

Abstract
A prominent characteristic of the inherited intellectual impairment disease fragile X syndrome (FXS) is neuronal hyperexcitability, resulting in a variety of symptoms, such as hyperactivity, increased sensitivity to sensory stimuli, and a high incidence of epileptic seizures. These symptoms account for a significant part of the disease pattern, but the underlying molecular mechanisms of neuronal hyperexcitability in FXS remain poorly understood. FXS is caused by loss of expression of fragile X mental retardation protein (FMRP), which regulates synaptic protein synthesis and is a key player to limit signaling pathways downstream of metabotropic glutamate receptors 1/5 (mGlu1/5). Recent findings suggest that FMRP might also directly regulate voltage-gated potassium channels. Here, we show that total and plasma membrane protein levels of Kv4.2, the major potassium channel regulating hippocampal neuronal excitability, are reduced in the brain of an FXS mouse model. Antagonizing mGlu5 activity with 2-methyl-6-(phenylethynyl)-pyridine (MPEP) partially rescues reduced surface Kv4.2 levels in Fmr1 knock-out (KO) mice, suggesting that excess mGlu1/5 signal activity contributes to Kv4.2 dysregulation. As an additional mechanism, we show that FMRP is a positive regulator of Kv4.2 mRNA translation and protein expression and associates with Kv4.2 mRNA in vivo and in vitro. Our results suggest that absence of FMRP-mediated positive control of Kv4.2 mRNA translation, protein expression, and plasma membrane levels might contribute to excess neuronal excitability in Fmr1 KO mice, and thus imply a potential mechanism underlying FXS-associated epilepsy.
Introduction
Patients suffering from fragile X syndrome (FXS), the most common inherited form of intellectual disability, show multiple signs of altered neuronal excitability. These include hyperactivity, attention deficit disorders, hypersensitivity to sensory stimuli, anxiety and in 20% of all cases development of epileptic seizures (Hagerman et al., 2009). Many of these symptoms are recapitulated in Fmr1 knock-out (KO) mice, and electrophysiological experiments have demonstrated that Fmr1 KO neurons are hyperexcitable (Chuang et al., 2005). However, the precise mechanisms leading to increased excitability of neurons in the absence of the fragile X mental retardation protein (FMRP) are unknown.
A possible mechanism underlying the heightened neuronal excitability in FXS might be direct regulation of neuronal ion channels by FMRP. Recently, evidence has been growing that FMRP might be involved in the function of potassium channels: FMRP was shown to regulate two different potassium channels, Kv3.1b and Slack, leading to altered potassium signaling of those channels in Fmr1 KO mice (Brown et al., 2010; Strumbos et al., 2010). However, neither of these potassium channels has been linked to epilepsy in humans or animal models, leaving the underlying mechanism of hyperexcitability in FXS elusive.
The major ion channel regulating neuronal excitability in the hippocampus is the A-type potassium channel Kv4.2, which decreases back-propagating action potentials in dendrites (Birnbaum et al., 2004; Chen et al., 2006). In several different animal models for epilepsy, seizures were shown to regulate hippocampal Kv4.2 protein phosphorylation, localization and levels (Francis et al., 1997; Lugo et al., 2008). Of note, a truncated mutation of Kv4.2 leads to temporal lobe epilepsy in humans (Singh et al., 2006), and Kv4.2 KO mice have a higher sensitivity to convulsant stimuli (Barnwell et al., 2009). Thus, a functional lack of either Kv4.2 channels or FMRP results in hyperexcitability and/or epilepsy. Based on these findings, we hypothesized that Kv4.2, a key player to dampen neuronal firing, might be dysregulated in FXS.
Here, we show that total, dendritic and cell surface expression of Kv4.2 are reduced in Fmr1 KO brains. The metabotropic glutamate receptor 5 (mGlu5) antagonist 2-methyl-6-(phenylethynyl)-pyridine (MPEP) partially rescues reduced surface levels in Fmr1 KO hippocampal slices, suggesting that excess mGlu receptor-signaling can modulate Kv4.2 dysregulation in FXS, which might be directly caused by loss of FMRP-mediated control. We show that FMRP associates with Kv4.2 mRNA, and that absence of FMRP decreases Kv4.2 mRNA translation and protein expression in vivo and in vitro, whereas FMRP overexpression increases protein levels of Kv4.2 3′-untranslated region (UTR) reporters. Our results suggest that reduced Kv4.2 protein expression in Fmr1 KO is not just a secondary effect of dysregulated neuronal transmission in FXS, but a direct effect of the absence of FMRP-mediated positive control of Kv4.2 mRNA translation. Loss of FMRP-mediated Kv4.2 mRNA regulation might thus contribute to the neuronal hyperexcitability and epileptic seizure phenotype observed in FXS.
Materials and Methods
Animals and quantitative real-time primers.
Male Fmr1 knock-out mice in C57BL/6J background and male wild-type (WT) littermates were used at 3 weeks (immunostainings, immunoprecipitation and biotinylation assays) and 8 weeks of age (Western blot analysis of cortical lysates). Small interfering RNAs(siRNAs) to knockdown FMRP in cells, and quantitative real-time (qRT)-PCR primers for GFP (GenBank accession number U55763), GluN1 [National Center for Biotechnology Information (NCBI) accession number NM_008169], and PSD95 (postsynaptic density-95 protein) (NCBI accession number NM_007864) have been described previously (Gross et al., 2010). qRT-PCR primers for Kv4.2 were as follows: Kv4.2 forward, gttctatggttgggctgtg; Kv4.2 reverse, gtggctctaactgtatctatg (NCBI accession number NM_019697).
Fluorescent immunostainings on cells and brain sections.
Brains from transcardially perfused (4% paraformaldehyde) mice were postfixed overnight, cryoprotected in 30% sucrose, and flash-frozen. Fluorescent immunostainings were performed on WT and Fmr1 KO brain sections (postnatal day 21) applying antigen retrieval methods, as described previously (Gabel et al., 2004; Christie et al., 2009), with the following modifications: the 10- to 12-μm-thick sections were mounted on microscope slides for processing, and all buffers were based on TBS (100 mm Tris-HCl, pH 7.4, 150 mm NaCl). Permeabilization was performed with 0.5% Triton X-100 for 20 min, and primary antibodies [Kv4.2, 1:200; Kv1.2, 1:500; Kv3.4, 1:1000 (UC Davis/NIH NeuroMab Facility); Kv4.2, 1:250 (Santa Cruz Biotechnology); GluN1, 1:200 (Millipore)] were incubated overnight at room temperature. Brain sections were imaged as z-stacks using a Zeiss LSM 510 Meta confocal microscope, deconvolved (Autoquant X, Media Cybernetics) and quantified using Imaris Software (Bitplane). Immunofluorescence on cells was performed as described previously (Antar et al., 2004). Cultured hippocampal neurons were fixed at 17 days in vitro (DIV), and primary antibody (Kv4.2, 1:2000, UC Davis/NIH NeuroMab Facility) was incubated at room temperature for 1 h. Cells were imaged and analyzed as described previously (Antar et al., 2004).
Western blot analysis.
Western blot analysis and quantification was performed as described previously (Gross et al., 2010). Antibodies for Kv4.2 were obtained from Millipore, 1:2000; for GluN1 from BD Pharmingen, 1:1000; for β-tubulin from Sigma, 1:500,000; and for green fluorescent protein (GFP) from Clontech, 1:1000.
Biotinylation assays.
Surface biotinylation of hippocampal slices was performed as described previously (Nosyreva and Huber, 2005), with the following modifications: Hippocampi were dissected into ice-cold artificial CSF (ACSF) (Chuang et al., 2005), and immediately cut into 350-μm-thick slices. Slices were equilibrated in prewarmed ACSF for 20 min (37°C, 5%CO2), treated with 50 μm MPEP or vehicle for 40 min, followed by biotinylation for 15 min with 2 mm EZ-Link sulfo-NHS-Biotin (Thermo Scientific) on ice, quenched for 10 min with 100 mm glycine, and washed once with ice-cold TBS before lysis. Cell surface biotinylation assay was performed as described previously (Ehlers, 2000) with the following modifications: high-density cultured cortical neurons were used at 17–21 DIV, and cells were incubated with 1.0 mg/ml EZ-Link sulfo-NHS-Biotin for 30 min.
Quantitative coimmuoprecipitation in vivo and in vitro.
FMRP-specific coimmunoprecipitations and quantitative real-time PCR were performed as described previously (Muddashetty et al., 2007).
Polysomal gradients on synaptoneurosomes.
Preparation and analysis of polysomal gradients from cortical synaptoneurosomes, and cell lysates were performed as described previously (Muddashetty et al., 2007; Gross et al., 2010).
Statistics.
All statistical analyses were performed in SPSS 17.0 and PASW Statistics 18. Data were tested for normality and homogeneity of variances, and appropriate tests were used as indicated for each figure.
Results
Decreased dendritic and membrane surface levels of the potassium channel Kv4.2 in Fmr1 KO mice
The molecular mechanisms underlying hyperexcitability of Fmr1 KO neurons or increased susceptibility to epileptic seizures in FXS patients have not been characterized. Here, we applied a candidate-based approach to investigate whether Kv4.2, the major ion channel regulating neuronal excitability in hippocampal dendrites, is dysregulated in the absence of FMRP and thus might cause hyperexcitability in FXS. Quantification of fluorescent immunostainings in dendritic areas of the dentate gyrus and CA1 in hippocampal sections (Fig. 1A) and in dendrites of cultured hippocampal neurons (Fig. 1D) demonstrated that dendritic Kv4.2 protein levels are significantly reduced in Fmr1 KO mice. In contrast, other Kv channels, such as Kv1.2 (Fig. 1B) and Kv3.4 (data not shown), as well as the NMDA receptor subunit GluN1 (Fig. 1C) were unchanged in Fmr1 KO hippocampus. A similar reduction of Kv4.2 protein could also be detected in total cortical homogenates by Western blot analysis (Fig. 2A). Analysis of surface proteins in cultured cortical neurons (Fig. 2B), and in hippocampal slices (Fig. 2C) demonstrated that not only total protein levels, but also surface levels of Kv4.2 were reduced in the absence of FMRP. This implies that loss of total Kv4.2 protein was not functionally compensated by altered ratios of surface/internalized channels. Furthermore, reduced surface ion channel levels were not a general phenotype of FXS neurons, as GluN1 membrane levels were not significantly different in either hippocampal slices, or cortical neurons. Pretreatment of hippocampal slices with the mGlu5 antagonist MPEP (50 μm, 40 min) partially increased, but not fully restored Kv4.2 plasma membrane levels in Fmr1 KO (Fig. 2C; 60% of WT before, 80% of WT after MPEP treatment). Of note, our results represent the first evidence for mGlu1/5-mediated regulation of potassium channels at the cell surface of hippocampal neurons, and suggest that excess mGlu1/5 signaling may contribute to reduced Kv4.2 expression in Fmr1 KO mice. However, we could not detect full rescue of Kv4.2 surface levels to wild-type levels with the same MPEP dose that previously had been shown to rescue several FXS-associated phenotypes (Chuang et al., 2005; Nakamoto et al., 2007; Gross et al., 2010). While we cannot exclude that higher MPEP doses would have led to a complete rescue, we sought to test the hypothesis that additional mechanisms besides dysregulated mGlu5 signaling might account for reduced Kv4.2 total and surface expression in Fmr1 KO hippocampus.
Figure 1.

Kv4.2 protein levels are reduced in Fmr1 KO hippocampal dendrites. A–C, Quantitative analysis of fluorescent immunohistochemical stainings in dendritic layers from dentate gyrus (DG) and CA1 shows significantly reduced dendritic levels of Kv4.2 (A) (DG: nWT = 27, nKO = 25, *p = 0.001, Mann–Whitney U test; CA1: nWT = 27, nKO = 25, *p = 0.034, independent t test), whereas Kv1.2 (B) (DG: nWT = 21, nKO = 18, p = 0.547; CA1: nWT = 22, nKO = 22, p = 0.461, independent t tests), and GluN1 (C) (DG: nWT = 14, nKO = 14, p = 0.943; CA1: nWT = 16, nKO = 16, p = 0.394, independent t tests) are not significantly different. Example images of the entire hippocampus (including 4′,6-diamidino-2-phenylindole (DAPI) staining to visualize cell nuclei), the CA1 region, and the DG for Kv4.2 and Kv1.2 are shown on the left; for GluN1, example images from DG are shown. High-magnification images were pseudocolored with the intensity map shown in A. Scale bar, 200 μm. D, Quantitative immunofluorescence analysis in dendrites of 17 DIV cultured hippocampal neurons shows that Kv4.2 protein levels are significantly reduced in KO (n = 30, *p = 0.0007, independent t test). Immunofluorescent images were pseudocolored with a 16-color intensity map (shown on the right). Scale bar, 10 μm. All error bars represent SEM.
Figure 2.

Reduced levels of Kv4.2 at neuronal cell surfaces in Fmr1 KO mice can be partially rescued by antagonizing mGlu5. A, Western blot analysis of total Kv4.2 protein shows reduced Kv4.2 levels in KO cortex (n = 5 each WT and KO, 2 independent litters, *p = 0.001, paired t test). B, Western blot analysis of surface biotinylation assays demonstrates reduced Kv4.2 surface levels in cultured cortical neurons (n = 4, *p = 0.03, paired t test), whereas GluN1 surface levels are unchanged (n = 4, p = 0.694, paired t test). Representative Western blots are shown above. Surface protein levels were normalized to β-tubulin input. C, In hippocampal slices from Fmr1 KO, Kv4.2, but not GluN1, surface levels are significantly reduced and can be partially rescued by pretreatment with the mGlu5 antagonist MPEP (50 μm, 40 min) [Kv4.2: n = 6, two-way ANOVA; treatment: p = 0.976, genotype: p = 0.01, interaction genotype–treatment: p = 0.01, Games-Howell post hoc analyses (unequal variances assumed): pwtctr-wtMPEP = 0.467, *pwtctr-koctr < 0.001, *pkoctr-koMPEP = 0.036, pwtctr-koMPEP = 0.053; GluN1: n = 6, two-way ANOVA; treatment: p = 0.936, genotype: p = 0.500, interaction genotype–treatment: p = 0.679). GluN1 and Kv4.2 surface levels were normalized to β-tubulin; representative Western blots are shown on the left. All error bars represent SEM.
Reduced association of Kv4.2 mRNA with translating polysomes in Fmr1 KO brain
FMRP has been shown to regulate mRNA translation of specific targets, as well as general protein synthesis (Bassell and Warren, 2008). To reveal a potential mechanism underlying reduced Kv4.2 protein levels, we therefore analyzed the levels and polysomal association of Kv4.2 mRNA in the absence of FMRP using qRT-PCR and sucrose gradient sedimentation profiling. Whereas total mRNA levels were unchanged in Fmr1 KO brain (Fig. 3A), the relative amount of Kv4.2 mRNA associated with puromycin-sensitive (i.e., actively translating) polysomes was reduced in cortical synaptic fractions (Fig. 3B,C), as well as in hippocampal lysates (data not shown). These results suggest that decreased levels of Kv4.2 protein are due to reduced translation of Kv4.2 mRNA in the FMRP-deficient brain. This is an unusual observation, as FMRP was shown to be a negative regulator of global translation in the brain and in synaptic fractions (Dölen et al., 2007; Gross et al., 2010). Furthermore, the great majority of FMRP target mRNAs was shown to be translated excessively in the absence of FMRP (Zalfa et al., 2003; Muddashetty et al., 2007; Gross et al., 2010). To analyze whether Kv4.2 mRNA might be one of the “unconventional” target mRNAs of FMRP that show reduced translation and protein levels in the FXS mouse model (Bechara et al., 2009; Fähling et al., 2009), we next examined whether Kv4.2 mRNA associates with FMRP in brain cortex. Using Kv4.2-specific quantitative real-time PCR analyses of FMRP-coimmunoprecipitates, we demonstrated that FMRP associates with Kv4.2 mRNA in mouse cortex (Fig. 3D,E). A previous study showed protein-protein interactions between FMRP and the potassium channel Slack (Brown et al., 2010), however we could not detect any association of FMRP with Kv4.2 protein (data not shown).
Figure 3.

Kv4.2 mRNA translation is reduced in synaptic fractions from Fmr1 KO cortices. A, qRT-PCR analysis of Kv4.2 mRNA levels in different brain regions: hippocampus (hippo) (n = 4, p = 0.648, paired t test), cortex (n = 4, p = 0.372, paired t test), and cerebellum (cb) (n = 4, p = 0.926, paired t test) shows no significant difference between WT and KO. mRNA levels were normalized to tubulin mRNA. B, Polysome association of Kv4.2 mRNA in cortical synaptoneurosomes is puromycin sensitive, suggesting that Kv4.2 mRNA in these fractions is actively translated (n = 4, *p = 0.026, paired t test). C, Association of Kv4.2 mRNA with puromycin-sensitive fractions is significantly reduced in Fmr1 KO (n = 6, *p = 0.04, paired t test). D, E, Kv4.2 mRNA associates with FMRP in vivo in forebrain. D, qRT-PCR analysis of FMRP-specific coimmunoprecipitations from WT brain shows that Kv4.2 mRNA and the positive control PSD95 are significantly enriched compared with IgG control, whereas the negative control GluN1 shows no significant enrichment (n = 3; Kv4.2, *p = 0.040; GluN1, p = 0.147; PSD95, *p = 0.007; paired t tests). E, Specific enrichment of Kv4.2 mRNA, but not GluN1 mRNA, was also detected in FMRP-specific immunoprecipitations from WT and Fmr1 KO lysates (n = 3; Kv4.2, *p = 0.015; GluN1, p = 0.84; paired t tests).
The 5′- and 3′-UTRs of Kv4.2 mRNA mediate association with and translational regulation by FMRP
Using recombinant GFP-constructs, we demonstrated that both the Kv4.2 5′- and 3′-UTRs were sufficient to mediate association with FMRP (Fig. 4A), suggesting a cooperative regulation of Kv4.2 mRNA by FMRP via several parts of the Kv4.2 mRNA sequence. In line with this assumption, analysis of Kv4.2 3′-UTR association with several FMRP deletion constructs suggests that the association of FMRP with Kv4.2 mRNA does not seem to be mediated by a single mRNA binding domain, but rather requires multiple domains of FMRP (Fig. 4B). The protein expression of the FMRP deletion constructs was similar for all constructs, as tested by Western blot (data not shown). Analysis of polysomal association and protein expression of GFP-constructs containing either the Kv4.2 5′-UTR or 3′-UTR further supported the hypothesis that FMRP-regulation of Kv4.2 mRNA occurs via both 5′- and 3′-UTRs. For GFP mRNA fused to either Kv4.2 UTRs, siRNA-mediated FMRP knockdown in cultured cell lines led to decreased polysomal association of mRNA (Fig. 4C–E), and reduced protein levels (Fig. 4F). Of note, reduced polysomal association was observed after siRNA-mediated knockdown of FMRP in HEK293T cells, which do not express mGlu1/5 (Gross et al., 2010). This strongly corroborates our hypothesis that there is an additional FMRP-dependent mechanism, apart from aberrant mGlu1/5 signaling, which leads to reduced Kv4.2 expression in Fmr1 KO mice. Overexpression of FMRP significantly increased the protein expression of recombinant constructs containing the Kv4.2 3′-UTR, but not for a construct just containing the 5′-UTR (Fig. 4G). Our results imply that FMRP is a positive regulator of Kv4.2 mRNA translation and protein expression, which is mediated by UTR sequences. Absence of FMRP leads to reduced Kv4.2 mRNA translation, protein expression and surface levels, which might partially underlie neuronal hyperexcitability in FXS.
Figure 4.
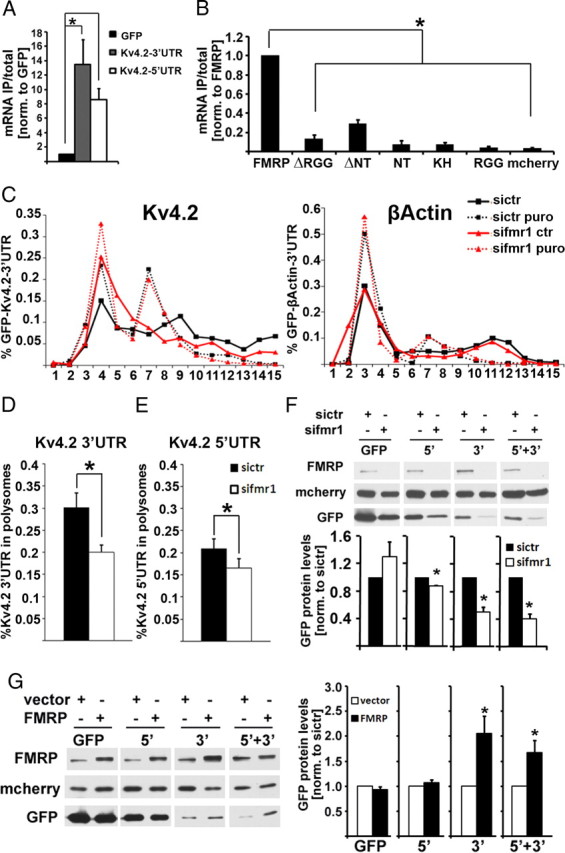
FMRP regulation of Kv4.2 mRNA is mediated by both 5′- and 3′-UTRs of Kv4.2 mRNA. A, The association of both GFP-Kv4.2 3′-UTR and Kv4.2 5′-UTR-GFP mRNAs with mcherry-FMRP protein is significantly higher than that with GFP mRNA alone (n = 4, one-way ANOVA with least significant difference post hoc tests: *pGFP 3′-UTR = 0.007, *pGFP 5′-UTR = 0.049). B, mcherry-FMRP shows significantly higher affinity to the 3′-UTR of Kv4.2 mRNA than does mcherry-FMRP lacking either the RGG box (ΔRGG) or the N terminus (ΔNT), or mcherry-fusion proteins containing either of the three known FMRP RNA-binding domains [N terminus (NT), KH-domain, or RGG-box, respectively] (n = 6, one-way ANOVA, *p < 0.0001; Tukey's honestly significant difference post hoc comparisons, *p ≤ 0.05). C, Association of recombinant GFP-Kv4.2 3′-UTR (16 h expression) with puromycin-sensitive polysome fractions is reduced in HEK293T cells after 48 h of siRNA-mediated depletion of FMRP (left), whereas the β-actin 3′-UTR is unchanged (right). D, Quantitative analysis shows a significant reduction of Kv4.2 3′-UTR in heavy polysomes from FMRP-knockdown cells (n = 4, *p = 0.0195, paired t test). Similar results were seen for a luciferase reporter, and in N2A cells (data not shown). E, Likewise, Kv4.2 5′-UTR was reduced in polysomes from FMRP-depleted HEK293T cells, but to a lesser extent compared with the 3′-UTR (n = 3, *p = 0.031, paired t test; 3′-UTR, 33%; 5′-UTR, 21% reduction). F, GFP protein levels are reduced when fused to either Kv4.2 5′-UTR (n = 3, *p = 0.003, paired t test), Kv4.2 3′-UTR (n = 3, *p = 0.021, paired t test), or both UTRs (n = 3, *p = 0.017, paired t test) in FMRP-knockdown cells compared with control knockdown cells; however, a GFP construct without any UTRs (n = 3, p = 0.297, paired t test) shows no significant expression difference (72 h of siRNA treatment, 16 h coexpression of GFP-constructs with mcherry). Representative Western blot images are shown above; mcherry was used as transfection control. G, Overexpression of FMRP leads to a significant increase of GFP protein levels fused to both UTRs or the Kv4.2 3′-UTR (n = 7; GFP, p = 0.301; 5′, p = 0.303; 3′, *p = 0.023; 5′ + 3′, *p = 0 .033, paired t test). Representative Western blots are shown on the left. All error bars represent SEM.
Discussion
Apart from moderate to severe intellectual disability, fragile X patients can have several other symptoms of neuronal dysfunctions, such as hyperactivity, anxiety, self-injurious behavior and epilepsy. While strong evidence supports the hypothesis that loss of FMRP-mediated regulation of neuronal mRNA translation leads to impaired synaptic plasticity and intellectual disability, the mechanisms underlying other symptoms observed in FXS are poorly understood. Our study provides initial insight into a potential molecular mechanism leading to excess neuronal excitability in FXS, which might underlie hyperactivity, sensory hypersensitivity and seizures. Here, we demonstrate that FMRP regulates translation and protein expression of the A-type potassium channel Kv4.2. Functional deletion of Kv4.2 has been shown to cause temporal lobe epilepsy in humans (Singh et al., 2006), and to increase seizure susceptibility in a mouse model (Barnwell et al., 2009). Based on these previous findings and our own results we hypothesize that Kv4.2 dysregulation in Fmr1 KO mice might represent a mechanistic link between FXS and epilepsy. Furthermore, our observation that MPEP increases Kv4.2 membrane levels in Fmr1 KO provides evidence for an mGlu1/5- and FMRP-dependent regulation of surface expression of a potassium channel in dendrites, similar to what was reported for the AMPA receptor GluA1 (Nakamoto et al., 2007). In the future, it will be interesting to investigate whether this partial increase of Kv4.2 cell surface levels after MPEP-treatment represents a possible molecular mechanism underlying the MPEP-mediated rescue of audiogenic seizures in Fmr1 KO mice (Yan et al., 2005).
Apart from neuronal hyperexcitability and FXS-associated epilepsy, dysregulated Kv4.2 expression might contribute to other symptoms of FXS, e.g., self-injurious behavior. Studies in an FXS mouse model have shown that nociception is defective in the absence of FMRP (Price et al., 2007), and Kv4.2 has been demonstrated to play an important role for mGlu1/5-mediated nociception in the spinal cord (Hu et al., 2007), yet a link between FMRP and Kv4.2 has not been shown previously. Future studies will have to analyze whether dysregulated Kv4.2 expression in the spinal cord might contribute to occurrence of self-injuries in FXS patients.
Our results demonstrate that Kv4.2 mRNA is an unusual FMRP target mRNA as absence of FMRP led to reduced translation and protein expression of Kv4.2. The vast majority of reports finds FMRP to be a translational inhibitor (Zalfa et al., 2003; Dölen et al., 2007; Muddashetty et al., 2007; Gross et al., 2010; Strumbos et al., 2010), but at least two other mRNAs, superoxide dismutase 1(SOD1) and human achaete-scute homolog-1 (hAsh), have been suggested to be positively regulated by FMRP (Bechara et al., 2009; Fähling et al., 2009). Our results suggest that Kv4.2 mRNA regulation by FMRP is mediated via a cooperative effect of large parts of the Kv4.2 mRNA that include the 3′-UTR, and association with several FMRP RNA binding domains, suggesting a different mechanism than proposed for SOD1, which was shown to bind to FMRP via a 64 nt stem-loop structure. However, our data neither supports nor rules out a direct interaction of FMRP with Kv4.2 mRNA, and future studies will have to further analyze the modalities of FMRP association with and translational regulation of Kv4.2 mRNA.
Together, the results of our study add to the variety of functions FMRP can have on specific potassium channels in different brain regions: FMRP alters gating properties of Slack via direct protein interaction (Brown et al., 2010) and negatively regulates protein expression of Kv3.1b via association with its mRNA (Strumbos et al., 2010) in the brainstem, affecting the function of these potassium channels in the auditory system. In contrast, here we show that FMRP associates with and positively regulates the translation of Kv4.2 mRNA in the hippocampus and cortex. Our study contributes to the emerging picture of FMRP as a diverse regulator of neuronal potassium conductance, and encourages further studies to investigate potassium channel modulators as potential treatment for epilepsy and other symptoms in fragile X syndrome.
Footnotes
This work was supported by a postdoctoral fellowship and a Conquer Fragile X research grant from the National Fragile X Foundation (NFXF) (C.G.), a NFXF-Sponsored William & Enid Rosen Summer Student Fellowship and an Emory University Summer Undergraduate Research Experience fellowship (D.L.P.), National Institutes of Health Grant MH085617 (G.J.B.), and Fragile X Center Grant 3P30HD024064 (G.J.B.). We thank Dr. Lei Xing and Sharon Swanger for discussions and technical assistance, Drs. Kim M. Huber (University of Texas Southwestern Medical Center) and Justin R. Fallon (Brown University) for providing protocols for surface biotinylation and immunostainings of mouse brain sections, and the Bassell laboratory for discussions.
References
- Antar LN, Afroz R, Dictenberg JB, Carroll RC, Bassell GJ. Metabotropic glutamate receptor activation regulates fragile X mental retardation protein and FMR1 mRNA localization differentially in dendrites and at synapses. J Neurosci. 2004;24:2648–2655. doi: 10.1523/JNEUROSCI.0099-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnwell LF, Lugo JN, Lee WL, Willis SE, Gertz SJ, Hrachovy RA, Anderson AE. Kv4.2 knockout mice demonstrate increased susceptibility to convulsant stimulation. Epilepsia. 2009;50:1741–1751. doi: 10.1111/j.1528-1167.2009.02086.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassell GJ, Warren ST. Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron. 2008;60:201–214. doi: 10.1016/j.neuron.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechara EG, Didiot MC, Melko M, Davidovic L, Bensaid M, Martin P, Castets M, Pognonec P, Khandjian EW, Moine H, Bardoni B. A novel function for fragile X mental retardation protein in translational activation. PLoS Biol. 2009;7:e16. doi: 10.1371/journal.pbio.1000016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnbaum SG, Varga AW, Yuan LL, Anderson AE, Sweatt JD, Schrader LA. Structure and function of Kv4-family transient potassium channels. Physiol Rev. 2004;84:803–833. doi: 10.1152/physrev.00039.2003. [DOI] [PubMed] [Google Scholar]
- Brown MR, Kronengold J, Gazula VR, Chen Y, Strumbos JG, Sigworth FJ, Navaratnam D, Kaczmarek LK. Fragile X mental retardation protein controls gating of the sodium-activated potassium channel Slack. Nat Neurosci. 2010;13:819–821. doi: 10.1038/nn.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Yuan LL, Zhao C, Birnbaum SG, Frick A, Jung WE, Schwarz TL, Sweatt JD, Johnston D. Deletion of Kv4.2 gene eliminates dendritic A-type K+ current and enhances induction of long-term potentiation in hippocampal CA1 pyramidal neurons. J Neurosci. 2006;26:12143–12151. doi: 10.1523/JNEUROSCI.2667-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie SB, Akins MR, Schwob JE, Fallon JR. The FXG: a presynaptic fragile X granule expressed in a subset of developing brain circuits. J Neurosci. 2009;29:1514–1524. doi: 10.1523/JNEUROSCI.3937-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang SC, Zhao W, Bauchwitz R, Yan Q, Bianchi R, Wong RK. Prolonged epileptiform discharges induced by altered group I metabotropic glutamate receptor-mediated synaptic responses in hippocampal slices of a fragile X mouse model. J Neurosci. 2005;25:8048–8055. doi: 10.1523/JNEUROSCI.1777-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dölen G, Osterweil E, Rao BS, Smith GB, Auerbach BD, Chattarji S, Bear MF. Correction of fragile X syndrome in mice. Neuron. 2007;56:955–962. doi: 10.1016/j.neuron.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD. Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron. 2000;28:511–525. doi: 10.1016/s0896-6273(00)00129-x. [DOI] [PubMed] [Google Scholar]
- Fähling M, Mrowka R, Steege A, Kirschner KM, Benko E, Förstera B, Persson PB, Thiele BJ, Meier JC, Scholz H. Translational regulation of the human achaete-scute homologue-1 by fragile X mental retardation protein. J Biol Chem. 2009;284:4255–4266. doi: 10.1074/jbc.M807354200. [DOI] [PubMed] [Google Scholar]
- Francis J, Jugloff DG, Mingo NS, Wallace MC, Jones OT, Burnham WM, Eubanks JH. Kainic acid-induced generalized seizures alter the regional hippocampal expression of the rat Kv4.2 potassium channel gene. Neurosci Lett. 1997;232:91–94. doi: 10.1016/s0304-3940(97)00593-4. [DOI] [PubMed] [Google Scholar]
- Gabel LA, Won S, Kawai H, McKinney M, Tartakoff AM, Fallon JR. Visual experience regulates transient expression and dendritic localization of fragile X mental retardation protein. J Neurosci. 2004;24:10579–10583. doi: 10.1523/JNEUROSCI.2185-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross C, Nakamoto M, Yao X, Chan CB, Yim SY, Ye K, Warren ST, Bassell GJ. Excess phosphoinositide 3-kinase subunit synthesis and activity as a novel therapeutic target in fragile X syndrome. J Neurosci. 2010;30:10624–10638. doi: 10.1523/JNEUROSCI.0402-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman RJ, Berry-Kravis E, Kaufmann WE, Ono MY, Tartaglia N, Lachiewicz A, Kronk R, Delahunty C, Hessl D, Visootsak J, Picker J, Gane L, Tranfaglia M. Advances in the treatment of fragile X syndrome. Pediatrics. 2009;123:378–390. doi: 10.1542/peds.2008-0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu HJ, Alter BJ, Carrasquillo Y, Qiu CS, Gereau RW., 4th Metabotropic glutamate receptor 5 modulates nociceptive plasticity via extracellular signal-regulated kinase-Kv4.2 signaling in spinal cord dorsal horn neurons. J Neurosci. 2007;27:13181–13191. doi: 10.1523/JNEUROSCI.0269-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugo JN, Barnwell LF, Ren Y, Lee WL, Johnston LD, Kim R, Hrachovy RA, Sweatt JD, Anderson AE. Altered phosphorylation and localization of the A-type channel, Kv4.2 in status epilepticus. J Neurochem. 2008;106:1929–1940. doi: 10.1111/j.1471-4159.2008.05508.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muddashetty RS, Kelić S, Gross C, Xu M, Bassell GJ. Dysregulated metabotropic glutamate receptor-dependent translation of AMPA receptor and postsynaptic density-95 mRNAs at synapses in a mouse model of fragile X syndrome. J Neurosci. 2007;27:5338–5348. doi: 10.1523/JNEUROSCI.0937-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamoto M, Nalavadi V, Epstein MP, Narayanan U, Bassell GJ, Warren ST. Fragile X mental retardation protein deficiency leads to excessive mGluR5-dependent internalization of AMPA receptors. Proc Natl Acad Sci U S A. 2007;104:15537–15542. doi: 10.1073/pnas.0707484104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosyreva ED, Huber KM. Developmental switch in synaptic mechanisms of hippocampal metabotropic glutamate receptor-dependent long-term depression. J Neurosci. 2005;25:2992–3001. doi: 10.1523/JNEUROSCI.3652-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price TJ, Rashid MH, Millecamps M, Sanoja R, Entrena JM, Cervero F. Decreased nociceptive sensitization in mice lacking the fragile X mental retardation protein: role of mGluR1/5 and mTOR. J Neurosci. 2007;27:13958–13967. doi: 10.1523/JNEUROSCI.4383-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh B, Ogiwara I, Kaneda M, Tokonami N, Mazaki E, Baba K, Matsuda K, Inoue Y, Yamakawa K. A Kv4.2 truncation mutation in a patient with temporal lobe epilepsy. Neurobiol Dis. 2006;24:245–253. doi: 10.1016/j.nbd.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Strumbos JG, Brown MR, Kronengold J, Polley DB, Kaczmarek LK. Fragile X mental retardation protein is required for rapid experience-dependent regulation of the potassium channel Kv3.1b. J Neurosci. 2010;30:10263–10271. doi: 10.1523/JNEUROSCI.1125-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan QJ, Rammal M, Tranfaglia M, Bauchwitz RP. Suppression of two major fragile X syndrome mouse model phenotypes by the mGluR5 antagonist MPEP. Neuropharmacology. 2005;49:1053–1066. doi: 10.1016/j.neuropharm.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Zalfa F, Giorgi M, Primerano B, Moro A, Di Penta A, Reis S, Oostra B, Bagni C. The fragile X syndrome protein FMRP associates with BC1 RNA and regulates the translation of specific mRNAs at synapses. Cell. 2003;112:317–327. doi: 10.1016/s0092-8674(03)00079-5. [DOI] [PubMed] [Google Scholar]