intrarenal superoxide production, which improved with administration of Tempol. That blood pressure improved with administration of Tempol in these animals suggested a pressor effect of superoxide independent of endothelium-derived NO.
The combined evidence supports the paradigm that the way in which the endothelium responds to excess salt intake dictates down stream events in the vasculature. As salt intake increases, the endothelium initially responds by increasing TGF-β, a matrigenic growth factor that promotes vascular stiffness. The increase in NO production counterbalances the effect of TGF-β, but over time other events, such as production of reactive oxygen species, interferes with this balance. The long-term consequences would be decreased vascular compliance, vasoconstriction and hypertension. These events would be exacerbated in patients with salt sensitivity, where excess salt is retained and NO production impaired.
Sodium is not alone: Importance of potassium in salt sensitivity
There is increasing appreciation for dietary potassium in the pathogenesis of salt sensitivity and related vascular disease. INTERSALT (The International Study of Salt and Blood Pressure) suggested that a decrease in potassium excretion by 50 mmol/d was associated with an increase in systolic pressure of 3.4 mmHg and an increase in diastolic pressure of 1.9 mmHg [42]. A meta-analysis of thirty-three randomized trials that evaluated effects of increased potassium intake on blood pressure concluded that potassium supplementation lowered systolic and diastolic blood pressures by small but significant amounts [43]. In a follow-up analysis of the Trials of Hypertension Prevention (TOHP I and TOHP II), a higher urinary sodium to potassium ratio was associated with increased risk of development of cardiovascular disease [44]. However, at least one recent study suggested that potassium supplementation for six weeks did not reduce blood pressure, improve endothelial dysfunction or decrease arterial stiffness in a small group of patients with mild hypertension [45].
In a recent randomized double-blind placebo-controlled study, He et al. [46], showed that supplementing potassium, either as potassium chloride or bicarbonate, in the diets of mildly hypertensive individuals did not promote reductions in blood pressure, determined by routine clinic blood pressure measurements, but did improve PWV, a measure of arterial stiffness, and endothelial function as determined by flow-mediated vasodilation. Therefore, while the literature on potassium is not completely consistent, along with numerous other studies, this well-conducted study provides strong evidence in favor of a role of potassium in cardiovascular protection.
A recent study by Ying and colleagues [47] provides insight into mechanism. Normotensive rats were given diets containing different amounts of sodium chloride and potassium for 4 days. As dietary potassium increased, salt-induced urinary active TGF-β fell. Inhibition of the large-conductance, calcium-activated potassium (BK) channel inhibited dietary salt-induced vascular production of TGF-β, but did not affect production of TGF-β by ring segments from rats on the low-salt diet. Increasing medium [K+] also inhibited vascular production of dietary salt-induced TGF-β. The findings demonstrated an interesting interaction between the dietary intake of potassium and salt and further showed the fundamental role of the endothelial BK channel in the vascular response to excess salt intake [47].
The use of atomic force microscopy has provided new insights into the function of endothelial cells as “salt sensors” [35]. Using bovine endothelial cells in culture, Oberleithner, et al. [48], demonstrated that addition of potassium to the medium swells and softens the endothelial cell and promotes release of NO. An increase in medium sodium concentration along with addition of aldosterone, which is required with sodium to promote stiffening of endothelial cells in culture [34], prevented the beneficial effects of potassium on endothelial cell production of NO. These fascinating in vitro studies support physiological studies suggesting the endothelium serves as a sensor of salt intake and an important role for potassium concentration to modulate the effect of salt.
Conclusions
It is well accepted that ingestion of excess sodium chloride is unequivocally associated with hypertension in some but not all individuals. However, the underlying mechanisms that promote salt sensitivity are complex and range from genetic to environmental influences. The phenotype of salt sensitivity is therefore heterogeneous with multiple mechanisms that potentially link high salt intake to increases in blood pressure. In addition, excess salt intake has functional and pathological effects on the vasculature that are independent of blood pressure. The ready availability of diuretics has perhaps promoted a lack of attention of physicians to dietary salt intake and recent data may provide new pharmacological tools that target end-organ damage from excess salt intake. At present, the findings of recent studies suggest that emphasizing a diet containing appropriate amounts of sodium chloride and potassium might have therapeutic benefits that equal or exceed those provided by medications. Further investigation into the molecular and cellular mechanisms of the direct actions of sodium and potassium on the vasculature will provide important insights into understanding salt sensitivity, and thus guide and augment personalized dietary strategies to prevent salt-induced blood vessel stiffness and hypertension.
Acknowledgments
Funding: National Institutes of Health grants, R01 DK046199 (PWS) and R01 HL092215 (YC), National Institutes of Health P30 DK079337 (George M. O'Brien Kidney and Urological Research Centers Program) (PWS) and the Office of Research and Development, Medical Research Service, Department of Veterans Affairs (YC and PWS), support this research.
References
- 1.Burt VL, Whelton P, Roccella EJ, et al. Prevalence of hypertension in the US adult population. Results from the Third National Health and Nutrition Examination Survey, 1988–1991. Hypertension. 1995 Mar;25(3):305–13. doi: 10.1161/01.hyp.25.3.305. [DOI] [PubMed] [Google Scholar]
- 2.Ezzati M, Lopez AD, Rodgers A, et al. Selected major risk factors and global and regional burden of disease. Lancet. 2002 Nov 2;360(9343):1347–60. doi: 10.1016/S0140-6736(02)11403-6. [DOI] [PubMed] [Google Scholar]
- 3.Weinberger MH, Miller JZ, Luft FC, et al. Definitions and characteristics of sodium sensitivity and blood pressure resistance. Hypertension. 1986;8(Suppl II):II127–II34. doi: 10.1161/01.hyp.8.6_pt_2.ii127. [DOI] [PubMed] [Google Scholar]
- 4.Weinberger MH, Fineberg NS, Fineberg SE, Weinberger M. Salt sensitivity, pulse pressure, and death in normal and hypertensive humans. Hypertension. 2001 Feb;37(2 Part 2):429–32. doi: 10.1161/01.hyp.37.2.429. [DOI] [PubMed] [Google Scholar]
- 5.Weinberger MH. Salt sensitivity of blood pressure in humans. Hypertension. 1996 Mar;27(3 Pt 2):481–90. doi: 10.1161/01.hyp.27.3.481. [DOI] [PubMed] [Google Scholar]
- 6•.Bibbins-Domingo K, Chertow GM, Coxson PG, et al. Projected effect of dietary salt reductions on future cardiovascular disease. N Engl J Med. 2010 Feb 18;362(7):590–9. doi: 10.1056/NEJMoa0907355. This prospective study investigated the effects of salt reduction on cardiovascular outcomes from a public health perspective and cost evaluation. The results of the trial showed substantial reductions in cardiovascular events and medical costs with dietary salt reduction to about 3 g per day. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7•.Machnik A, Neuhofer W, Jantsch J, et al. Macrophages regulate salt-dependent volume and blood pressure by a vascular endothelial growth factor-C-dependent buffering mechanism. Nat Med. 2009 May;15(5):545–52. doi: 10.1038/nm.1960. This study unveils a new physiopathologic pathway in salt-sensitive hypertension. The study demonstrated that high salt diet led to interstitial hypertonic sodium accumulation in skin, resulting in increased density and hyperplasia of the lymphcapillary network. the pathophysiologic changes leading this lymphocapillary remodelling involved activation of tonicity-responsive enhancer binding protein (TonEBP) in mononuclear phagocyte system cells infiltrating the interstitium of the skin and VEGF-C secretion by macrophages. MPS cell depletion or VEGF-C trapping augments interstitial hypertonic volume retention, decreases endothelial nitric oxide synthase expression and elevates blood pressure. [DOI] [PubMed] [Google Scholar]
- 8.Ying W-Z, Sanders PW. Dietary salt enhances glomerular endothelial nitric oxide synthase through TGF-β1. Am J Physiol. 1998;275(1 Pt 2):F18–F24. doi: 10.1152/ajprenal.1998.275.1.F18. [DOI] [PubMed] [Google Scholar]
- 9.Ying W-Z, Sanders PW. Dietary salt increases endothelial nitric oxide synthase and TGF-β1 in rat aortic endothelium. Am J Physiol. 1999;277(4 Pt 2):H1293–H8. doi: 10.1152/ajpheart.1999.277.4.H1293. [DOI] [PubMed] [Google Scholar]
- 10.Elmarakby AA, Quigley JE, Imig JD, et al. TNF-alpha inhibition reduces renal injury in DOCA-salt hypertensive rats. Am J Physiol Regul Integr Comp Physiol. 2008 Jan;294(1):R76–83. doi: 10.1152/ajpregu.00466.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gu JW, Tian N, Shparago M, et al. Renal NF-kappaB activation and TNF-alpha upregulation correlate with salt-sensitive hypertension in Dahl salt-sensitive rats. Am J Physiol Regul Integr Comp Physiol. 2006 Dec;291(6):R1817–24. doi: 10.1152/ajpregu.00153.2006. [DOI] [PubMed] [Google Scholar]
- 12•.Mazor R, Itzhaki O, Sela S, et al. Tumor necrosis factor-alpha: a possible priming agent for the polymorphonuclear leukocyte-reduced nicotinamide-adenine dinucleotide phosphate oxidase in hypertension. Hypertension. 2010 Feb;55(2):353–62. doi: 10.1161/HYPERTENSIONAHA.109.144154. This pre-clinical study adds new knowledge in that priming of PMNLs are important in induction of salt-sensitive hypertension. The authors showed that TNF-alpha behaves as the initiator of the priming of PMNLs in Sabra salt sensitive hypertension rat model. These rats had already increased levels of TNF-alpha before the development of hypertension and salt-loding further increased the levels of this cytokine. [DOI] [PubMed] [Google Scholar]
- 13.Guzik TJ, Hoch NE, Brown KA, et al. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. 2007 Oct 1;204(10):2449–60. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14•.Viel EC, Lemarie CA, Benkirane K, et al. Immune regulation and vascular inflammation in genetic hypertension. Am J Physiol Heart Circ Physiol. 2010 Mar;298(3):H938–44. doi: 10.1152/ajpheart.00707.2009. In this important experimental study, the authors hypothesized that under the influence of chromosome 2, T lymphocytes contribute vascular inflammation in genetic salt-sensitive hypertension. The authors used consomic mice in which chromosome 2 has been transferred from Brown Norway to Dahl rats. The results of the study showed that transfer of chromosome 2 from Brown Norway rats led to reduction in blood pressure, inflammatory markers and mediators compared to Dahl salt-sensitive rats. These results suggest a central regulatory role of chromosome 2 in vascular homeostasis possibly by impacting on the balance of regulatory and helper subsets of T lymphocytes. [DOI] [PubMed] [Google Scholar]
- 15.Border WA, Okuda S, Languino LR, et al. Suppression of experimental glomerulonephritis by antiserum against transforming growth factor β1. Nature. 1990;346:371–4. doi: 10.1038/346371a0. [DOI] [PubMed] [Google Scholar]
- 16.Derynck R, Jarrett JA, Chen EY, et al. Human transforming growth factor-beta complementary DNA sequence and expression in normal and transformed cells. Nature. 1985;316:701–5. doi: 10.1038/316701a0. [DOI] [PubMed] [Google Scholar]
- 17.Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFbeta activation. J Cell Sci. 2003 Jan 15;116(Pt 2):217–24. doi: 10.1242/jcs.00229. [DOI] [PubMed] [Google Scholar]
- 18.Zacchigna L, Vecchione C, Notte A, et al. Emilin1 links TGF-beta maturation to blood pressure homeostasis. Cell. 2006 Mar 10;124(5):929–42. doi: 10.1016/j.cell.2005.12.035. [DOI] [PubMed] [Google Scholar]
- 19.Dahly AJ, Hoagland KM, Flasch AK, et al. Antihypertensive effects of chronic anti-TGF-β antibody therapy in Dahl S rats. Am J Physiol Regul Integr Comp Physiol. 2002;283:R757–R67. doi: 10.1152/ajpregu.00098.2002. [DOI] [PubMed] [Google Scholar]
- 20.Ying W-Z, Sanders PW. Dietary salt modulates renal production of transforming growth factor-β in rats. Am J Physiol. 1998;274(4 Pt 2):F635–F41. doi: 10.1152/ajprenal.1998.274.4.F635. [DOI] [PubMed] [Google Scholar]
- 21.Chen PY, Sanders PW. L-arginine abrogates salt-sensitive hypertension in Dahl/Rapp rats. J Clin Invest. 1991;88:1559–67. doi: 10.1172/JCI115467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen PY, Sanders PW. Role of nitric oxide synthesis in salt-sensitive hypertension in Dahl/Rapp rats. Hypertension. 1993;22:812–8. doi: 10.1161/01.hyp.22.6.812. [DOI] [PubMed] [Google Scholar]
- 23.Deng X, Welch WJ, Wilcox CS. Renal vasoconstriction during inhibition of NO synthase: Effects of dietary salt. Kidney Int. 1994;46:639–46. doi: 10.1038/ki.1994.316. [DOI] [PubMed] [Google Scholar]
- 24.Shultz PJ, Tolins JP. Adaptation to increased dietary salt intake in the rat: role of endogenous nitric oxide. J Clin Invest. 1993;91:642–50. doi: 10.1172/JCI116244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bech JN, Nielsen CB, Ivarsen P, et al. Dietary sodium affects systemic and renal hemodynamic response to NO inhibition in healthy humans. Am J Physiol. 1998;274(5 Pt 2):F914–F23. doi: 10.1152/ajprenal.1998.274.5.F914. [DOI] [PubMed] [Google Scholar]
- 26.Ying W-Z, Sanders PW. The interrelationship between TGF-β1 and nitric oxide is altered in salt-sensitive hypertension. Am J Physiol Renal Physiol. 2003 Nov;285(5):F902–F8. doi: 10.1152/ajprenal.00177.2003. [DOI] [PubMed] [Google Scholar]
- 27.Ying WZ, Aaron K, Sanders PW. Dietary Salt Activates an Endothelial Proline-Rich Tyrosine Kinase 2/c-src/Phosphatidylinositol 3-Kinase Complex to Promote Endothelial Nitric Oxide Synthase Phosphorylation. Hypertension. 2008 Dec;52(6):1134–41. doi: 10.1161/HYPERTENSIONAHA.108.121582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu HCM, Burrell LM, Black MJ, et al. Salt induces myocardial and renal fibrosis in normotensive and hypertensive rats. Circulation. 1998;98:2621–8. doi: 10.1161/01.cir.98.23.2621. [DOI] [PubMed] [Google Scholar]
- 29.O'Rourke MF, Safar ME. Relationship between aortic stiffening and microvascular disease in brain and kidney: cause and logic of therapy. Hypertension. 2005 Jul;46(1):200–4. doi: 10.1161/01.HYP.0000168052.00426.65. [DOI] [PubMed] [Google Scholar]
- 30•.Todd AS, Macginley RJ, Schollum JB, et al. Dietary salt loading impairs arterial vascular reactivity. Am J Clin Nutr. 2010 Mar;91(3):557–64. doi: 10.3945/ajcn.2009.28645. This small intervention study adds to the current knowledge that salt loading in hypertensive subjects produced significant increases in pulse wave velocity and blood pressure. To draw the conclusion that salt-loading caused deleterious effects on vascular structures independent of blood pressure elevation was difficult due to small number of participants of the study. [DOI] [PubMed] [Google Scholar]
- 31.Gates PE, Tanaka H, Hiatt WR, Seals DR. Dietary sodium restriction rapidly improves large elastic artery compliance in older adults with systolic hypertension. Hypertension. 2004 Jul;44(1):35–41. doi: 10.1161/01.HYP.0000132767.74476.64. [DOI] [PubMed] [Google Scholar]
- 32•.Dickinson KM, Keogh JB, Clifton PM. Effects of a low-salt diet on flow-mediated dilatation in humans. Am J Clin Nutr. 2009 Feb;89(2):485–90. doi: 10.3945/ajcn.2008.26856. This randomised cross-over study in obese and overweight normotensive adults showed that low salt diet enabled beneficial effects on the vasculature assessed by flow mediated dilation. This trial showed beneficial effects of salt reduction on the endothelium independent of blood pressure reduction. [DOI] [PubMed] [Google Scholar]
- 33•.He FJ, Marciniak M, Visagie E, et al. Effect of modest salt reduction on blood pressure, urinary albumin, and pulse wave velocity in white, black, and Asian mild hypertensives. Hypertension. 2009 Sep;54(3):482–8. doi: 10.1161/HYPERTENSIONAHA.109.133223. Authors of this randomized double-blind crossover trial showed that even modest reductions in dietary salt intake led to reductions in albuminuria, blood pressure and arterial stiffness. [DOI] [PubMed] [Google Scholar]
- 34.Oberleithner H, Riethmuller C, Schillers H, et al. Plasma sodium stiffens vascular endothelium and reduces nitric oxide release. Proc Natl Acad Sci U S A. 2007 Oct 9;104(41):16281–6. doi: 10.1073/pnas.0707791104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oberleithner H, Kusche-Vihrog K, Schillers H. Endothelial cells as vascular salt sensors. Kidney Int. 2010 Mar;77(6):490–4. doi: 10.1038/ki.2009.490. [DOI] [PubMed] [Google Scholar]
- 36.He FJ, Markandu ND, Sagnella GA, et al. Plasma sodium: ignored and underestimated. Hypertension. 2005 Jan;45(1):98–102. doi: 10.1161/01.HYP.0000149431.79450.a2. [DOI] [PubMed] [Google Scholar]
- 37.Soucy KG, Ryoo S, Benjo A, et al. Impaired shear stress-induced nitric oxide production through decreased NOS phosphorylation contributes to age-related vascular stiffness. J Appl Physiol. 2006 Dec;101(6):1751–9. doi: 10.1152/japplphysiol.00138.2006. [DOI] [PubMed] [Google Scholar]
- 38.Tolins JP, Shultz PJ. Endogenous nitric oxide synthesis determines sensitivity to the pressor effect of salt. Kidney Int. 1994;46:230–6. doi: 10.1038/ki.1994.264. [DOI] [PubMed] [Google Scholar]
- 39.Fujihara CK, Michellazzo SM, De Nucci G, Zatz R. Sodium excess aggravates hypertension and renal parenchymal injury in rats with chronic NO inhibition. Am J Physiol. 1994;266(5 Pt 2):F697–F705. doi: 10.1152/ajprenal.1994.266.5.F697. [DOI] [PubMed] [Google Scholar]
- 40•.Ma S, Ma L, Yang D, et al. Uncoupling protein 2 ablation exacerbates high-salt intake-induced vascular dysfunction. Am J Hypertens. 2010 Aug;23(8):822–8. doi: 10.1038/ajh.2010.73. This pre-clinical study presented novel data regarding the role of Uncoupling protein 2 on determination of salt sensitivity. Uncoupling protein 2 knock-out mice developed hypertension while on high-salt diet in contrast to wild-type controls. The underlying mechanism was lack of suppression of oxidative stress markers andlack of nitric oxide. [DOI] [PubMed] [Google Scholar]
- 41•.Kopkan L, Hess A, Huskova Z, et al. High salt intake enhances superoxide activity in eNOS knockout mice leading to the development of salt-sensitivity. Am J Physiol Renal Physiol. 2010 Jul 7; doi: 10.1152/ajprenal.00047.2010. The results of this experimental study in eNOS knock-out mice revealed the importance of increased oxidative stress, here in the form of superoxide, in induction of salt sensitive hypertension. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Intersalt: an international study of electrolyte excretion and blood pressure. Results for 24 hour urinary sodium and potassium excretion. Intersalt Cooperative Research Group. BMJ. 1988 Jul 30;297(6644):319–28. doi: 10.1136/bmj.297.6644.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Whelton PK, He J, Cutler JA, et al. Effects of oral potassium on blood pressure. Meta-analysis of randomized controlled clinical trials. JAMA. 1997 May 28;277(20):1624–32. doi: 10.1001/jama.1997.03540440058033. [DOI] [PubMed] [Google Scholar]
- 44.Cook NR, Obarzanek E, Cutler JA, et al. Joint effects of sodium and potassium intake on subsequent cardiovascular disease: the Trials of Hypertension Prevention follow-up study. Arch Intern Med. 2009 Jan 12;169(1):32–40. doi: 10.1001/archinternmed.2008.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45•.Berry SE, Mulla UZ, Chowienczyk PJ, Sanders TA. Increased potassium intake from fruit and vegetables or supplements does not lower blood pressure or improve vascular function in UK men and women with early hypertension: a randomised controlled trial. Br J Nutr. 2010 Aug 2;:1–9. doi: 10.1017/S0007114510002904. This randomised controlled cross-over trial showed no beneficial effects of dietary potassium supplement in terms of blood pressure, arterial stiffness, endothelial function, and urinary and plasma isoprostane and C-reactive protein concentrations in patients with early stages of hypertension. [DOI] [PubMed] [Google Scholar]
- 46•.He FJ, Marciniak M, Carney C, et al. Effects of potassium chloride and potassium bicarbonate on endothelial function, cardiovascular risk factors, and bone turnover in mild hypertensives. Hypertension. 2010 Mar;55(3):681–8. doi: 10.1161/HYPERTENSIONAHA.109.147488. This 12-week randomized, double-blind, placebo-controlled study found that potassium supplementation may be beneficial in terms of improving endothelial dysfunction and reducing albuminuria. [DOI] [PubMed] [Google Scholar]
- 47•.Ying WZ, Aaron K, Wang PX, Sanders PW. Potassium inhibits dietary salt-induced transforming growth factor-beta production. Hypertension. 2009 Nov;54(5):1159–63. doi: 10.1161/HYPERTENSIONAHA.109.138255. This experimental study showed an important role of the endothelial calcium-activated potassium channel in the vascular response to excess salt intake. They also showed increased dietary intake of potassium could reduce the production of TGF-beta in high salt-fed rats. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48•.Oberleithner H, Callies C, Kusche-Vihrog K, et al. Potassium softens vascular endothelium and increases nitric oxide release. Proc Natl Acad Sci U S A. 2009 Feb 24;106(8):2829–34. doi: 10.1073/pnas.0813069106. The authors demonstrated that an acute increase of potassium in the physiological range swelled and softened the endothelial cell and increased the release of nitric oxide. A high physiological sodium concentration, in the presence of aldosterone, prevented these changes. [DOI] [PMC free article] [PubMed] [Google Scholar]