

Abstract
Based on the promising drug resistance profile and potent anti-HIV activity of β-d-3′-azido-2′,3′-dideoxyguanosine, a series of purine modified nucleosides were synthesized by a chemical transglycosylation reaction and evaluated for their antiviral activity, cytotoxicity, and intracellular metabolism. Among the synthesized compounds, several show potent and selective anti-HIV activity in primary lymphocytes.
Keywords: HIV-1, Nucleoside, Antiviral, Transglycosylation
HIV-1 has spread worldwide and causes excessive morbidity and mortality in the absence of treatment. Nucleoside reverse transcriptase inhibitors (NRTIs) are the oldest and most successful class of agents used to treat HIV-1 infection. NRTIs continue to be the cornerstone of combination antiretroviral therapy (ART)1 that suppresses viremia and improves longevity and quality of life for persons infected with HIV-1. After long-term treatment, however, drug toxicity and the emergence of drug-resistant viral variants can limit the effectiveness of ART.2 Thus, the discovery of novel anti-HIV agents with low toxicity and favorable resistance profiles is necessary for continued success and further advances in ART. Structural variations in the pyrimidine and purine moiety of NRTI may impart different activity, resistance and toxicity profiles.3
3′-Azido-3′-deoxythymidine (AZT, Zidovudine) is a widely utilized anti-HIV drug despite its bone marrow toxicity which occurs in about 5% of the patients (Fig. 1)4 We recently showed that the corresponding purine derivative 3′-azido-2′,3′-dideoxyguanosine (AZG) with the same 3′-azido sugar5 was a potent and selective inhibitor of HIV-1 with a superior resistance profile compared to AZT6 This discovery led to the evaluation of the antiviral activity, resistance profile, intracellular metabolism and toxicity of a series of 3′-azido-2′,3′-dideoxypurine nucleosides. It should be noted that a previous study reported on the anti-HIV-1 activity of a limited number of purine 3′-azido-nucleosides in MT-4 cells.7 Our work differs from this study in that we synthesized a larger number of base modified analogs and correlated antiviral activity in human lymphocytes to cellular pharmacology and toxicity. In this regard, previous discovery efforts in this area have mostly neglected the importance of pharmacology and toxicity in NRTI development.
Figure 1.
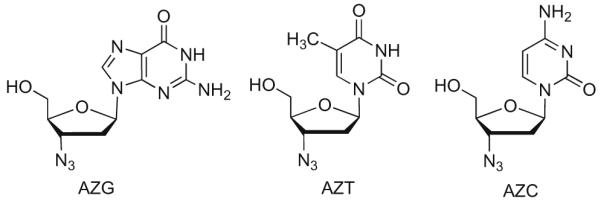
Some important 3′-azido nucleosides.
A previous study of 3′-azido-purine nucleosides found the chemical transglycosylation reaction to be a poor synthetic strategy resulting in α/β mixture of nucleosides in less than 25% yield.8 Herein, we report the development of a transglycosylation approach to efficiently prepare various 3′-azido-2′,3′-dideoxypurine nucleoside analogs. Further we evaluated the anti-HIV activity and cellular pharmacology in human peripheral blood mononuclear (PBM) cells and cytotoxicity in PBM, CEM (a human-T-cell-derived cell line), and Vero (kidney epithelial cells from the African green monkey) cells.
Initially, the 3′-azido purine analogs were prepared by a multistep traditional sugar modification method.9 Briefly, AZG was prepared from guanosine 1, by protection of the 2-amino group as isobutyl amide 2, followed by 5′-hydroxy protection to give benzoyl ester 3 (Scheme 1). Inversion of the 3′-hydroxyl group was found to be problematic due to benzoyl group migration10 to the 3′-hydroxy group and formation of N3-3′ cyclic adduct, but compound 4 could be obtained after a difficult chromatographic separation. Activation as the mesylate and displacement by azide anion gave protected 3′-azido nucleoside, 5, which was deprotected with sodium methoxide to give AZG.9,11
Scheme 1.

Reagents and conditions: (a) TMSCl, (i-BuCO)2O, pyridine, rt, 4 h, 75%; (b) Bz2O, DMF, Et3N, rt 2 h, 74%; (c) (i) Tf2O, DCM, pyridine, 0 °C, (ii) H2O, (iii) MeOH, NaHCO3; 32%; (d) (i) MsCl, DCM, DMAP, 0 °C, 40 min, (ii) NaN3, DMF, 120 °C, 2 h, 65%, two steps; (e) NaOCH3/MeOH, DCM 45 °C, 4 h, 82%.
After much optimization to simplify purifications and improve yields, AZG was obtained in a 9% yield over six steps from guanosine, 1. In order to prepare analogs which vary the substituent at the 6-position of the purine ring, we initially prepared the 6-chloro-3′-azido purine by treatment with phosphorous oxychloride, but found the reaction to be unreliable providing yields ranging from 8% to 32%.10
Conversion of the 6-oxo group of 5 to the tosylate 6 was accomplished in 99% yield and subsequent displacement by various nucleophiles occurred without incident (Scheme 2). Several 6-substituted 2-amino 3′-azido-2′,3′-dideoxy purines, 7, were synthesized in good yield by this approach from intermediate 6. An alternate 10-step nucleoside modification methodology to AZG was also explored, but was found to suffer from most of the same shortcomings described above.12 Thus, side reactions, difficult purifications, low overall yield, and the lengthiness of the synthesis of AZG led us to pursue a more efficient and concise methodology.
Scheme 2.

Reagents and conditions: (a) TosCl or TPPSCl, Et3N, DMAP, DCM, rt, 18 h 99%; 80%; (b) (i) R1H, (ii) NaOMe/MeOH, DCM 45 °C, 18 h, 55–73%.
The transglycosylation reaction is a two-step, one-pot transformation that involves the glycosyl transfer from a nucleoside to a heterocyclic base in an intermolecular fashion. It is most often utilized to transfer the sugar moiety of a more readily available pyrimidine nucleoside to the corresponding purine base. The chemical transglycosylation with AZT has been reported with adenosine13a and availability of AZT made this approach attractive. A later investigation of the chemical transglycosylation with AZT11b concluded that the resulting mixture of α and β anomers were obtained in less than 25% yield and the desired β anomer was difficult to obtain in high purity.
Our initial efforts, following literature precedent,13 involved presilylation of purine bases and AZT with bistrimethylsilyl acetamide (BSA), followed by TMSOTf-promoted transglycosylation that resulted in very low yields. 5′-Hydroxy protection with benzoyl chloride,14 silylation of purine bases such as 9 with bistrimethylsilyl acetamide, and transglycosylation in acetonitrile in the presence of TMSOTf under Vorbruggen type conditions at 70 °C for 12–24 h resulted in a much improved 30% yield of 1:1.1 mixture of β and α anomers as an inseparable mixture. After deprotection of the 5′-position, the anomers were readily separated by column chromatography on silica gel. However, the inability to differentiate the electrophilicity of the 5′-benzoyl removal versus 6-chloro displacement limited the usefulness of this approach.
A switch to TBS protection of the 5′-position of AZT gave 8 that greatly broadened the scope of the nucleophile tolerated for 6-chloro displacement (Scheme 3). Silylation with BSA and transglycosylation with 2-amino-6-chloropurine 9 gave the anomeric mixture of 10 and 11 that was inseparable by silica gel chromatography although obtained in slightly better 40% yield. Deprotection of the 5′-TBS group of nucleosides 10 and 11 provided a more easily separated β anomer 10, which was readily converted to nucleosides such as 2,6-diamino purine 12.15 Direct transglycosylation from TBS protected AZT with silylated purine bases was found to be problematic as trace AZT (with an EC50 = 5–50 nM vs HIV-1LAI in PBM cells) often led to false hits. However, this two-step approach involving two column purifications was useful for larger scale re-synthesis of interesting 3′-azido-purine nucleosides.
Scheme 3.

Reagents and condition: (a) (i) BSA, CH3CN, 78 °C, 30 min, then TMSOTf, 75 °C, 18 h, 43%, (ii) TBAF, THF, rt, 4 h; (b) (i) anomer separation on silica gel, 43%, (ii) NH3/CH3OH, 110 °C, 18 h, 71%.
We sought a readily available 3′-azido nucleoside with significantly reduced anti-HIV activity as a glycosyl transfer agent. As outlined in Scheme 4,5′-TBS protected AZU (AzddU) 13, was found to meet these requirements in that it was available in three steps based on a known procedure16 and it had a weaker potency against HIV in cell culture than AZT (AZU; EC50 = 0.2 μM; HIV-1LAI; human PBM cells).
Scheme 4.

Reagents and condition: (a) (i) TBSCl, imidazole, DMF, rt, overnight, 80%, (ii) DIAD, TPP, 80 °C, 30 min, 75%, (iii) LiN3, DMF, 100 °C, 24 h, 70%; (b) purine base, BSA, TMSOTf, CH3CN, 70 °C, 18 h, 34–61%; (c) (i) TBAF/acetic acid (v/v: 1:0.2), THF, rt, 4–6 h, (ii) anomer separation on silica gel, (iii) nucleophilic reagents in the case of 2- and/or 6-chloro purines in one to three steps in 20–95% yield.
Transglycosylation of the dU derived 13 proceeded smoothly to give a 1:1.1 ratio of the mixture of β and α anomers 14 and 15, again, as an inseparable mixture (Scheme 4). During the course of reaction, monitoring by LC/MS revealed not only the α and β anomers, but also other isomers of the same molecular weight. We observed this with both AZT and AZU under transglycosylation reactions. These isomers were tentatively assigned as N-7 and/or possibly N-3 attachment of the sugar unit to the purine base. Over the course of extended heating these minor isomers decomposed or were converted to the desired N-9 isomers. The use of 2-amino-6-chloropurine17 (43%) or 2,6-dichloropurine (41%) as the glycosyl acceptor in the transglycosylation allows for subsequent displacement of the chlorine atom(s) by appropriate nucleophile(s) and preparation of diversity at the 2 and 6-position.
Anti-HIV activity of the synthesized 3′-azido-purine nucleoside analogs was evaluated against HIV-1LAI in human PBM cells and cytotoxicity was determined in PBM, CEM, and Vero cells.18 The results are summarized in Table 1 along with the synthetic scheme utilized to prepare each analog. A significant number of the synthesized compounds showed good anti-HIV activity without enhancement of cytotoxicity compared to the natural purine bases analogs 3′-azido-2′,3′-dideoxyadenosine (AZA) and AZG.
Table 1.
Anti-HIV activity and cytotoxicity of selective synthesized compounds in cellular assay for compounds 10–40
| Compds | R1 | R2 | Anti-HIV activity (μM)18 | Cytotoxicity; IC50 (μM) | Synthesis | |||
|---|---|---|---|---|---|---|---|---|
| Based on 16 | EC50 | EC90 | PBM | CEM | Vero | Scheme | ||
| AZT | – | – | 0.005 | 0.02 | >100 | 14.3 | 50.6 | NA |
| AZU | – | – | 0.16 | 0.6 | >100 | 69.3 | 26 | NA |
| AZG | OH | NH2 | 0.18 | 2.0 | 100 | >100 | >100 | 1 |
| AZA | NH2 | H | 0.4 | 2.7 | 74.3 | 86.2 | >100 | 4 via 27 |
| 10 | Cl | NH2 | 0.1 | 0.8 | >100 | >100 | >100 | 3; 4 |
| 12 | NH2 | NH2 | 0.05 | 0.2 | 54.0 | >100 | >100 | 3 via 10 |
| 17 | OCH3 | NH2 | 0.57 | 1.4 | 32.1 | >100 | >100 | 2; 4 |
| 18 | N(CH3)2 | NH2 | 0.21 | 1.1 | >100 | >100 | >100 | 2 |
| 19 | NHc-pr | NH2 | 1.5 | 8.8 | >100 | >100 | >100 | 2; 4 via 10 |
| 20 | NHallyl | NH2 | 0.5 | 4.3 | >100 | >100 | >100 | 2; 4 via 10 |
| 21 | N(Me)allyl | NH2 | 0.4 | 1.5 | >100 | 27.0 | 32.0 | 2 |
| 22 | NHpropargyl | NH2 | 1.7 | 8.5 | >100 | >100 | >100 | 4 via 10 |
| 23 | NH(CH2)5OH | NH2 | 86.5 | >10 | >100 | >100 | >100 | 2; 4 via 10 |
| 24 | NHC(CH3)2CH2OH | NH2 | 52.5 | >10 | >100 | >100 | >100 | 2 |
| 25 | NHCH(CH3)CH2OH | NH2 | 59.9 | >10 | >100 | >100 | >100 | 2 |
| 26 | NH(CH2)2OH | NH2 | 2.1 | 16.2 | >100 | >100 | >100 | 2 |
| 27 | Cl | H | 0.4 | 1.3 | 23.3 | 19.9 | 67.9 | 4 |
| 28 | NHallyl | H | 0.3 | 1.5 | 51.1 | >100 | >100 | 2 |
| 29 | Cl | Cl | 9.3 | 26.3 | 4.2 | 2.1 | 27.1 | 4 |
| 30 | OCH3 | Cl | 55.3 | >100 | >100 | >100 | >100 | 3 |
| 31 | NH2 | Cl | 50.3 | >100 | 35.8 | >100 | >100 | 3 |
| 32 | NHCH3 | Cl | >100 | >100 | >100 | >100 | >100 | 3 |
| 33 | NHc-pr | Cl | 15.0 | 51.5 | 35.3 | >100 | >100 | 3 |
| 34 | N(CH3)2 | Cl | >100 | >100 | >100 | >100 | >100 | 3 |
| 35 | NH2 | F | 45.8 | 94.7 | 30.1 | 30.1 | 34.4 | 4 |
| 36 | OCH3 | F | >100 | >100 | 9.4 | >100 | >100 | 4 |
| 37 | Cl | F | 2.6 | 10.0 | 7.2 | 13.3 | 21.8 | 4 |
| 38 | NH2 | OCH3 | 12.4 | 39.6 | 35.0 | >100 | >100 | 3 via 31 |
| 39 | OCH3 | OCH3 | 13.2 | >100 | >100 | >100 | >100 | 3 via 30 |
| 40 | NHc-pr | OCH3 | 59.7 | >100 | >100 | >100 | >100 | 3 via 33 |
Compounds 10, 12, and 17–19 were previously studied in MT4 cells against HIV-1IIIB and were reported to have low μM anti-HIV activity with little or no toxicity toward the MT4 cells. We observed a significant increase in activity in PBM cells and some mild to moderate toxicity was also noted in PBM, CEM, and Vero cells. We made seven additional analogs by variation of the substituent at the 6-position while holding the 2-NH2 group constant. The 6-NH-allyl, 20, and 6-NCH3-allyl, 21, displayed good activity with an EC50 of 0.5 and 0.4 μM, however, the NCH3-allyl, 21 showed increased toxicity in both CEM and Vero cells. The related 6-NH-propargyl analog, 22 and four 6-alkyl hydroxy analogs 23–26 were significantly less active and displayed no toxicity.
Such broad activity across this series of compounds led us to investigate whether these 6-substituted analogs could be converted to 3′-azido guanosine (AZG) in the PBM cells used for the anti-HIV assay. To test this hypothesis, PBM cells were exposed to the nucleoside analog at 50 μM for 4 h at 37 °C in a 5% CO2 atmosphere. After washing the cells and extraction of metabolites the supernatant was dried and analyzed by LC/MS/MS. For each sample attempts to detect the nucleoside analog triphosphate (TP) along with the parent AZG-TP were made. 6-Cl (10), 6-NH2 (12), 6-OMe (17), 6-N(CH3)2 (18), 6-NHcyclopropyl (19), and 6-NH-allyl (20), were all found to be converted to AZG-TP under these conditions with such efficiency that we were unable to detect the nucleoside analog in the TP form except in the case of 12, where we detected low levels of 12-TP along with significant AZG-TP.
To determine if these 6-substituted compounds are converted to AZG by the enzyme adenosine deaminase, nucleoside analogs were subjected to 0.2 units per mL of commercially available purified enzyme for 120 min and measured spectrophotometrically at 265 or 285 nm.19 Nucleosides 10, 12, 17, 19, and 20 were found to be substantially or completely converted to AZG by adenosine deaminase. Compound 18, the 6-N,N-dimethyl analog was found to be resistant to adenosine deaminase under the conditions tested.
We next investigated analogs of AZA with variation of substituents at the 2 and 6-position of the purine base. The 6-Cl (27) and 6-NH-allyl (28) had a similar anti-HIV profile to AZA; however, 27 displayed more cytotoxicity. Nine AZA analogs, 29–37, were prepared with a halogen at the 2-position, but did not have any significant anti-HIV activity. Two of the 2-fluoro analogs, 35 and 37 and the 2,6-dichloro analog 29 showed an increase in cytotoxicity. The 2-methoxy analogs 38–40 showed the best overall anti-HIV activity of the 2-substituents tested to date in the AZA series with the 2,6-bis-methoxy analog 39, displaying an anti-HIV EC50 of 13.2 μM and no cytotoxicity up to 100 μM.
Many of the synthesized 3′-azido purine nucleosides showed potent anti-HIV activity with low or no toxicity. In addition, a number of the compounds proved to be acting as prodrugs for AZG in PBM cells. Any potential benefit of these various prodrugs to a future clinical candidate will be the subject of future studies. In this regard, the 2,6-diamino compound 12 deserves attention. This compound is between 4 times (EC50) and 10 times (EC90) more potent than AZG (see Table 1), but it is substantially converted to AZG by adenosine deaminase. Based on these promising results, further exploration of purine modifications on 3′-azido nucleosides and their effects on anti-HIV activity, cellular pharmacology, and cytotoxicity are underway.
Acknowledgments
This study was supported by grants from the National Institutes of Health (R01-AI-071846 to J. W. M and 5P30-AI-50409 and 5R37-AI-041980 to R. F. S.) and by the Department of Veterans Affairs (R. F. S.). R. F. S. is a founder and major shareholder of RFS Pharma, LLC. J. W. M. is a consultant to RFS Pharma and owns share options in RFS Pharma. All other authors have no competing interests.
References and notes
- 1.Kaufmann GR, Cooper DA. Curr. Opin. Microbiol. 2000;3:508. doi: 10.1016/s1369-5274(00)00131-4. [DOI] [PubMed] [Google Scholar]
- 2 (a).Lewis W. Antiviral Ther. 2005;10:M13. [PubMed] [Google Scholar]; (b) Pillay D, Taylor S, Richman DD. Rev. Med. Virol. 2000;10:231. doi: 10.1002/1099-1654(200007/08)10:4<231::aid-rmv290>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]; (c) Ruane PJ, DeJesus EJ. Acquir. Immun. Defic. Syndr. 2004;37:S21. doi: 10.1097/01.qai.0000137003.25258.76. [DOI] [PubMed] [Google Scholar]; (d) Palacios R, Santos J, Camino X, Arazo P, Perea RT, Echevarrfa S, Ribera E, de la Rosa RS, Guillen SM. Curr. Ther. Res. 2005;66:117. doi: 10.1016/j.curtheres.2005.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3 (a).For example see: Lee K, Choi Y, Gumina G, Zhou W, Schinazi RF, Chu CK. J. Med. Chem. 2002;45:1313. doi: 10.1021/jm010418n.; Schinazi RF, Lloyd RM, Jr., Nguyen MH, Cannon DL, McMillan A, Ilksoy N, Chu CK, Liotta DC, Bazmi HZ, Mellors JW. Antimicrob. Agents Chemother. 1993;37:875. doi: 10.1128/aac.37.4.875.; Feng JY, Shi J, Schinazi RF, Anderson KS. FASEB J. 1999;13:1511. doi: 10.1096/fasebj.13.12.1511.
- 4.Mitsuya H, Weinhold KJ, Furman PA, St. Clair MH, Lehrman SN, Gallo RC, Barry DW, Broker S. Proc. Natl. Acad. Sci. U.S.A. 1985;82:7096. doi: 10.1073/pnas.82.20.7096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pathak T. Chem. Rev. 2002;102:1623. doi: 10.1021/cr0104532. [DOI] [PubMed] [Google Scholar]
- 6.Sluis-Cremer N, Arion D, Parikh U, Koontz D, Schinazi RF, Mellors JW, Parniak MA. J. Biol. Chem. 1995;280:29047. doi: 10.1074/jbc.M503166200. [DOI] [PubMed] [Google Scholar]
- 7.Freeman GA, Shaver SR, Rideout JL, Short SA. Bioorg. Med. Chem. 1995;3:447. doi: 10.1016/0968-0896(95)00030-k. [DOI] [PubMed] [Google Scholar]
- 8.Robins MJ, Wood SG, Dalley NK, Herdewijn P, Balzarini J, De Clercq E. J. Med. Chem. 1989;32:1763. doi: 10.1021/jm00128a017. [DOI] [PubMed] [Google Scholar]
- 9 (a).Almond MR, Collins JL, Reitter BE, Rideout JL, Freeman GA, St. Clair MH. Tetrahedron Lett. 1991;32:5745. [Google Scholar]; (b) Herdewijn P, Balzarini J, Baba M, Pauwels R, Van Aerschot A, Janssen G, De Clercq E. J. Med. Chem. 1988;31:2040. doi: 10.1021/jm00118a033. [DOI] [PubMed] [Google Scholar]
- 10.Herdewijn PAM. J. Org. Chem. 1988;53:5050. [Google Scholar]
- 11 (a).Dyatkina NB, Kraevskii AA, Azhaev AV. Bioorg. Khim. 1986;12:1048. [Google Scholar]; (b) Eckstein F, Hunsmann G, Hartmann H. WO 8803804 A2. PCT Int. Appl. 1988:31. 19880602.; (c) Rideout JL, Averett DR, Freeman GA. EP 505181 A1. Eur. Pat. Appl. 1992:14. 19920923.; (d) Rideout JL, Freeman GA, Short SA, Almond MR, Collins JL. EP 421739 A1. Eur. Pat. Appl. 1991:45. 19910410.
- 12.Timoshchuk VA, Hogrefe RI, Vaghefi M. Nucleosides Nucleotides Nucleic Acids. 2004;23:171. doi: 10.1081/ncn-120027826. [DOI] [PubMed] [Google Scholar]
- 13 (a).Imazawa M, Eckstein F. J. Org. Chem. 1978;43:3044.; (b) An enzymatic transglycosylation approach from AZT is shown in Ref. 7.
- 14 (a).Mag M, Engels JW. Tetrahedron. 1994;50:10225. [Google Scholar]; (b) Wigerinck P, Van Aerschot A, Janssen G, Claes P, Balzarini J, De Clercq E, Herdewijn PAM. J. Med. Chem. 1990;33:868. doi: 10.1021/jm00164a063. [DOI] [PubMed] [Google Scholar]
- 15.Herdewijn P, Van Aerschot A. Tetrahedron Lett. 1989;30:855. [Google Scholar]
- 16 (a).Colla L, Herdewijn P, De Clercq E, Balzarini J, Vanderhaeghe H. Eur. J. Med. Chem. 1985;20:295. [Google Scholar]; (b) Moharram S, Zhou A, Wiebe LI, Knaus EE. J. Med. Chem. 2004;47:1840. doi: 10.1021/jm030544m. [DOI] [PubMed] [Google Scholar]
- 17.9-((2R/S,4S,5S)-4-Azido-5-((tert-butyldimethylsilyloxy)-methyl)tetrahydrofuran-2-yl)-6-chloro-9H-purin-2-amine, 14/15 (R1 = Cl; R2 = NH2): To a solution of 2-amino-6-chloropurine (250 mg, 1.47 mmol) and 5′-TBSAZU, 13 (200 mg, 0.54 mmol) in acetonitrile (6 mL) in oven dried glassware was added BSA (2 mL, 8.17 mmol). The resulting heterogeneous mixture was heated at 85 °C under positive nitrogen pressure for 30 min or until the reaction mixture became homogeneous. The reaction mixture was cooled to 0 °C, TMSOTf (0.51 mL, 2.79 mmol) was added, and the solution was heated at 85 °C under positive nitrogen pressure for 18 h. The reaction mixture was cooled to rt, poured into saturated NaHCO3 (50 mL), extracted with ethyl acetate (10 mL × 2), and the resulting combined organic layers were dried over sodium sulfate. The solvent was concentrated and the resulting residue was purified by silica gel chromatography with ethyl acetate/hexane (1:1) to give 100 mg (43%) of white solid as a 1.1:1 mixture of α:β anomers. LC/MS calcd for C16H26ClN8O2Si (M+1): 425.2; Found: 425.2. β-Isomer: 1H NMR (CDCl3, 400 MHz): δ 8.07 (s, 1H), 6.22 (t, J = 6.0 Hz, 1H), 5.08 (s, 2H), 4.39 (m, 1H), 4.02 (q, J = 3.6 Hz, 1H), 3.87 (dd, J = 3.2 Hz, J = 11.2 Hz, 1H), 3.79 (dd, J = 3.2 Hz, J = 10 Hz, 1H), 2.70 (m, 1H), 2.48 (m, 1H), 0.89 (s, 9H), 0.08 (s, 6H). α-Isomer: 1H NMR (CDCl3, 400 MHz): δ 8.05 (s, 1H), 6.24 (dd, J = 2.8 Hz, J = 7.2 Hz, 1H), 5.10 (s, 2H), 4.30 (m, 2H), 3.76 (dd, J = 3.2 Hz, J = 11.2 Hz, 1H), 3.71 (dd, J = 3.6 Hz, J = 11.6 Hz, 1H), 2.82 (m, 1H), 2.61 (dt, J = 2.8 Hz, J = 14.4 Hz, 1H), 0.90 (s, 9H), 0.08 (s, 6H).
- 18 (a).Schinazi RF, Sommadossi JP, Saalmann V, Cannon DL, Xie M-W, Hart GC, Smith GA, Hahn EF. Antimicrob. Agents Chemother. 1990;34:1061. doi: 10.1128/aac.34.6.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Stuyver LJ, Lostia S, Adams M, Mathew J, Pai BS, Grier J, Tharnish P, Choi Y, Chong Y, Choo H, Chu CK, Otto MJ, Schinazi RF. Antimicrob. Agents Chemother. 2002;46:3854. doi: 10.1128/AAC.46.12.3854-3860.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reaction conditions were 50 mM potassium phosphate, pH 7.4, with 50 μM nucleoside analog in 0.5 mL at 25 °C. Reaction time was 7 min with 0.002 units of enzyme and 120 min with 0.2 units of enzyme (the unit definition of adenosine deaminase is one unit will deaminate 1.0 μmol of adenosine to inosine per min at pH 7.5 at 25 °C). 2′-Deoxyadenosine was the positive control which was 59% deaminated to 2′-deoxyinosine under the given conditions in 7 min with 0.002 units of enzyme. 2′-Deoxyguanosine was the negative control.