

Abstract
Objective
Cleaved high molecular weight kininogen (HKa), an activation product of the plasma kallikrein-kinin system, inhibits endothelial cell functions. We questioned whether HKa affects the function of endothelial progenitor cells (EPCs) and accelerates their senescence.
Methods and Results
Treatment with HKa for two weeks markedly inhibited the formation of large colonies and proliferation of EPCs on collagen surfaces, whereas HKa did not affect collagen-mediated EPC adhesion and survival. Concomitantly, treated EPCs displayed flattened and giant cell morphological changes with formation of intracellular vacuoles. As determined by acidic β-galactosidase staining, HKa increased senescent EPCs by 2 fold and >3 fold after culture for one and two weeks, respectively. Additionally, HKa suppressed the telomerase activity of EPCs. HKa concentration-dependently increased the generation of intracellular reactive oxygen species (ROS), and markedly upregulated p38 kinase phosphorylation and prosenescence molecule p16INK4a expression. SB203580, a p38 inhibitor attenuated the level of HKa-enhanced p16INK4a expression. Either quenching of ROS or inhibition of p38 kinase prevented HKa-induced EPC senescence.
Conclusion
HKa accelerates the onset of EPC senescence by activating the ROS - p38 kinase - p16INK4a signaling cascade. This novel activity of HKa points out the likelihood for HKa serving as an endogenous inducer of EPC senescence.
Keywords: kininogen, endothelial progenitor cells, senescence, reactive oxygen species
Introduction
Endothelial progenitor cells (EPCs) are precursors to mature endothelial cells that display distinct characteristics1. These cells have the ability to be mobilized to the site of vascular injury or tissue ischemia, and differentiate into mature endothelial cells, promoting re-endothelialization and neovascularization. Thus, EPCs play an important role in endothelial cell and vessel maintenance. However, in patients with atherosclerosis and cardiovascular diseases, the number and function of EPCs are negatively correlated with the atherosclerotic risk factors, which may contribute to vascular dysfunction2. For example, the frequency of circulating EPCs is reduced 50% in patients with coronary artery disease and patient EPCs display an impaired migratory response3. Moreover, the clinical administration of EPCs to patients with cardiovascular diseases has had limited efficacy, whereas in animal models EPCs successfully restore endothelial function and enhance angiogenesis after tissue ischemia4. Very likely, EPCs are targets of endogenous angiogenic inhibitors elaborated in the setting of atherosclerosis4. Therefore, understanding the factors that affect EPC function and number is important, because it could improve the specific therapies to ultimately correct EPC dysfunction and prevent progression of atherosclerosis.
The plasma kallikrein–kinin system (KKS) consists of the proteins factor XII, prekallikrein, and high molecular weight kininogen (HK)5. This system displays multiple physiologic and pathophysiological activities, such as blood pressure adjustment, modulation of thrombosis, and regulation of endothelial cell function and angiogenesis. Plasma HK is a major component of the KKS and is responsible for the association of this system with the cell surface. The membrane surface of endothelial cells is an important site for the assembly and activation of the KKS 5. Activation of the KKS is triggered in vivo by tissue destruction or by thrombus development6, and results in cleavage of HK by kallikrein into two-chain HK (HKa). Compared with HK, HKa exposes its domain 5 to the surface upon cleavage, thereby acquiring a function of anti-adhesion7. This antiadhesive property enables HKa to inhibit endothelial cell proliferation and to induce endothelial apoptosis on extracellular proteins8,9.
On the basis of these investigations, we examined in this study whether HKa targets EPCs and regulates their function and number. Our data demonstrate that HKa inhibited clonogenic and proliferative capacities of EPCs and accelerated the onset of EPC senescence. HKa significantly increased intracellular reactive oxygen species (ROS) generation and p38 kinase phosphorylation. Moreover, HKa suppressed EPC telomerase activity and upregulated prosenescence molecule p16INK4a expression. Our results provide an improved understanding of the contribution of the KKS cascade to EPC dysfunction in vitro and suggest candidate pathways for investigation of EPC dysfunction in subjects with atherosclerosis.
Materials and methods
EPC Preparation
In this study those EPCs referred to as endothelial colony-forming cells (ECFCs) were isolated from peripheral blood and their progenitor cell capacities were validated as previously described10, 11. To be defined as an EPC, ECFC were examined for evidence of clonal proliferative potential, endothelial cell surface phenotype, and in vitro capillary lumen formation. In brief, after informed consent was obtained in accordance with the Declaration of Helsinki, human blood was collected from healthy volunteer donors. After dilution at 1:1 with Hank’s Buffered Salt Solution, blood was overlaid onto Ficoll-Paque and centrifuged at 740g for 30 minutes. Buffy coat mononuclear cells were collected and resuspended in complete endothelial growth culture medium -2 (EGM-2, Cambrex) with additives (Bullet Kit) provided by the manufacturer and 10% fetal bovine serum (FBS). The cells were cultured in a 6-well tissue culture plate precoated with type I collagen (BD Bioscience) at 37°C. Colonies appearing between 5 and 22 days of culture were identified as a well circumscribed monolayer of cobblestone-appearing cells. EPCs at early passages (passage 1 to 3) were examined for the phenotypic and functional validation analysis.
Lentiviral Transduction of EPCs and Single Cell-based Colony Formation Assay
Lentivirus expressing enhanced green fluorescent protein (EGFP) was generated as previously described 12. The initial mononuclear cells on collagen-coated plates were infected with the supernatant containing Lentivirus expressing EGFP diluted 1:1 with complete EGM-2. The culture medium was replaced every two days. After three weeks, EGFP-positive EPCs (EGFP-EPCs) were observed under fluorescence microscope and sorted by fluorescence cytometry. One single EGFP-EPC was placed in one well of a 96-well tissue culture plate precoated with type 1 collagen containing EGM-2 media. After 24 hours, individual wells were examined under a fluorescence microscope and the wells that contained only one cell were scored in the assay. The cell culture media was changed every 2 days. On day 14, each well was examined under a fluorescence microscope and those wells in which 2 or more endothelial cells were enumerated were scored positive for proliferation.
Data Analysis
Unless stated otherwise, the results shown are from a single experiment representative of at least three separate experiments. The data were calculated as average ± SEM from experiments done at least 3 times, and statistically analyzed by Students’ t test (two groups only) or One Way Analysis of variance (ANOVA) and Student-Newman-Keuls test (multiple groups) 13. Differences with probability values below 0.05 were considered significant.
The supplemental data provide a complete description of materials and methods used for Reagents, Cell Proliferation Assay, Measurement of mRNA Expression by Reverse Transcription-Polymerase Chain Reaction (RT-PCR), Immunoblot Analysis and Adhesion Assay, Senescence-associated β-galactosidase Activity Assay, Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)-based Apoptosis Detection Assay, Intracellular ROS Production, Telomerase activity assay
Results
HKa suppresses clonogenic capacity of EPCs
As described in our previous studies10-12, EPCs have been isolated and expanded ex vivo from adult peripheral blood mononuclear cells. We analyzed clonogenic capacity of EPCs by using a single cell assay previously described 12. Within the first week after isolation, mononuclear cells were cultivated in the EGM-2 containing Lentivirus expressing EGFP, followed by culture in EGM2. A single colony of EPCs uniformly expressing EGFP appeared in most wells (Figure 1A), and the colonies were collected for subsequent studies. To test whether HKa affects the clonal expansion potential of EPCs, a single cell suspension of EGFP-EPCs was seeded into 96-well culture plate precoated with collagen. Because it has been observed that more than 30 % of plasma HK was cleaved in the patients with sepsis and autoimmune diseases14,15, in this study we chose the concentrations of HKa between 30 and 100 nM, 5 to 15% of plasma concentration (660 nM). The culture medium EGM-2 was replaced every two days. After culture for 14 days, EGFP-EPCs formed large colonies (Figure1B), and about 85% of colonies contained more than 200 cells (Large colony, Figure1C). However, in the presence of 50 nM HKa, the percentage of large colonies was markedly reduced to < 6% (p<0.001, Figure1B and C), indicating that HKa strongly inhibits clonogenic capacity of EPCs.
Figure 1. HKa inhibits the colony-forming capacity of EPC colonies on collagen surfaces.
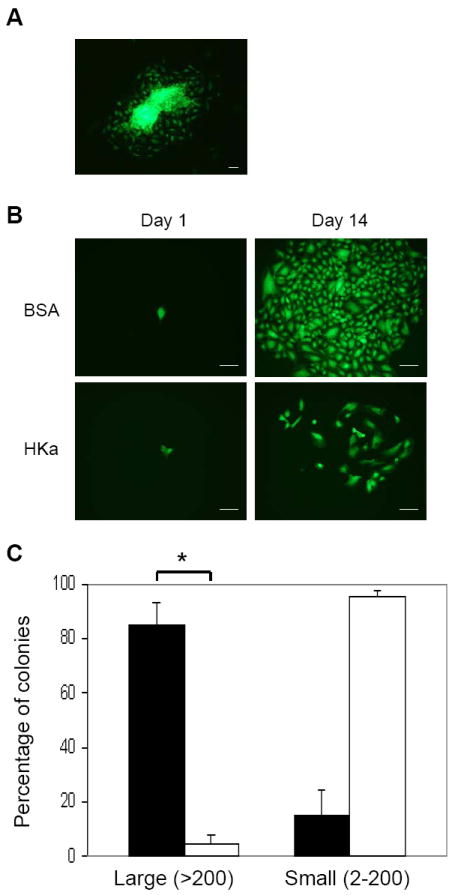
(A) Fluorescent microscopic image of single colony expansion of initial EPCs expressing EGFP. (B) Colony forming assay. On day 1, single cell suspension was seeded on a collagen-coated plate, and cultured in the presence of 0.1% BSA or 50 nM HKa for 14 days. Representative photomicrographs of different endothelial cell clusters (2 - 200 cells) or colonies (more than 200 cells) derived from a single EPC. Scale bar represents 50 μm. (C) The percentage of single EPC-derived colonies undergoing at least 1 cell division after 14 days of culture. Large: colonies of more than 200 cells; Small: colonies of 2-200 cells. ■, BSA, □, HKa. Data represent the results of 3 independent experiments. *P < 0.001.
HKa inhibits EPC proliferation without induction of apoptosis
HKa inhibition of EPC colony formation suggested that HKa suppresses proliferation of EPCs. We thus examined the effect of HKa on proliferative capacity of EPCs using a BrdU incorporation assay. Figure 2A indicates that the treatment of EPCs with HKa for 72 hours significantly inhibited VEGF-stimulated BrdU incorporation into EPCs and the inhibition was concentration-dependent, whereas the significant inhibitory effect of HKa was only detectable at 100 nM after treatment for 48 hours. In contrast to significant effects on proliferation, HKa at the same concentrations did not inhibit EPC adhesion to collagen (Figure 2B), suggesting that HKa inhibition of EPC proliferation does not result from an anti-adhesion activity. Since HKa induces apoptosis of differentiated endothelial cells on vitronectin coated surfaces 9, we examined the effect of HKa on EPC apoptosis. Figure 3 indicates that HKa did not induce EPC apoptosis on collagen surfaces, although its effect on induction of EPCs apoptosis on vitronectin-coated plates was significant. Therefore, HKa inhibition of EPC colony formation and proliferation is not subject to anti-adhesive activity and induction of apoptosis.
Figure 2. HKa inhibits proliferation of EPCs on collagen surfaces.
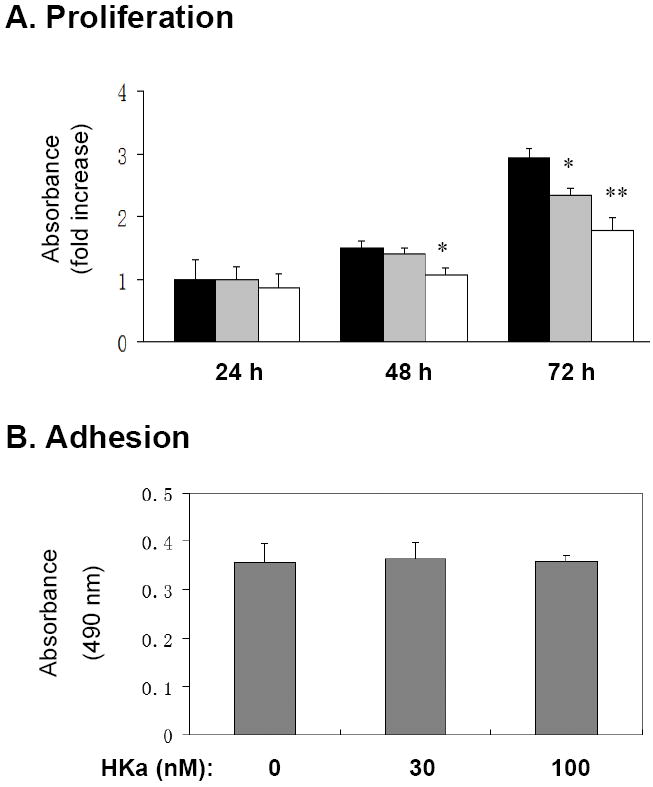
(A) EPCs were cultured in 10 ng/ml VEGF with or without added HKa for the indicated time periods. Black columns, 0.1% BSA; Grey columns, 30 nM HKa; Open columns, 100 nM HKa. Cell proliferation was estimated by the incorporation of BrdU and demonstrated as fold increase compared with the control (0 nM HKa, at 24 hour). *P < 0.05, **P < 0.01 vs. control (BSA). (B) EPCs were cultured on collagen-coated plates with 0, 30 or 100 nM HKa for 2 hours (n=4). After washing with PBS, the remaining adherent cell number was determined as described in the Materials and Methods.
Figure 3. HKa fails to induce apoptosis of EPCs on collagen-coated surface.
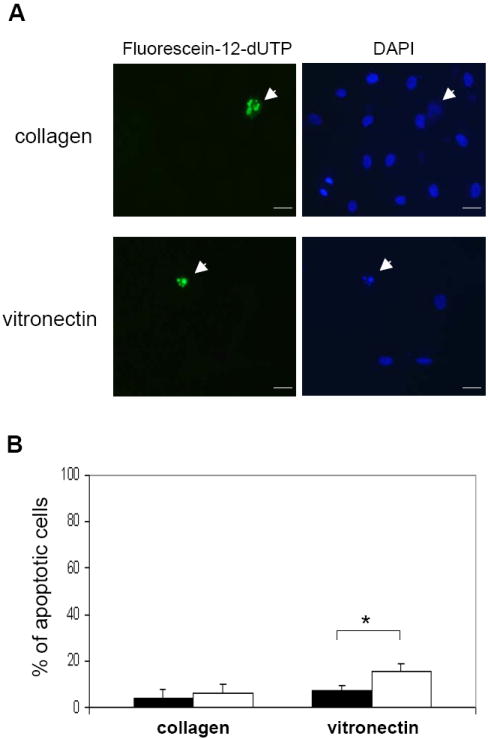
EPCs were cultured on plates precoated with collagen or vitronectin in the presence or absence of 50 nM HKa. After 72 hours, cell apoptosis was analyzed with the TUNEL method as described in the Materials and Methods. (A) Representative fluorescent micrographs of Fluorescein-12-dUTP-positive cells (left) and counterstaining with DAPI (right) in HKa-treated EPCs. Scale bar represents 25 μm. (B) EPCs were cultured with 0.1% BSA or 50 nM HKa on collagen or vitronectin surfaces for 72 hours, which was followed by TUNEL assay. Percentage of apoptotic cells is shown. Closed column, BSA; Open column, HKa. *P<0.01 v.s. BSA.
HKa accelerates the onset of EPC senescence and suppresses telomerase activity
Reduction of EPCs in number and activity has been previously associated with EPC senescence16. The common features of senescent endothelial cells include the enhanced presence of acidic β-galactosidase activity, an increase in lysosomal mass, and formation of autophagic vacuoles17. To determine whether HKa induces EPC senescence, we measured the activity of acidic β-galactosidase, referred as SA-β-galactosidase (SA-β-gal). Although a small portion of EPCs were positive for SA-β-gal staining after culture for 14 days (Figure 4A and B), in the presence of 50 nM HKa, the majority of EPCs became positive for SA-β-gal staining (Figure 4). Moreover, EPCs treated with 50 nM HKa displayed a unique flattened and enlarged morphology, and formed intracellular vacuoles (Figure 4A). Figure 4B demonstrates that HKa treatment significantly resulted in an increase in SA-β-gal-positive cells, 17.2±2.6% v.s. 52.3±3.7% on day 7, and 22.3±2.7% v.s. 85.5±7.9% on day 14. Acceleration of the onset of EPC senescence is known to be critically influenced by the level of telomerase activity, which elongates telomeres, thereby counteracting telomere length reduction induced by each cell division18. Thus, we tested whether HKa treatment was capable of regulating telomerase activity in EPCs. Figure 4C demonstrates that treatment of EPCs with 50 nM HKa for 14 days markedly diminished telomerase activity by > 60% (p<0.005), serving as additional evidence for HKa acceleration of EPC senescence.
Figure 4. HKa accelerates the onset of EPC senescence and suppresses telomerase activity.
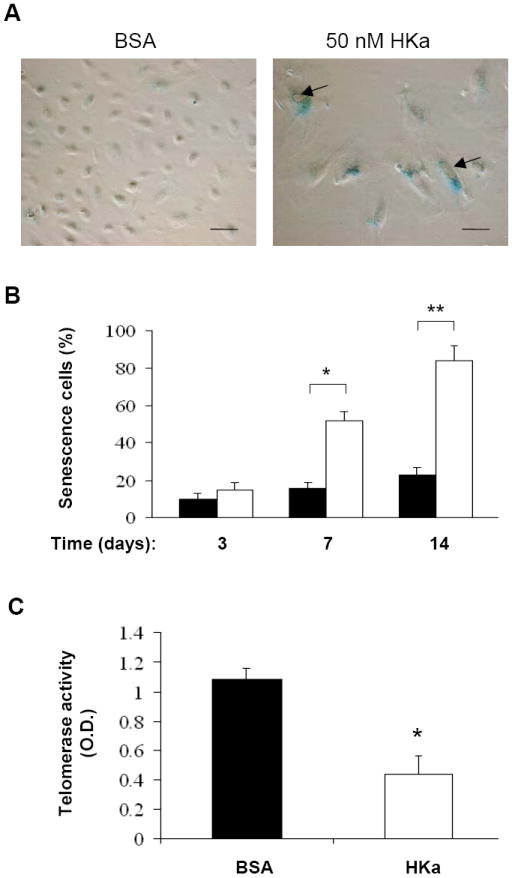
(A) Representative photomicrographs of SA-β-gal-positive EPCs (40 × magnifications). EPCs were cultured in the presence of 0.1% BSA or 50 nM HKa for 14 days. Culture medium was replaced every two days. SA-β-galactosidase activity of EPCs was analyzed as described under Materials and Methods. Scale bar represents 50 μm. Arrows indicate intracellular vacuoles. (B) Percentage of senescent cells was shown. EPCs were cultured in the presence of 0.1% BSA (open columns) or 50 nM HKa (grey columns) for the indicated period of time. The number of SA-β-gal-positive EPCs (senescent cells) was counted (n=6). *P < 0.05, **P < 0.01 v.s. BSA. (C) Effect of HKa on EPC telomerase activity. After EPCs were cultured in the presence of 0.1% BSA or 50 nM HKa for 14 days, telomerase activity was measured as described under the Materials and Methods. Data are mean ± SEM, n=4. *P < 0.001 vs. BSA.
HKa increases intracellular ROS production, contributing to EPC senescence
It has been well documented that cell senescence is tightly associated with intracellular ROS production18. We tested whether HKa exposure increases intracellular ROS in EPCs using the H2DCF-DA labeling assay. As shown in Figure 5A, in a concentration-dependent manner, HKa significantly increased H2DCF-DA oxidation level in exposed EPCs. Thus, we hypothesized that HKa accelerates EPC senescence via ROS production, and performed an inhibition assay using ROS scavengers, Mn(III)tetrakis(4-Benzoic acid)porphyrin Chloride (MnTBAP) and N-Acetyl-Cysteine (NAC). MnTBAP is a cell permeable superoxide dismutase mimetic and peroxynitrite scavenger. NAC is an efficient free radical scavenger and contributes to production of other antioxidant species. Treatment with either 100 μM NAC or 10 μM MnTBAP significantly attenuated the increase in ROS production in EPCs treated with 100 nM HKa for 12 hours (supplemental Figure I). Moreover, 100 μM NAC or 10 μM MnTBAP significantly reduced both the percentage of senescent cells and ROS production in EPCs treated with 50 nM HKa for 14 days (Figure 5B and C). These observations suggest that HKa generation of ROS is involved in the acceleration of EPC senescence.
Figure 5. HKa stimulates generation of ROS in EPCs.
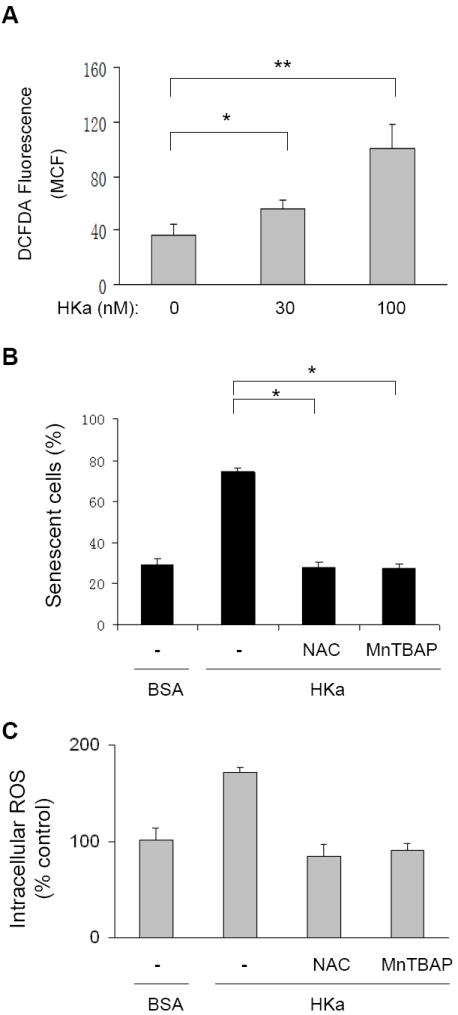
(A) H2DCF-DA oxidation in EPCs. As described under Materials and Methods, EPCs were preincubated with 10 μM H2DCF-DA for 30 minutes, and then treated with or without HKa for 12 hours. H2DCF-DA oxidation was analyzed by flow cytometry. The data are expressed as the mean channel fluorescence (MCF) of H2DCF-DA and represent the average of three separate experiments (± SEM).*P<0.05, **P < 0.001. (B and C) Effect of NAC and MnTBAP on HKa-induced EPC senescence (B) and ROS production (C). EPCs were cultured in the absence or presence of 50 nM HKa for 14 days. As indicated, EPCs in separate groups were also treated with 0.1% BSA (-), 100 μM NAC or 10 μM MnTBAP. Percentage of senescent cells was calculated and depicted (B), and intracellular ROS level are expressed as percent of controls (*P < 0.001, v.s. BSA; n=3).
HKa upregulates p38 kinase phosphorylation and p16INK4a expression
To investigate the mechanism for HKa acceleration of EPC senescence, we measured the levels of p38 kinase activation and prosenescence molecule p16INK4a expression, which are downstream of ROS generation in the process of cellular senescence 19. Immunoblotting analysis indicated that during the seven day experimental period, phosphorylation of p38 kinase was increased by HKa at 30 and 100 nM, respectively (Figure 6A). Concomitantly, HKa upregulated p16INK4a expression at the protein level (Figure 6A). Figure 6B shows an increase in p16INK4a mRNA expression in HKa-treated EPCs. To determine the connection between p38 kinase activation and p16INK4a expression, EPCs were treated with SB203580, a specific p38 kinase inhibitor. Figure 6C demonstrates that 10 μM SB203580 markedly suppressed HKa-induced p16INK4a expression, suggesting that HKa upregulates p16INK4a expression via its activation of p38 kinase. Moreover, 10 μM SB203580 significantly attenuated HKa-mediated EPC senescence (Figure 6D).
Figure 6. HKa increases p38 kinase activity and p16INK4a expression in EPCs.
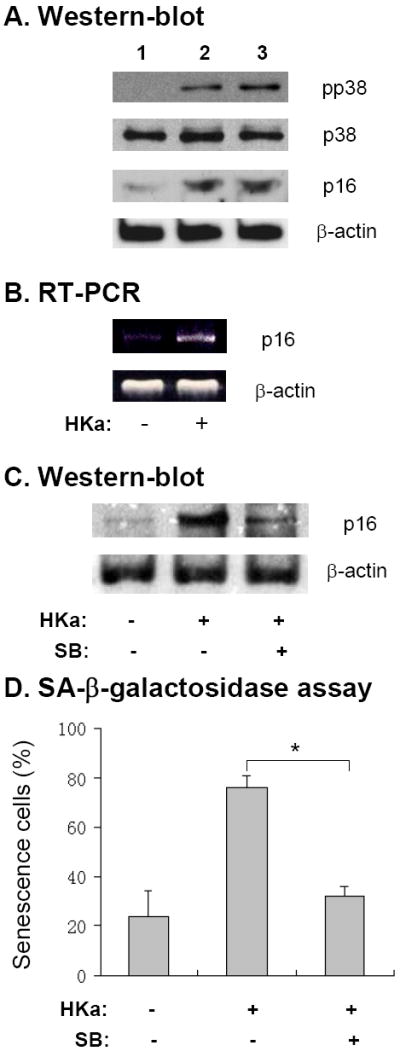
(A) EPCs were cultured in the presence of 0.1% BSA (lane 1), 30 nM HKa (lane 2) or 100 nM HKa (lane 3) for 14 days. Representative immunoblot of phosphorylated p38 kinase and p16INK4a (p16) level in EPCs is shown. The blots for p38 kinase and β-actin serve as loading control. (B) RT-PCR analysis of p16INK4a (p16) mRNA expression in EPCs, which were cultured with or without 50 nM HKa for 14 days. Representative agarose gel image from three experiments is shown. (C) EPCs were cultured in the presence or absence of 50 nM HKa with or without 10 μM SB203580 for 14 days. The level of p16 expression was analyzed by immunoblotting. Representative result from three experiments is shown. The blot for β-actin serves as loading control. (D) EPCs were cultured as described in the panel C. Percentage of senescent EPCs was calculated by SA-β-galactosidase assay (* p<0.01; n=3).
Discussion
Our current study for the first time reveals that HKa accelerates the onset of EPC senescence and induces typical morphological changes of EPC senescence. Consistently, HKa inhibited telomerase activity, and the clonogenic and proliferative capacities of EPCs. HKa treatment increased intracellular ROS generation, and the quenching of ROS by NAC and MnTBAP prevented HKa-induced EPC senescence. HKa treatment enhanced p38 kinase phosphorylation and upregulated p16INK4a expression in the exposed EPC. Inhibition of p38 kinase by SB203580 attenuated HKa-increased p16INK4a expression and EPC senescence. Thus, this study demonstrates novel activities of HKa in regulating several key elements of EPC biology.
Aging is associated with an increased risk for atherosclerosis, and among the many possible causes, insufficient repair of damaged vascular walls by a diminished number or dysfunction in EPC is one hypothesis3. A reduction in EPC number and activity has been associated with EPC senescence14. Because senescence limits the ability of EPCs to sustain ischemic tissue repair, a full characterization of the pathophysiological factors leading to EPC senescence as well as the related underlying mechanisms is clearly important. Our current study demonstrating HKa acceleration of EPC senescence not only expands our understanding of the KKS activation in the regulation of vascular biology, but also reveals a potential novel endogenous inducer of EPC senescence.
The in vivo activation of the KKS and cleavage of HKa has been widely detected in numerous pathophysiological conditions, such as thrombosis6, arthritis20, inflammatory bowel disease21, vasculitis22, sepsis23, systemic amyloidosis24, and preeclampsia25. However, our understanding of how the KKS activation products initiate and regulate downstream effects remains elusive. It has been well documented that HKa inhibits the function of differentiated endothelial cells, such as HUVECs in vitro, as well as perturbs in vivo angiogenesis 26, 27. In this study our results have provided new evidence that HKa targets EPCs and accelerates the onset of their senescence. The plasma concentration of HK is 660 nM. In the patients with sepsis and autoimmune diseases, more than 30 % of plasma HK was cleaved 14,15. Since the minimal concentration of HKa that significantly induced EPC senescence was 30 nM (data not shown), the circulating levels of HKa in the pathological settings could affect EPC function. This HKa-mediated effect was associated with profoundly impaired EPC clonal expansion potential and resulted in a low overall proliferative capacity of the EPC progeny. Since HK is localized at sites of vascular injury, such as atherosclerotic lesions28, it might become cleaved over the abundant negative-charged surfaces to release HKa. The observation that HKa potently inhibited the clonogenic capacity of EPCs suggests that HKa is possibly involved in the vascular dysfunction by blocking EPCs aggregation and expansion. Since HKa inhibition of EPC function is not dependent on its anti-adhesion activity, HKa in plasma may attack circulating EPCs. Although this study provides the first evidence for prosenescent activity of HKa, whether the blockade of HK cleavage prevents EPC senescence remains to be determined using an in vivo model.
EPC senescence can be triggered by a variety of factors, such as proinflammatory cytokines, DNA damage, hyperoxia and hyperglycemia 18. All these factors increase oxidative stress, by which they induce EPC senescence and suppress telomerase activity 16. The mechanism by which HKa accelerates the onset of senescence of EPCs seems to be tightly associated with ROS production. NAC and MnTBAP, which quench ROS, significantly attenuated HKa-induced EPCs senescence (Figure 5). A previous study has shown that HKa inhibits Akt phosphorylation and eNOS phosphorylation 29, which may inhibit the production of nitric oxide (NO). Although NO prevents endothelial cell senescence 30, the NO donor S-nitrosopenicillamine did not prevent HKa-accelerated onset of EPCs senescence (data not shown). Thus, HKa regulation of EPC senescence appears independent of the NO pathway. Accumulation of ROS in most cell types is associated with high expression of p16INK4a, which leads to the arrest of the cell cycle at the G1 phase and accelerates senescence of cells 31. The expression of p16INK4a increases with age and contributes to age-dependent stem and progenitor cell senescence 32. It can be deduced that accumulative ROS exposure, caused by various atherosclerotic risk factors, may also increase the expression of p16INK4a in EPCs, contributing to EPC senescence. We found that HKa increased expression of p16INK4a at the mRNA and protein level (Figure 6), demonstrating that HKa accelerates EPC senescence by the regulation of p16INK4a expression. We previously have found that p38 kinase is responsible for mediating the effect of HKa in other cells 33. Our observation in this study indicates that HKa enhances phosphorylation of p38 kinase, and the inhibition of p38 kinase prevented both HKa-induced p16INK4a expression and EPC senescence. Therefore, HKa-induced ROS seems to act through p38 kinase to upregulate prosenescence molecule p16INK4a expression, which may further regulate telomerase activity and result in EPC senescence.
In conclusion, the present study demonstrates that HKa accelerates the onset of EPC senescence by the activation of the ROS/p38 kinase/p16INK4A signaling cascade. This activity of HKa reveals a novel link between KKS activation and vascular dysfunction, and expands our understanding of the additional pathophysiological activities of this system. Although it remains to be determined whether and how HKa inhibition of EPCs is involved in the disordered vascular remodeling that occurs in vivo, our novel recognition that components of plasma KKS modulate EPC function allows for focused determination of the pathophysiologic activities of plasma KKS in atherosclerosis in future studies.
Supplementary Material
Acknowledgments
Sources of Funding This study was supported in part by the grants from NIH (AR057542 and AR051713).
Footnotes
Disclosures None.
References
- 1.Hirschi KK, Ingram DA, Yoder MC. Assessing identity, phenotype, and fate of endothelial progenitor cells. Arterioscler Thromb Vasc Biol. 2008;28:1584–1595. doi: 10.1161/ATVBAHA.107.155960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hill JM, Zalos G, Halcox JP, Schenke WH, Waclawiw MA, Quyyumi AA, Finkel T. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med. 2003;348:593–600. doi: 10.1056/NEJMoa022287. [DOI] [PubMed] [Google Scholar]
- 3.Schmidt-Lucke C, Rossig L, Fichtlscherer S, Vasa M, Britten M, Kamper U, Dimmeler S, Zeiher AM. Reduced number of circulating endothelial progenitor cells predicts future cardiovascular events: proof of concept for the clinical importance of endogenous vascular repair. Circulation. 2005;111:2981–2987. doi: 10.1161/CIRCULATIONAHA.104.504340. [DOI] [PubMed] [Google Scholar]
- 4.Werner N, Nickenig G. Endothelial progenitor cells in health and atherosclerotic disease. Ann Med. 2007;39:82–90. doi: 10.1080/07853890601073429. [DOI] [PubMed] [Google Scholar]
- 5.Colman RW, Schmaier AH. Contact system: a vascular biology modulator with anticoagulant, profibrinolytic, antiadhesive, and proinflammatory attributes. Blood. 1997;90:3819–3843. [PubMed] [Google Scholar]
- 6.Merkulov S, Zhang WM, Komar AA, Schmaier AH, Barnes E, Zhou Y, Lu X, Iwaki T, Castellino FJ, Luo G, McCrae KR. Deletion of murine kininogen gene 1 (mKng1) causes loss of plasma kininogen and delays thrombosis. Blood. 2008;111:1274–1281. doi: 10.1182/blood-2007-06-092338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weisel JW, Nagaswami C, Woodhead JL, DeLa Cadena RA, Page JD, Colman RW. The shape of high molecular weight kininogen. Organization into structural domains, changes with activation, and interactions with prekallikrein, as determined by electron microscopy. J Biol Chem. 1994;269:10100–10106. [PubMed] [Google Scholar]
- 8.Guo YL, Wang S, Colman RW. Kininostatin, an angiogenic inhibitor, inhibits proliferation and induces apoptosis of human endothelial cells. Arterioscler Thromb Vasc Biol. 2001;21:1427–1433. doi: 10.1161/hq0901.095277. [DOI] [PubMed] [Google Scholar]
- 9.Guo YL, Wang S, Cao DJ, Colman RW. Apoptotic effect of cleaved high molecular weight kininogen is regulated by extracellular matrix proteins. J Cell Biochem. 2003;89:622–632. doi: 10.1002/jcb.10536. [DOI] [PubMed] [Google Scholar]
- 10.Yoder MC, Mead LE, Prater D, Krier TR, Mroueh KN, Li F, Krasich R, Temm CJ, Prchal JT, Ingram DA. Redefining endothelial progenitor cells via clonal analysis and hematopoietic stem/progenitor cell principals. Blood. 2007;109:1801–1809. doi: 10.1182/blood-2006-08-043471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu Y, Dai J, Schmuckler NG, Bakdash N, Yoder MC, Overall CM, Colman RW. Cleaved high molecular weight kininogen inhibits tube formation of endothelial progenitor cells via suppression of matrix metalloproteinase 2. J Thromb Haemost. 2010;8:185–193. doi: 10.1111/j.1538-7836.2009.03662.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ingram DA, Mead LE, Tanaka H, Meade V, Fenoglio A, Mortell K, Pollok K, Ferkowicz MJ, Gilley D, Yoder MC. Identification of a novel hierarchy of endothelial progenitor cells using human peripheral and umbilical cord blood. Blood. 2004;104:2752–2760. doi: 10.1182/blood-2004-04-1396. [DOI] [PubMed] [Google Scholar]
- 13.Wu Y, Rizzo V, Liu Y, Sainz IM, Schmuckler NG, Colman RW. Kininostatin associates with membrane rafts and inhibits alpha(v)beta3 integrin activation in human umbilical vein endothelial cells. Arterioscler Thromb Vasc Biol. 2007;27:1968–1975. doi: 10.1161/ATVBAHA.107.148759. [DOI] [PubMed] [Google Scholar]
- 14.Schmaier AH, Farber A, Schein R, Sprung C. Structural changes of plasma high molecular weight kininogen after in vitro activation and in sepsis. J Lab Clin Med. 1988;112:182–192. [PubMed] [Google Scholar]
- 15.Weiser P, Qian Y, Pan J, Zhou X, Lu H, Studelska DR, Shih FF, Zhang L. Activated contact system and abnormal glycosaminoglycans in lupus and other auto- and non-autoimmune diseases. Prog Mol Biol Transl Sci. 2010;93:443–472. doi: 10.1016/S1877-1173(10)93019-6. [DOI] [PubMed] [Google Scholar]
- 16.Imanishi T, Tsujioka H, Akasaka T. Endothelial progenitor cells dysfunction and senescence: contribution to oxidative stress. Curr Cardiol Rev. 2008;4:275–286. doi: 10.2174/157340308786349435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thijssen DH, Torella D, Hopman MT, Ellison GM. The role of endothelial progenitor and cardiac stem cells in the cardiovascular adaptations to age and exercise. Front Biosci. 2009;14:4685–4702. doi: 10.2741/3560. [DOI] [PubMed] [Google Scholar]
- 18.Imanishi T, Tsujioka H, Akasaka T. Endothelial progenitor cell senescence--is there a role for estrogen? Ther Adv Cardiovasc Dis. 2010;4:55–69. doi: 10.1177/1753944709353173. [DOI] [PubMed] [Google Scholar]
- 19.Chang J, Li Y, Huang Y, Lam KS, Hoo RL, Wong WT, Cheng KK, Wang Y, Vanhoutte PM, Xu A. Adiponectin Prevents Diabetic Premature Senescence of Endothelial Progenitor Cells and Promotes Endothelial Repair by Suppressing the p38 MAP kinase/p16INK4A Signaling Pathway. Diabetes. 2010 doi: 10.2337/db10-0582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sainz IM, Isordia-Salas I, Castaneda JL, Agelan A, Liu B, DeLa Cadena RA, Pixley RA, Adam A, Sartor RB, Colman RW. Modulation of inflammation by kininogen deficiency in a rat model of inflammatory arthritis. Arthritis Rheum. 2005;52:2549–2552. doi: 10.1002/art.21202. [DOI] [PubMed] [Google Scholar]
- 21.Isordia-Salas I, Pixley RA, Sainz IM, Martinez-Murillo C, Colman RW. The role of plasma high molecular weight kininogen in experimental intestinal and systemic inflammation. Arch Med Res. 2005;36:87–95. doi: 10.1016/j.arcmed.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 22.Kahn R, Hellmark T, Leeb-Lundberg LM, Akbari N, Todiras M, Olofsson T, Wieslander J, Christensson A, Westman K, Bader M, Muller-Esterl W, Karpman D. Neutrophil-derived proteinase 3 induces kallikrein-independent release of a novel vasoactive kinin. J Immunol. 2009;182:7906–7915. doi: 10.4049/jimmunol.0803624. [DOI] [PubMed] [Google Scholar]
- 23.Asmis LM, Asmis R, Sulzer I, Furlan M, Lammle B. Contact system activation in human sepsis -47kD HK, a marker of sepsis severity? Swiss Med Wkly. 2008;138:142–149. doi: 10.4414/smw.2008.11788. [DOI] [PubMed] [Google Scholar]
- 24.Maas C. Misfolded proteins activate Factor XII in humans, leading to kallikrein formation without initiating coagulation. The Journal of Clinical Investigation. 2008;118:3208–3218. doi: 10.1172/JCI35424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamada H, Atsumi T, Amengual O, Koike T, Furuta I, Ohta K, Kobashi G. Anti-beta2 glycoprotein-I antibody increases the risk of pregnancy-induced hypertension: a case-controlled study. J Reprod Immunol. 2010;84:95–99. doi: 10.1016/j.jri.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 26.Cao DJ, Guo YL, Colman RW. Urokinase-type plasminogen activator receptor is involved in mediating the apoptotic effect of cleaved high molecular weight kininogen in human endothelial cells. Circ Res. 2004;94:1227–1234. doi: 10.1161/01.RES.0000126567.75232.46. [DOI] [PubMed] [Google Scholar]
- 27.Colman RW, Jameson BA, Lin Y, Johnson D, Mousa SA. Domain 5 of high molecular weight kininogen (kininostatin) down-regulates endothelial cell proliferation and migration and inhibits angiogenesis. Blood. 2000;95:543–550. [PubMed] [Google Scholar]
- 28.Peerschke EI, Minta JO, Zhou SZ, Bini A, Gotlieb A, Colman RW, Ghebrehiwet B. Expression of gC1q-R/p33 and its major ligands in human atherosclerotic lesions. Mol Immunol. 2004;41:759–766. doi: 10.1016/j.molimm.2004.04.020. [DOI] [PubMed] [Google Scholar]
- 29.Katkade V, Soyombo AA, Isordia-Salas I, Bradford HN, Gaughan JP, Colman RW, Panetti TS. Domain 5 of cleaved high molecular weight kininogen inhibits endothelial cell migration through Akt. Thromb Haemost. 2005;94:606–614. doi: 10.1160/TH04-12-0834. [DOI] [PubMed] [Google Scholar]
- 30.Hayashi T, Matsui-Hirai H, Miyazaki-Akita A, Fukatsu A, Funami J, Ding QF, Kamalanathan S, Hattori Y, Ignarro LJ, Iguchi A. Endothelial cellular senescence is inhibited by nitric oxide: implications in atherosclerosis associated with menopause and diabetes. Proc Natl Acad Sci U S A. 2006;103:17018–17023. doi: 10.1073/pnas.0607873103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takahashi A, Ohtani N, Yamakoshi K, Iida S, Tahara H, Nakayama K, Nakayama KI, Ide T, Saya H, Hara E. Mitogenic signalling and the p16INK4a-Rb pathway cooperate to enforce irreversible cellular senescence. Nat Cell Biol. 2006;8:1291–1297. doi: 10.1038/ncb1491. [DOI] [PubMed] [Google Scholar]
- 32.Heiss C, Keymel S, Niesler U, Ziemann J, Kelm M, Kalka C. Impaired progenitor cell activity in age-related endothelial dysfunction. J Am Coll Cardiol. 2005;45:1441–1448. doi: 10.1016/j.jacc.2004.12.074. [DOI] [PubMed] [Google Scholar]
- 33.Khan MM, Bradford HN, Isordia-Salas I, Liu Y, Wu Y, Espinola RG, Ghebrehiwet B, Colman RW. High-molecular-weight kininogen fragments stimulate the secretion of cytokines and chemokines through uPAR, Mac-1, and gC1qR in monocytes. Arterioscler Thromb Vasc Biol. 2006;26:2260–2266. doi: 10.1161/01.ATV.0000240290.70852.c0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.