

Abstract
Smoking is a significant health concern and strongly correlated with clinical depression. Depression is associated with decreased extracellular NE concentrations in brain. Smokers may be self-medicating and alleviating their depression through nicotine stimulated norepinephrine (NE) release. Several antidepressants inhibit NE transporter (NET) function, thereby augmenting extracellular NE concentrations. Antidepressants, such as bupropion, also inhibit nicotinic receptor (nAChR) function. The current study determined if a recently discovered novel nAChR antagonist, N,N′-dodecane-1,12-diyl-bis-3-picolinium dibromide (bPiDDB), inhibits nicotine-evoked NE release from superfused rat hippocampal slices. Previous studies determined that bPiDDB potently (IC50 = 2 nM) inhibits nicotine-evoked striatal [3H]dopamine (DA) release in vitro, nicotine-evoked DA release in nucleus accumbens in vivo, and nicotine self-administration in rats. In the current study, nicotine stimulated [3H]NE release from rat hippocampal slices (EC50 = 50 μM). bPiDDB inhibited (IC50 = 430 nM; Imax = 90%) [3H]NE release evoked by 30 μM nicotine. For comparison, the nonselective nAChR antagonist, mecamylamine, and the α7 antagonist, methyllycaconitine, also inhibited nicotine-evoked [3H]NE release (IC50 = 31 and 275 nM, respectively; Imax = 91% and 72%, respectively). Inhibition by bPiDDB and mecamylamine was not overcome by increasing nicotine concentrations; Schild regression slope was different from unity, consistent with allosteric inhibition. Thus, bPiDDB was 200-fold more potent inhibiting nAChRs mediating nicotine-evoked [3H]DA release from striatum than those mediating nicotine-evoked [3H]NE release from hippocampus.
Keywords: antidepressants, nicotine, nicotinic acetylcholine receptor, norepinephrine release, smoking cessation
1. Introduction
Tobacco dependence, the most preventable cause of death in the US, is described as a chronic, relapsing disorder in which compulsive drug seeking and taking persist despite negative consequences [1, 2]. Approximately 80% of those who attempt to quit smoking relapse within the first month, and only 3% remain abstinent 6 months after cessation [3]. Clinical studies reveal a strong correlation between incidence of tobacco smoking and mood disorders [3, 4]. Individuals with clinical depression are more likely to use tobacco, to be nicotine dependent and to have difficulty quitting, with greater withdrawal symptoms upon cessation [5-7]. Smokers undergoing cessation experience symptoms of depression, which occur more frequently among those with a history of major depression [8].
Astute observations that clinically depressed patients treated with bupropion as an antidepressant spontaneously reduced or quit tobacco use, led to a controlled clinical trial investigating the ability of bupropion to decrease smoking in non-depressed individuals [9]. The findings supported the introduction of bupropion as the first non-nicotine tobacco use cessation product, and provided rationale for the evaluation of other antidepressants as potential tobacco use cessation agents [10, 11]. Bupropion inhibits dopamine (DA) and norepinephrine (NE) transporters (DAT and NET, respectively) and inhibits nicotinic acetylcholine receptors (nAChRs) that mediate nicotine-evoked striatal [3H]DA and hippocampal [3H]NE release [12, 13]. Unfortunately, bupropion has limited efficacy as a tobacco use cessation agent and is associated with high relapse rates [11], revealing a need for more efficacious pharmacotherapies.
The antidepressant, nortriptyline, a relatively selective NET inhibitor, has been reported to increase smoking cessation rates [14, 15], providing evidence for the involvement of NE systems in nicotine addiction. Reboxetine, another antidepressant and NET inhibitor [16, 17], inhibits nAChRs mediating nicotine-evoked [3H]NE release from hippocampus, but not nAChRs mediating nicotine-evoked [3H]DA release from striatum [18]. While the effects of reboxetine on smoking cessation have not been reported, pre-clinical studies have demonstrated that reboxetine decreases nicotine self-administration in rats [19], providing evidence that NE is involved in nicotine reward. Thus, nAChRs mediating nicotine-evoked NE release may constitute an unexplored target for development of treatments for nicotine addiction.
The nonselective nAChR antagonist, mecamylamine, reverses both the positive and negative subjective effects of intravenous nicotine in smokers [20]. In a randomized, double-blind placebo-controlled study, mecamylamine combined with a nicotine transdermal patch improved smoking cessation outcome for up to 1 year compared to nicotine alone [2, 21, 22], providing further evidence for the use of nAChR antagonists as smoking cessation agents. However, due to the lack of selectivity at nAChRs and the inhibition of peripheral nAChRs, the clinical utility of mecamylamine as a smoking cessation agent is limited by anticholinergic side effects (e.g. constipation, hypotension).
Our laboratory has previously demonstrated that the novel nAChR antagonist, N,N′-dodecane-1,12-diyl-bis-3-picolinium dibromide (bPiDDB), potently (IC50 = 2 nM) inhibits nicotine-evoked striatal [3H]DA release in vitro [23] and nicotine-evoked accumbal DA release in vivo [24], and decreases intravenous nicotine self-administration in rats [25]. However, the effects of bPiDDB on nicotine-evoked NE release have yet to be determined. Thus, the current study sought to determine if bPiDDB inhibits nicotine-evoked hippocampal [3H]NE release in vitro and to elucidate the mechanism of inhibition.
2. Methods
2.1. Chemicals
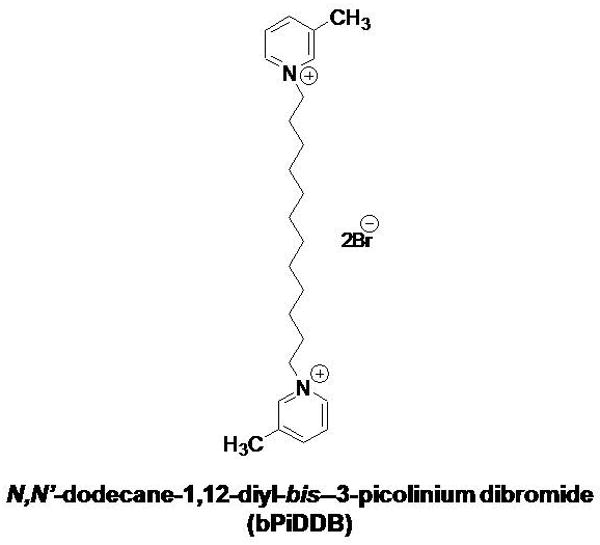
S(-)-Nicotine ditartrate, pargyline HCl, mecamylamine HCl, methyllycaconitine (MLA) and α-D-glucose were purchased from Sigma-Aldrich (St. Louis, MO). TS-2 Tissue solubilizer was purchased from Research Products International (Mount Prospect, IL). [3H]NE (specific activity, 14.0 Ci/mmol) and [3H]DA (3,4-ethyl-2-[N-3H]dihydroxyphenylethylamine; specific activity 28.0 Ci/mmol) were purchased from Perkin Elmer Life and Analytical Sciences (Boston, MA). All other chemicals were purchased from Fisher Scientific (Pittsburgh, PA). bPiDDB was synthesized as previously described [26] and the structure was verified by 1H- and 13C-NMR spectroscopy, mass spectrometry and X-ray crystallography. The chemical structure of bPiDDB is illustrated in Fig. 1.
Fig. 1. The chemical structure of N,N′-dodecane-1,12-diyl-bis-3-picolinium dibromide (bPiDDB).
2.2. Animals
Male Sprague-Dawley rats (200–225 g) were obtained from Harlan (Indianapolis, IN) and housed two per cage with ad libitum access to food and water in the Division of Laboratory Animal Resources (University of Kentucky, Lexington, KY). All experimental animal protocols were approved by the Institutional Animal Care and Use Committee at the University of Kentucky.
2.3. [3H]Neurotransmitter Overflow Assay
Hippocampal slices (500 μm, 3-5 mg) were incubated in Krebs' buffer (118 mM NaCl, 4.7 mM KCl, 1.2 mM MgCl2, 2.5 mM CaCl2, 1.0 mM NaH2PO4, 11.1 mM α–D-glucose, 25 mM NaHCO3, 0.11 mM L-ascorbic acid and 4.0 μM disodium ethylenediamine tetraacetate; pH 7.4, and saturated with 95% O2/5% CO2) in a metabolic shaker at 34°C for 30 min. Slices were incubated in fresh buffer (7-8 slices/3 ml) containing [3H]NE (0.1 μM, final concentration) for an additional 30 min. After rinsing in fresh buffer, each slice was transferred to 1 of 7 glass superfusion chambers maintained at 34°C and superfused (1 ml/min) with oxygenated Krebs' buffer containing the monoamine oxidase inhibitor pargyline (10 μM), to prevent metabolism of NE and to assure that the [3H] collected in superfusate primarily represents the parent neurotransmitter. For each experiment, slices were superfused initially for a 60-min period and then three 5-min samples (5 ml/sample) were collected to determine basal [3H]NE outflow. To determine the concentration- dependent effect of nicotine to evoke [3H]NE release, a series of experiments was conducted in which each hippocampal slice from an individual rat was superfused for 30 min in the absence (control) or presence of a single concentration of nicotine (1 – 300 μM), which remained in the buffer throughout the remainder of the experiment. Based on this concentration response for nicotine, a concentration of 30 μM was chosen for the subsequent inhibition studies.
In separate series of experiments, the concentration-dependent inhibition produced by mecamylamine, MLA and bPiDDB on nicotine-evoked [3H]NE overflow was determined. After collection of the third basal sample, each hippocampal slice from an individual rat was superfused in the absence (control) or presence of a single concentration of one of the three inhibitors, mecamylamine (1 nM – 1 μM), MLA (0.1 – 10 μM) or bPiDDB (1 nM – 10 μM), and superfusate samples were collected every 5 min for 30 min. Inhibitor remained in the buffer until the end of the experiment. Subsequently, nicotine (30 μM) was added to the buffer, and slices were superfused for 30 min and samples collected every 5 min. A control slice in each experiment was superfused for 30 min with buffer in the absence of inhibitor, followed by superfusion with nicotine (30 μM), to determine nicotine-evoked [3H]NE overflow. At the end of each experiment, slices were solubilized with TS-2 tissue solubilizer, and the [3H]-content of the tissue and samples was determined using liquid scintillation spectroscopy.
The concentration-dependent inhibition produced by MLA on nicotine-evoked [3H]DA overflow from coronal striatal slices (500 μm; 6–8 mg/slice, 7-8 slices/3 ml) was also determined using reported methods [27, 28]. Incubation buffer contained [3H]DA (0.1 μM, final concentration) and superfusion buffer contained pargyline (10 μM) and nomifensine (10 μM), a DA reuptake inhibitor. After 60 min of superfusion, three 5-min samples (5 ml/sample) were collected to determine basal [3H]DA outflow followed by superfusion in the absence (control) or presence of a single concentration of MLA (0.1 – 10 μM), which remained in the buffer until the end of the experiment. Then, nicotine (10 μM) was added to the buffer, and slices were superfused for 45 min and samples collected every 5 min, and [3H]-content of the tissue and samples was determined.
Schild analysis was used to evaluate the mechanism by which mecamylamine inhibited nicotine-evoked [3H]NE overflow. The same experimental design was used to determine the mechanism by which bPiDDB inhibits nicotine-evoked [3H]NE overflow. In each experiment, the concentration response for nicotine (1 – 300 μM) was determined in the absence and presence of a single concentration of inhibitor using hippocampal slices from a single rat. Inhibition of the effect of nicotine was determined at five concentrations of mecamylamine (0.001-10 μM) and three concentrations of bPiDDB (0.01-1.0 μM). After collection of three basal samples to determine outflow, slices were superfused in the absence or presence of a single concentration of inhibitor, which remained in the buffer throughout the experiment. Then, one of six concentrations of nicotine (1- 300 μM) was added to the buffer, and superfusion continued for an additional 30 min. Each hippocampal slice from an individual rat was exposed to only one concentration of nicotine and one concentration of inhibitor. Thus, nicotine concentration was a within-subjects factor, whereas inhibitor concentration was a between-groups factor.
2.4. Data Analysis
Fractional release for each superfusate sample was calculated by dividing the amount of tritium in each 5-min sample by the total tissue-[3H] at the time of sample collection. Basal [3H]outflow was calculated as the average fractional release in the three samples just before addition of inhibitor to the superfusion buffer. Total [3H]overflow was calculated by summing the increases in fractional release above basal [3H]outflow resulting from exposure to nicotine, either in the absence or presence of inhibitor and subtracting [3H]outflow for an equivalent period of inhibitor exposure. For analysis of inhibitor concentration response, data were fit by nonlinear least-squares regression using a variable slope, sigmoidal function. EC50, Emax, IC50 and Imax values were determined using Prism 5.0 (GraphPad Software Inc., San Diego, CA). Statistical analyses were conducted using SPSS (version 15.0; SPSS Inc., Chicago, IL). Two-way analysis of variance (ANOVA) was used to analyze the effect of the inhibitors on fractional [3H]NE release, with inhibitor concentration and time as within-subjects factors. One-way repeated-measures ANOVAs were used to analyze the concentration dependent effect of nicotine to evoke [3H]NE overflow and the concentration-dependent effect of each inhibitor on nicotine-evoked [3H]NE overflow. For the Schild analyses, nicotine concentration-response curves in the absence and presence of mecamylamine or bPiDDB were generated by fit of the data to a sigmoidal dose-response equation (variable slope): response = Bt + (Tp - Bt)/[1 + 10(log EC50 – X)n], where X is the logarithm of the nicotine concentration and n is the Hill slope. For each experiment, the dose ratio (dr) for each concentration of inhibitor was calculated as that producing an equivalent response in the absence and presence of inhibitor. The log of dr – 1 was plotted as a function of log inhibitor concentration to provide the Schild regression. These data were fit by linear regression, the slope determined, and linearity was assessed using Prism 5.0. Post hoc analyses were performed using Dunnett's test. Statistical significance was declared at p < 0.05.
3. Results
3.1. Nicotine evokes [3H]NE overflow from superfused rat hippocampal slices in a concentration-dependent manner
Nicotine increased fractional [3H]NE release from superfused rat hippocampal slices across a range of concentrations (1 – 300 μM). Analysis of fractional release by two-way repeated-measures ANOVA revealed main effects of nicotine concentration (F6, 120 = 57.6, p < 0.001) and time (F8, 160 = 287.1, p < 0.001), and a significant nicotine concentration × time interaction (F48, 960 = 59.9, p < 0.001; Fig. 2 top). The effect of nicotine to increase fractional release peaked within 5 min of the addition of nicotine to the superfusion buffer and returned to basal levels within 20 min, despite the continued presence of nicotine in the buffer. One-way ANOVA of nicotine-evoked [3H]NE overflow revealed a concentration-dependent effect (F6,133 = 61.7, p < 0.0001; Fig. 2, bottom). Post hoc analysis revealed that each concentration of nicotine increased [3H]NE overflow above the buffer control. Using nonlinear regression, a significant fit to a single-site model (R2 = 0.73, p < 0.05) was obtained for the nicotine concentration response curve. The EC50 for nicotine-evoked [3H]NE overflow was 50.1 ± 8.1 μM, with 100 μM and 300 μM nicotine producing a maximal effect.
Fig. 2. Time course and concentration dependence of nicotine-evoked fractional [3H]NE release (top) and nicotine-evoked [3H]NE overflow (bottom) from superfused rat hippocampal slices.

Hippocampal slices were superfused in the absence or presence of a single concentration (1-300 μM) of nicotine for 30 min. Arrow indicates the time point at which nicotine was added to the superfusion buffer. Each experiment included a buffer control condition in which one slice was superfused with buffer only and fractional [3H]NE release (top) and [3H]NE overflow (bottom) determined. Fractional release data are expressed as a percentage of basal (mean ± SEM), n = 19 rats. Basal fractional release was 0.44 ± 0.004 as percentage of tissue-[3H] content. Fractional release data were used to calculate [3H]NE overflow data, which are expressed as mean ± S.E.M. total [3H]NE overflow as a percentage of tissue-[3H] content. n = 19. The concentration-response curve for nicotine was generated using nonlinear regression. * indicates difference from buffer control, p < 0.05.
3.2. Nicotine-evoked [3H]NE overflow is inhibited by mecamylamine, MLA and bPiDDB
From the nicotine concentration response, a concentration of 30 μM was chosen to determine the ability of mecamylamine, MLA and bPiDDB to inhibit nicotine-evoked [3H]NE overflow from rat hippocampal slices. Mecamylamine and MLA inhibition of nicotine-evoked [3H]NE overflow were determined initially. Superfusion with either mecamylamine or MLA alone did not alter [3H]NE overflow (total evoked [3H]NE overflow was ≤0.04 % tissue-[3H] content for each concentration of inhibitor). The time course for mecamylamine-induced inhibition of nicotine-evoked fractional [3H]NE release is illustrated in Fig. 3 (top). Repeated measures two-way ANOVA revealed main effects of concentration (F6, 336 = 10.28, p < 0.01) and time (F8, 336 = 14.86, p < 0.0001), and a concentration × time interaction (F48, 336 = 6.33, p < 0.0001). The effect of mecamylamine to inhibit nicotine-evoked [3H]NE overflow was analyzed by one-way ANOVA, which revealed concentration-dependent inhibition (F6, 40 = 18.18, p < 0.0001; Fig. 3, bottom), and post hoc analysis revealed that mecamylamine inhibited nicotine-evoked [3H]NE overflow at concentrations ≥ 3 nM. Using nonlinear regression, a significant fit to a single-site model (R2 = 0.71, p < 0.05) was obtained for the mecamylamine concentration response curve. Mecamylamine had an IC50 value of 31.1 ± 11.4 nM and an Imax of 91 ± 2%. Thus, nicotine-evoked [3H]NE overflow was completely inhibited by mecamylamine.
Fig. 3. Time course and concentration dependence of mecamylamine inhibition of nicotine-evoked fractional [3H]NE release (top) and nicotine-evoked [3H]NE overflow (bottom) from superfused rat hippocampal slices.

Hippocampal slices were superfused in the absence or presence of a single concentration of mecamylamine (MEC; 1 nM – 1 μM) for 30 min, and then superfused for an additional 30 min with nicotine (30 μM) added to the buffer. Arrow indicates the time point at which nicotine was added to the superfusion buffer. Fractional release data are expressed as a percentage of basal (mean ± SEM), n = 12 rats. Basal fractional release was 0.46 ± 0.012 as percentage of tissue-[3H] content. Fractional release data were used to calculate [3H]NE overflow data, which are expressed as mean ± S.E.M. total [3H]NE overflow as a percentage of tissue-[3H] content. n = 12. Control [3H]NE overflow represents response to 30 μM nicotine in the absence of mecamylamine. The mecamylamine concentration-response curve was generated using nonlinear regression. * indicates difference from control, p < 0.05.
With respect to the time course of the MLA-induced inhibition of the effect of nicotine to stimulate fractional [3H]NE release (Fig. 4, top), repeated measures two-way ANOVA revealed main effects of concentration (F5, 240 = 2.90, p < 0.01) and time (F8, 240 = 115.65, p < 0.001), and a concentration × time interaction (F40, 240 = 7.35, p < 0.0001). The concentration-dependent effect of MLA to inhibit nicotine-evoked [3H]NE overflow was evaluated using one-way ANOVA, which revealed concentration-dependent inhibition (F5, 27 = 14.30, p < 0.0001; Fig. 4, bottom). MLA inhibited [3H]NE overflow at all concentrations employed. Using nonlinear regression, a significant fit to a single-site model (R2 = 0.70, p < 0.05) was obtained for the MLA concentration response curve. IC50 and Imax values of 275 ± 156 nM and 72 ± 4% were obtained. Thus, the effect of nicotine to stimulate [3H]NE overflow may involve α7 nAChRs.
Fig. 4. Time course and concentration dependence of MLA inhibition of nicotine-evoked fractional [3H]NE release (top) and nicotine-evoked [3H]NE overflow (bottom) from superfused rat hippocampal slices.

Hippocampal slices were superfused in the absence or presence of a single concentration of MLA (0.1–10 μM) for 30 min, and then superfused for an additional 30 min with nicotine (30 μM) added to the buffer. Arrow indicates the time point at which nicotine was added to the superfusion buffer. Fractional release data are expressed as a percentage of basal (mean ± SEM), n = 6 rats. Basal fractional release was 0.40 ± 0.006 as a percentage of tissue-[3H] content. Time course data for MLA-induced inhibition of nicotine-evoked fractional [3H]NE release were used to generate the [3H]NE overflow data, expressed as mean ± S.E.M. total [3H]NE overflow as a percentage of tissue-[3H] content. n = 6. Control [3H]NE overflow represents response to 30 μM nicotine in the absence of MLA. The MLA concentration-response curve was generated using nonlinear regression. * indicates difference from control, p < 0.05.
Similar to mecamylamine and MLA, bPiDDB alone did not evoke [3H]NE release (total evoked [3H]NE overflow was ≤0.04 % tissue-[3H] content for each bPiDDB concentration). The ability of bPiDDB to inhibit nicotine-evoked fractional [3H]NE release from superfused rat hippocampal slices was determined across a range of concentrations (1 nM – 10 μM; Fig. 5, top). Repeated measures two-way ANOVA revealed main effects of concentration (F5, 25 = 13.0, p < 0.001) and time (F8, 40 = 148.3, p < 0.001), and a significant bPiDDB concentration × time interaction (F40, 200 = 8.9, p < 0.001). One-way ANOVA revealed a concentration-dependent inhibition of nicotine-evoked [3H]NE overflow by bPiDDB (F5, 25 = 20.0, p < 0.001; Fig. 5, bottom). Post hoc analysis revealed that bPiDDB inhibited nicotine-evoked [3H]NE overflow at concentrations of 1 and 10 μM. Using nonlinear regression, a significant fit to a single-site model (R2 = 0.74, p < 0.05) was obtained for the bPiDDB concentration response curve. bPiDDB potently and completely inhibited nicotine-evoked [3H]NE overflow, with an IC50 of 430 ± 21 nM and an Imax of 90 ± 2%.
Fig. 5. Time course and concentration dependence of bPiDDB inhibition of nicotine-evoked fractional [3H]NE release (top) and nicotine-evoked [3H]NE overflow (bottom) from superfused rat hippocampal slices.

Hippocampal slices were superfused in the absence or presence of a single concentration of bPiDDB (1 nM – 10 μM) for 30 min, and then superfused for an additional 30 min with nicotine (30 μM) added to the buffer. Time course data for bPiDDB-induced inhibition of nicotine-evoked fractional [3H]NE release were used to generate [3H]NE overflow data. Arrow indicates the time point at which nicotine was added to the superfusion buffer. Fractional release data are expressed as a percentage of basal (mean ± SEM), n = 6 rats. Basal fractional release was 0.33 ± 0.006 as a percentage of tissue-[3H] content. [3H]NE overflow data are expressed as mean ± S.E.M. total [3H]NE overflow as a percentage of tissue-[3H] content. n = 6. Control [3H]NE overflow represents response to 30 μM nicotine in the absence of bPiDDB. The bPiDDB concentration-response curve was generated using nonlinear regression. * indicates difference from control, p < 0.05.
In addition, while the concentration response curves for nicotine, mecamylamine and MLA may indicate a trend towards biphasic curves in each of these series of experiments, an attempt to fit the data points using nonlinear regression would not converge, indicating a poor fit. This may represent a limitation of these experiments, in that 6-7 data points per concentration response curve may be too few to detect a significant fit to a two-site model. Nevertheless, the significant fit obtained when these data points were fit to a single-site model (R2 = 0.73, 0.71, 0.70, for nicotine, mecamylamine and MLA, respectively, p < 0.05) suggests that this is an appropriate model for these experiments. Thus, the effect of nicotine to stimulate [3H]NE overflow from hippocampal slices was inhibited completely by bPiDDB and mecamylamine, whereas MLA only attenuated the effect of nicotine.
3.3. MLA does not inhibit nicotine-evoked [3H]DA overflow from striatum
To provide a more comprehensive assessment of the inhibitory effects of bPiDDB, mecamylamine and MLA in our [3H]NE and [3H]DA release assay systems, we determined if MLA inhibits nicotine-evoked [3H]DA overflow from superfused rat striatal slices to assess the role of the α7 subtype in this response to nicotine. These experiments were undertaken considering the above observation that MLA partially and bPiDDB completely inhibited nicotine-evoked [3H]NE overflow, whereas we previously found that maximal inhibition by bPiDDB of nicotine-evoked [3H]DA overflow was incomplete [23]. A repeated measures two-way ANOVA on the data expressed as fractional [3H]DA release across time revealed that main effects of MLA concentration and time, and the MLA concentration × time interaction were not significant (p > 0.05; data not shown). One-way ANOVA of the [3H]DA overflow data also revealed no concentration-dependent inhibition produced by MLA (p > 0.05; Table 1). Thus, the lack of MLA-induced inhibition indicates α7* nAChRs are not involved in nicotine-evoked [3H]DA release from striatum and that nicotine evokes DA and NE release via different nAChR subtypes.
Table 1. MLA does not inhibit nicotine-evoked [3H]DA overflow from superfused rat striatal slices.
| Compound | Concentration (μM) | |||||
|---|---|---|---|---|---|---|
| Control | 0.1 | 0.3 | 1 | 3 | 10 | |
| MLA | 4.02 ± 0.64 | 5.70 ± 0.04 | 4.2 ± 0.95 | 5.1 ± 1.34 | 3.47 ± 0.74 | 4.81 ± 1.22 |
MLA (0.1 – 10 μM) did not inhibit (p > 0.05) nicotine (10 μM)-evoked [3H]DA overflow during the 45 min period of superfusion of striatal slices. Control represents response to 10 μM nicotine in the absence of MLA. Data are mean total [3H]DA overflow expressed as a percentage of tissue-[3H] content ± SEM. n = 5 rats.
3.4. Mecamylamine and bPiDDB inhibit nicotine-evoked [3H]NE overflow through an allosteric mechanism of action
Schild analysis of the inhibition of nicotine-evoked [3H]NE overflow was performed with the known allosteric inhibitor, mecamylamine (Fig. 6). Rightward and downward shifts in the nicotine concentration-response curves were evident with increasing concentrations of mecamylamine (1 nM – 10 μM). Moreover, mecamylamine-induced inhibition was not surmounted by increasing concentrations of nicotine, consistent with its generally accepted mechanism of action as an allosteric antagonist at nAChRs. The lowest concentration (1 nM) of mecamylamine did not inhibit nicotine-evoked [3H]NE overflow, whereas the three highest concentrations (0.1, 1 and 10 μM) all completely inhibited the effect of nicotine across its concentration response. As a result, the dose ratio for the Schild regression was not obtained.
Fig. 6. Schild analysis for mecamylamine inhibition of nicotine-evoked [3H]NE overflow from superfused rat hippocampal slices.

After collection of the third basal sample, slices were superfused with buffer in the absence and presence of mecamylamine (MEC; 1 nM-10 μM; between-groups factor) for 30 min before the addition of nicotine (1 – 300 μM; within subjects factor) to the buffer, and superfusion continued for an additional 30 min. Control is the concentration-response for nicotine in the absence of mecamylamine, and the nicotine concentration response was determined contemporaneously for each concentration of mecamylamine. Concentration-response curves were generated using nonlinear regression. Curves illustrated for the 0.1 and 1 μM mecamylamine are superimposed. Data are presented as mean ± S.E.M. total [3H]NE overflow during the 30-min exposure to nicotine in the absence or presence of mecamylamine; n = 4-5 rats/mecamylamine concentration; control, n = 16 rats.
Schild analysis was performed to determine the mechanism of action of bPiDDB inhibition of nicotine-evoked [3H]NE overflow. Rightward and downward shifts in the nicotine concentration-response curve were evident with increasing concentrations (0.01-1.0 μM) of bPiDDB (Fig. 7). The inhibition produced by bPiDDB was not surmounted by increasing concentrations of nicotine, consistent with allosteric inhibition. Further, a linear fit (r2 = 0.95) to the Schild-transformed data revealed a slope (0.63 ± 0.15) significantly different from unity (t10 = 13.27, p < 0.0001; Fig. 7, inset), also consistent with allosteric inhibition.
Fig. 7. Schild analysis for bPiDDB inhibition of nicotine-evoked [3H]NE overflow from superfused rat hippocampal slices.

After collection of the third basal sample, slices were superfused with buffer in the absence and presence of bPiDDB (0.01-1.0 μM; between-groups factor) for 30 min before the addition of nicotine (1 – 300 μM; within subjects factor) to the buffer, and superfusion continued for an additional 30 min. Control is the concentration-response for nicotine in the absence of bPiDDB, and the nicotine concentration response was determined contemporaneously for each concentration of bPiDDB. Concentration-response curves were generated using nonlinear regression. Data are presented as mean ± S.E.M. total [3H]NE overflow during the 30-min exposure to nicotine in the absence or presence of mecamylamine; n = 5-7 rats/bPiDDB concentration; control, n = 12 rats. Inset shows the Schild regression in which the log of dr – 1 was plotted as a function of log of bPiDDB concentration and data were fit by linear regression.
4. Discussion
Previous research from our laboratory has shown that the novel bis-azaaromatic quaternary ammonium analog, bPiDDB, inhibits nicotine-evoked [3H]DA release from rat striatal slices in vitro [23]; and following subcutaneous administration, bPiDDB inhibits nicotine-evoked accumbal DA release in vivo [24] and decreases intravenous nicotine self-administration in rats [25]. Schild analysis of bPiDDB inhibition of nicotine-evoked [3H]DA release revealed rightward shifts in the nicotine concentration-response curves that were surmounted by increasing concentrations of nicotine. A linear fit to the Schild-transformed data revealed a slope not different from unity, consistent with the hypothesis that bPiDDB inhibits nAChRs mediating nicotine-evoked [3H]DA overflow in an orthosteric manner [23]. The current results extend our previous work and demonstrate that bPiDDB inhibits (IC50 = 430 nM; Imax= 90%) nicotine-evoked [3H]NE release from superfused rat hippocampal slices. Inhibition of nicotine-evoked [3H]NE release by bPiDDB was not overcome by increasing nicotine concentrations; and Schild regression slope was different from unity, consistent with allosteric inhibition. Thus, bPiDDB was 200-fold more potent inhibiting nAChRs mediating nicotine-evoked [3H]DA release from striatum compared with nAChRs mediating nicotine-evoked [3H]NE release from hippocampus, and bPiDDB appears to inhibit these nAChR subtypes via different mechanisms.
Neuronal nAChRs are pentameric complexes and the presence of nine nAChR subunits (α2-α7, β2-β4) in mammalian brain and a diverse number of nAChR subtypes are expressed [29]. A recent molecular genetics study reported that six different nAChR subtypes mediate nicotine-evoked DA release, i.e., α4β2* and α4α5β2*, which are insensitive to the α6* nAChR-selective antagonist α-conotoxin MII (α-CtxMII), and α6β2*, α6β2β3*, α4α6β2* and α4α6β2β3*, which are α-CtxMII-sensitive [30]. Our previous results demonstrated that concomitant exposure to maximally effective concentrations of bPiDDB and α-CtxMII did not produce inhibition of nicotine-evoked [3H]DA release greater than that observed by either antagonist alone, suggesting that, like α-CtxMII, bPiDDB inhibits α6-containing nAChRs [23].
Nicotine (0.1-300 μM) evokes NE release from rat hippocampal [31-35] and cortical [36, 37] synaptosomes and slices. Nicotine-induced NE release also modulates DA function, and may contribute also to nicotine addiction through this indirect mechanism [38]. The concentration of nicotine (30 μM) used to evoke [3H]NE release from hippocampus in the current study is higher than the plasma nicotine concentrations observed in smokers (10 – 50 ng/ml; ∼0.1-0.5 μM, [39,40]). Plasma nicotine concentrations likely do not provide an accurate estimate of brain concentrations after nicotine exposure, since the nicotine metabolite cotinine is the predominant species appearing in plasma. Brain nicotine concentrations in smokers have not been determined; however, in animal models, peripheral administration of nicotine both intermittently (0.3 mg/kg/day for 10 days, sc) and continuously via osmotic minipump (0.8 mg/kg/day for 21 days) results in brain nicotine concentrations up to 8-fold higher in brain compared to blood [41]. The current research employed higher concentrations (1 – 300 μM) of nicotine than those found in smokers' plasma in order to detect a reproducible increase in NE release from the superfused rat hippocampal slice preparation. Importantly, mecamylamine completely inhibited (Imax= 91%) nicotine (30 μM)-evoked [3H]NE release, indicating mediation by nAChRs.
α3, α4, α5, α6, α7, β2, β3 and β4 subunit mRNAs have been localized to locus coeruleus neurons, providing potential subtype diversity in NE cell body and terminal regions [42-46]. Previous reports suggest that the β4 nAChR subunit is required for nicotine-evoked NE release from rat hippocampus [47]. α-Conotoxin AuIB, which blocks both α3β4* and α6β4* nAChRs [48], inhibited ∼50% of nicotine (100 μM)-evoked [3H]NE release from rat hippocampal synaptosomes, suggesting that these two populations of nAChRs contribute to nicotine-evoked NE release from hippocampus. However, α-CtxMII does not inhibit nicotine-evoked [3H]NE release from rat hippocampal synaptosomes [48,49], suggesting a lack of involvement of α6-containing nAChRs. Collectively, these findings suggest that α3β4* nAChRs mediate at least 50% of nicotine-evoked NE release from rat hippocampus. With respect to the current findings, bPiDDB inhibition of nicotine-evoked [3H]NE release from rat hippocampal slices suggests that bPiDDB inhibits α3β4* nAChRs in hippocampus in addition to α6-containing nAChRs in striatum. Given the high sequence identity shared by the α6 and α3 nAChR subunits [50], it is not surprising that bPiDDB also interacts with α3-containing nAChRs, particularly at higher concentrations.
With regard to the classical nAChR antagonists, mecamylamine potently inhibited (IC50 = 31 nM) nicotine-evoked [3H]NE release, and was an order of magnitude more potent than bPiDDB in this regard. Similar to bPiDDB, maximal inhibition produced by mecamylamine was complete (Imax = 91%). The classical α7 nAChR antagonist, MLA, also potently (IC50 = 275 nM) inhibited nicotine-evoked [3H]NE, but maximal inhibition was incomplete (Imax = 72%). The current results are consistent with a recent in vitro study which found that MLA (10 μM) inhibits nicotine (100 μM)-evoked [3H]NE from rat hippocampal synaptosomes [51], and with in vivo microdialysis studies showing that MLA (microdialysis probe concentrations of 0.4 – 32 nM) inhibits nicotine-evoked NE release from rat hippocampus [52]. α7* nAChRs are thought to mediate NE release from hippocampus through an indirect mechanism, i.e., α7*-mediated stimulation of glutamate release, which in turn promotes NE release [53]. Also, activation of α7 nAChRs on GABAergic neurons in hippocampus evokes GABA release, resulting in disinhibition of noradrenergic neurons [53,54]. Further, bPiDDB has been shown to inhibit both α3β4 and α7 nAChRs expressed in Xenopus oocytes, although with a ∼40-fold greater potency for α3β4 than for α7 nAChRs [55]. Given the localization of α7* nAChRs in rat hippocampus, and the observation that bPiDDB inhibits α7 nAChRs [55], a contributory role for α7* nAChRs in mediating bPiDDB inhibition of nicotine-evoked NE release must be given serious consideration.
The results of the current study show that MLA (0.1 – 10 μM) does not inhibit nicotine-evoked DA release from rat striatal slices, suggesting a lack of involvement of α7* nAChRs in mediating nicotine-evoked [3H]DA release in rat striatum. These results are in agreement with a previous in vitro study showing that MLA (0.1 pM – 10 nM) does not inhibit nicotine (10 μM)-evoked [3H]DA release from rat striatal or prefrontal cortical slices [56]. Thus, while α7* nAChRs may play an indirect role in mediating nicotine-evoked NE release from rat hippocampal slices, this receptor subtype does not appear to mediate nicotine-evoked DA release from rat striatal slices, supporting the hypothesis that different nAChR subtypes are responsible for mediating nicotine-evoked NE and DA release.
In contrast, others have reported that at concentrations > 40 nM, MLA inhibits nicotine-evoked [3H]DA release from rat striatal synaptosomes [57]. One explanation for these discrepant results is the difference in preparation, i.e., striatal slices in which the associated circuitry is intact and synaptosomes in which the circuitry is disrupted. Concurrent MLA and α-CtxMII inhibition of nicotine-evoked [3H]DA release from rat striatal synaptosomes was not additive, indicating that MLA also interacts with α3/α6* nAChRs and is not selective for α7 nAChRs. Thus, an alternative interpretation of the observed MLA inhibition of nicotine-evoked [3H]NE release from hippocampal slices is that MLA may be inhibiting α3β4* nAChRs to produce this response.
Regarding the differences in the interaction of bPiDDB with nAChR subtypes mediating DA and NE release, bPiDDB was 2-orders of magnitude more potent (IC50 = 2 nM) inhibiting nicotine-evoked [3H]DA release than nicotine-evoked [3H]NE release (IC50 = 430 nM), indicating that bPiDDB is 215-fold more selective for α6-containing nAChRs mediating nicotine [3H]DA release from striatum than for α3β4* nAChRs mediating nicotine-evoked [3H]NE release from hippocampus. Also, maximal bPiDDB-induced inhibition of nicotine-evoked [3H]DA release was incomplete (Imax = 64%), whereas bPiDDB completely inhibited nicotine-evoked [3H]NE release (Imax = 90%), indicating that more than one nAChR subtype mediates nicotine-evoked DA release, whereas nicotine-evoked NE release may be mediated via a single nAChR subtype. Finally, the mechanism by which bPiDDB inhibits these subtypes appears to be different. In the present study, Schild analysis of bPiDDB inhibition of nicotine-evoked [3H]NE release revealed rightward and downward shifts in the nicotine concentration-response curve that could not be surmounted with increasing concentrations of nicotine, and the slope of the Schild regression was different from unity, consistent with allosteric inhibition. As expected, mecamylamine, an allosteric and nonselective inhibitor of nAChRs also produced a rightward and downward shift in nicotine concentration-response curves that could not be overcome with increasing nicotine concentrations. Thus, bPiDDB inhibits nicotine-evoked [3H]NE release through an allosteric mechanism of action in contrast to the orthosteric inhibition produced by bPiDDB at nAChRs mediating nicotine-evoked DA release.
Importantly, the pharmacokinetics of bPiDDB after sc administration have been studied in detail in rats [58]. In spite of the fact that bPiDDB is a polar, cationic molecule, it has been demonstrated to enter brain from the periphery by active transport via the blood-brain barrier choline transporter and achieves behaviorally-relevant brain concentrations. An important factor to consider is that while plasma concentrations of bPiDDB in nicotine self-administering rats have not been examined, bPiDDB at a dose (3.0 mg/kg, sc) that decreases nicotine self-administration [25] results in a maximum plasma concentration of 0.33 μg/ml (640 nM [58]), which exceeds the IC50 values for both bPiDDB-induced inhibition of nicotine-evoked [3H]DA and [3H]NE release (2 and 430 nM, respectively; [23 and current findings]). However, as with nicotine, the brain concentration of bPiDDB, which is actively transported into the CNS by the blood-brain barrier choline transporter, is probably more relevant than the plasma bPiDDB concentration. Since bPiDDB decreases nicotine self-administration and has greater nAChR subtype selectivity than mecamylamine, it may prove to be more beneficial as a therapeutic for smoking cessation. Regardless, these results suggest that bPiDDB inhibition of nicotine self-administration may be mediated by inhibition of both nicotine-evoked DA and NE release.
In conclusion, inhibition of nicotine-evoked [3H]NE release by bPiDDB appears to be mediated by an allosteric mechanism at α3β4* nAChRs in rat hippocampus. These results extend previous research demonstrating that bPiDDB inhibits nicotine-evoked [3H]DA release from rat striatal slices through an orthosteric interaction with α6-containing nAChRs [23]. Taken together, bPiDDB is greater than 200-fold more selective for nAChRs mediating nicotine-evoked DA release than those mediating nicotine-evoked NE release, suggesting that bPiDDB is selective for α6-containing nAChRs. Thus, bPiDDB represents a novel small molecule that can be used as a pharmacologic tool to differentiate between those nAChR subtypes mediating nicotine-evoked DA and NE release, both of which likely play a role in nicotine reward. Importantly, nicotine self-administration in rats is decreased by peripherally administered bPiDDB [25]. Therefore, bPiDDB can be considered a lead compound in the search for subtype-selective nAChR antagonists as novel therapeutics for tobacco use cessation.
Acknowledgments
This research was supported by NIH grant U19 DA17548 and T32 DA007304.
The University of Kentucky holds patents on N,N′-dodecane-1,12-diyl-bis-3-picolinium dibromide. A potential royalty stream to L.P.D. and P.A.C. may occur consistent with University of Kentucky policy.
Nonstandard Abbreviations
- bPiDDB
N,N′-dodecane-1,12-diyl-bis-3-picolinium dibromide
- *
indicates putative nAChR subtype assignment.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Le Foll B, Goldberg SR. Effects of nicotine in experimental animals and humans: an Update on Addictive Properties. Handb Exp Pharmacol. 2009;192:335–67. doi: 10.1007/978-3-540-69248-5_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rose JE. Disrupting nicotine reinforcement: from cigarette to brain. Ann NY Acad Sci. 2008;1141:233–56. doi: 10.1196/annals.1441.019. [DOI] [PubMed] [Google Scholar]
- 3.Benowitz NL. Pharmacology of nicotine: addiction, smoking-induced disease, and therapeutics. Ann Rev Pharmacol Toxicol. 2009;49:57–71. doi: 10.1146/annurev.pharmtox.48.113006.094742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Glassman AH, Helzer JE, Covey LS, Cottler LB, Stetner F, Tipp JE, et al. Smoking, smoking cessation, and major depression. JAMA. 1990;264:1546–9. [PubMed] [Google Scholar]
- 5.Berlin I, Covey LS. Pre-cessation depressive mood predicts failure to quit smoking: the role of coping and personality traits. Addiction. 2006;101:1814–21. doi: 10.1111/j.1360-0443.2006.01616.x. [DOI] [PubMed] [Google Scholar]
- 6.Covey LS, Glassman AH, Jiang H, Fried J, Masmela J, LoDuca C, et al. A randomized trial of bupropion and/or nicotine gum as maintenance treatment for preventing smoking relapse. Addiction. 2007;102:1292–302. doi: 10.1111/j.1360-0443.2007.01887.x. [DOI] [PubMed] [Google Scholar]
- 7.Covey LS. Tobacco cessation among patients with depression. Prim Care. 1999;26:691–706. doi: 10.1016/s0095-4543(05)70124-x. [DOI] [PubMed] [Google Scholar]
- 8.Covey LS, Glassman AH, Stetner F. Major depression following smoking cessation. Am J Psychiatry. 1997;154:263–5. doi: 10.1176/ajp.154.2.263. [DOI] [PubMed] [Google Scholar]
- 9.Ferry LH, Burchette RJ. Bupropion for smoking cessation in non-depressed smokers. J Addict Dis. 1994;13:249. [Google Scholar]
- 10.Benowitz NL, Peng MW. Non-nicotine pharmacotherapy for smoking cessation. CNS Drugs. 2000;13:265–85. [Google Scholar]
- 11.Hughes JR, Stead LF, Lancaster T. Antidepressants for smoking cessation. Cochrane Database Syst Rev. 2007;(1) doi: 10.1002/14651858.CD000031.pub3. [DOI] [PubMed] [Google Scholar]
- 12.Miller DK, Sumithran SP, Dwoskin LP. Bupropion inhibits nicotine-evoked [(3)H]overflow from rat striatal slices preloaded with [3H]dopamine and from rat hippocampal slices preloaded with [3H]norepinephrine. J Pharmacol Exp Ther. 2002;302:1113–22. doi: 10.1124/jpet.102.033852. [DOI] [PubMed] [Google Scholar]
- 13.Sidhpura N, Redfern P, Wonnacott S. Comparison of the effects of bupropion on nicotinic receptor-evoked [(3)H]dopamine release from rat striatal synaptosomes and slices. Eur J Pharmacol. 2007;567:102–9. doi: 10.1016/j.ejphar.2007.03.052. [DOI] [PubMed] [Google Scholar]
- 14.da Costa CL, Younes RN, Lourenco MT. Stopping smoking: a prospective, randomized, double-blind study comparing nortriptyline to placebo. Chest. 2002;122:403–8. doi: 10.1378/chest.122.2.403. [DOI] [PubMed] [Google Scholar]
- 15.Hughes JR, Stead LF, Lancaster T. Nortriptyline for smoking cessation: a review. Nicotine Tob Res. 2005;7:491–9. doi: 10.1080/14622200500185298. [DOI] [PubMed] [Google Scholar]
- 16.Montgomery SA. Is there a role for a pure noradrenergic drug in the treatment of depression? Eur Neuropsychopharmacol. 1997;7 Suppl 1:S3–9. doi: 10.1016/s0924-977x(97)00414-8. discussion S71-3. [DOI] [PubMed] [Google Scholar]
- 17.Wong EH, Sonders MS, Amara SG, Tinholt PM, Piercey MF, Hoffmann WP, et al. Reboxetine: a pharmacologically potent, selective, and specific norepinephrine reuptake inhibitor. Biol Psychiatry. 2000;47:818–29. doi: 10.1016/s0006-3223(99)00291-7. [DOI] [PubMed] [Google Scholar]
- 18.Miller DK, Wong EH, Chesnut MD, Dwoskin LP. Reboxetine: functional inhibition of monoamine transporters and nicotinic acetylcholine receptors. J Pharmacol Exp Ther. 2002;302:687–95. doi: 10.1124/jpet.302.2.687. [DOI] [PubMed] [Google Scholar]
- 19.Rauhut AS, Mullins SN, Dwoskin LP, Bardo MT. Reboxetine: attenuation of intravenous nicotine self-administration in rats. J Pharmacol Exp Ther. 2002;303:664–72. doi: 10.1124/jpet.303.2.664. [DOI] [PubMed] [Google Scholar]
- 20.Lundahl LH, Henningfield JE, Lukas SE. Mecamylamine blockade of both positive and negative effects of IV nicotine in human volunteers. Pharmacol Biochem Behav. 2000;66:637–43. doi: 10.1016/s0091-3057(00)00252-5. [DOI] [PubMed] [Google Scholar]
- 21.Rose JE. Nicotine and nonnicotine factors in cigarette addiction. Psychopharmacology. 2006;184:274–85. doi: 10.1007/s00213-005-0250-x. [DOI] [PubMed] [Google Scholar]
- 22.Rose JE, Behm FM, Westman EC, Levin ED, Stein RM, Ripka GV. Mecamylamine combined with nicotine skin patch facilitates smoking cessation beyond nicotine patch treatment alone. Clin Pharmacol Ther. 1994;56:86–99. doi: 10.1038/clpt.1994.105. [DOI] [PubMed] [Google Scholar]
- 23.Dwoskin LP, Wooters TE, Sumithran SP, Siripurapu KB, Joyce BM, Lockman PR, et al. N,N′-Alkane-diyl-bis-3-picoliniums as nicotinic receptor antagonists: inhibition of nicotine-induced dopamine release and hyperactivity. J Pharmacol Exp Ther. 2008;326:563–76. doi: 10.1124/jpet.108.136630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rahman S, Neugebauer NM, Zhang Z, Crooks PA, Dwoskin LP, Bardo MT. The effects of a novel nicotinic receptor antagonist N,N-dodecane-1,12-diyl-bis-3-picolinium dibromide (bPiDDB) on acute and repeated nicotine-induced increases in extracellular dopamine in rat nucleus accumbens. Neuropharmacology. 2007;52:755–63. doi: 10.1016/j.neuropharm.2006.09.012. [DOI] [PubMed] [Google Scholar]
- 25.Neugebauer NM, Zhang Z, Crooks PA, Dwoskin LP, Bardo MT. Effect of a novel nicotinic receptor antagonist, N,N′-dodecane-1,12-diyl-bis-3-picolinium dibromide, on nicotine self-administration and hyperactivity in rats. Psychopharmacology. 2006;184:426–34. doi: 10.1007/s00213-005-0163-8. [DOI] [PubMed] [Google Scholar]
- 26.Dwoskin LP, Sumithran SP, Zhu J, Deaciuc AG, Ayers JT, Crooks PA. Subtype-selective nicotinic receptor antagonists: potential as tobacco use cessation agents. Bioorg Med Chem Lett. 2004;14:1863–7. doi: 10.1016/j.bmcl.2003.10.073. [DOI] [PubMed] [Google Scholar]
- 27.Grinevich VP, Crooks PA, Sumithran SP, Haubner AJ, Ayers JT, Dwoskin LP. N-n-alkylpyridinium analogs, a novel class of nicotinic receptor antagonists: selective inhibition of nicotine-evoked [3H] dopamine overflow from superfused rat striatal slices. J Pharmacol Exp Ther. 2003;306:1011–20. doi: 10.1124/jpet.103.051789. [DOI] [PubMed] [Google Scholar]
- 28.Wilkins LH, Jr, Haubner A, Ayers JT, Crooks PA, Dwoskin LP. N,N′-alkylnicotinium analogs, a novel class of nicotinic receptor antagonist: inhibition of S(-)-nicotine-evoked [3H]dopamine overflow from superfused rat striatal slices. J Pharmacol Exp Ther. 2002;301:1088–96. doi: 10.1124/jpet.301.3.1088. [DOI] [PubMed] [Google Scholar]
- 29.Dani JA, Bertrand D. Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Ann Rev Pharmacol Toxicol. 2007;47:699–729. doi: 10.1146/annurev.pharmtox.47.120505.105214. [DOI] [PubMed] [Google Scholar]
- 30.Salminen O, Murphy KL, McIntosh JM, Drago J, Marks MJ, Collins AC, et al. Subunit composition and pharmacology of two classes of striatal presynaptic nicotinic acetylcholine receptors mediating dopamine release in mice. Mol Pharmacol. 2004;65:1526–35. doi: 10.1124/mol.65.6.1526. [DOI] [PubMed] [Google Scholar]
- 31.Amtage F, Neughebauer B, McIntosh JM, Freiman T, Zentner J, Feuerstein TJ, et al. Characterization of nicotinic receptors inducing noradrenaline release and absence of nicotinic autoreceptors in human neocortex. Brain Res Bull. 2004;62:413–23. doi: 10.1016/j.brainresbull.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 32.Clarke PB, Reuben M. Release of [3H]-noradrenaline from rat hippocampal synaptosomes by nicotine: mediation by different nicotinic receptor subtypes from striatal [3H]-dopamine release. Br J Pharmacol. 1996;117:595–606. doi: 10.1111/j.1476-5381.1996.tb15232.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leslie FM, Gallardo KA, Park MK. Nicotinic acetylcholine receptor-mediated release of [3H]norepinephrine from developing and adult rat hippocampus: direct and indirect mechanisms. Neuropharmacology. 2002;42:653–61. doi: 10.1016/s0028-3908(02)00019-9. [DOI] [PubMed] [Google Scholar]
- 34.Sacaan AI, Dunlop JL, Lloyd GK. Pharmacological characterization of neuronal acetylcholine gated ion channel receptor-mediated hippocampal norepinephrine and striatal dopamine release from rat brain slices. J Pharmacol Exp Ther. 1995;274:224–30. [PubMed] [Google Scholar]
- 35.Sershen H, Balla A, Lajtha A, Vizi ES. Characterization of nicotinic receptors involved in the release of noradrenaline from the hippocampus. Neuroscience. 1997;77:121–30. doi: 10.1016/s0306-4522(96)00425-3. [DOI] [PubMed] [Google Scholar]
- 36.Balfour DJ, Fagerstrom KO. Pharmacology of nicotine and its therapeutic use in smoking cessation and neurodegenerative disorders. Pharmacol Ther. 1996;72:51–81. doi: 10.1016/s0163-7258(96)00099-x. [DOI] [PubMed] [Google Scholar]
- 37.Summers KL, Giacobini E. Effects of local and repeated systemic administration of (-)nicotine on extracellular levels of acetylcholine, norepinephrine, dopamine, and serotonin in rat cortex. Neurochem Res. 1995;20:753–9. doi: 10.1007/BF01705545. [DOI] [PubMed] [Google Scholar]
- 38.Linner L, Endersz H, Ohman D, Bengtsson F, Schalling M, Svensson TH. Reboxetine modulates the firing pattern of dopamine cells in the ventral tegmental area and selectively increases dopamine availability in the prefrontal cortex. J Pharmacol Exp Ther. 2001;297:540–6. [PubMed] [Google Scholar]
- 39.Benowitz NL, Jacob P., 3rd Daily intake of nicotine during cigarette smoking. Clin Pharmacol Ther. 1984;35:499–504. doi: 10.1038/clpt.1984.67. [DOI] [PubMed] [Google Scholar]
- 40.Matta SG, Balfour DJ, Benowitz NL, Boyd RT, Buccafusco JJ, Caggiula AR, et al. Guidelines on nicotine dose selection for in vivo research. Psychopharmacology. 2007;190:269–319. doi: 10.1007/s00213-006-0441-0. [DOI] [PubMed] [Google Scholar]
- 41.Ghosheh OA, Dwoskin LP, Miller DK, Crooks PA. Accumulation of nicotine and its metabolites in rat brain after intermittent or continuous peripheral administration of [2′-(14)C]nicotine. Drug metabolism and disposition: the biological fate of chemicals. 2001;29:645–51. [PubMed] [Google Scholar]
- 42.Dineley-Miller K, Patrick J. Gene transcripts for the nicotinic acetylcholine receptor subunit, beta4, are distributed in multiple areas of the rat central nervous system. Brain Res Mol Brain Res. 1992;16:339–44. doi: 10.1016/0169-328x(92)90244-6. [DOI] [PubMed] [Google Scholar]
- 43.Le Novere N, Zoli M, Changeux JP. Neuronal nicotinic receptor alpha 6 subunit mRNA is selectively concentrated in catecholaminergic nuclei of the rat brain. Eur J Neurosci. 1996;8:2428–39. doi: 10.1111/j.1460-9568.1996.tb01206.x. [DOI] [PubMed] [Google Scholar]
- 44.Lena C, de Kerchove D'Exaerde A, Cordero-Erausquin M, Le Novere N, del Mar Arroyo-Jimenez M, Changeux JP. Diversity and distribution of nicotinic acetylcholine receptors in the locus ceruleus neurons. Proc Natl Acad Sci U S A. 1999;96:12126–31. doi: 10.1073/pnas.96.21.12126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vincler MA, Eisenach JC. Immunocytochemical localization of the alpha3, alpha4, alpha5, alpha7, beta2, beta3 and beta4 nicotinic acetylcholine receptor subunits in the locus coeruleus of the rat. Brain Res. 2003;974:25–36. doi: 10.1016/s0006-8993(03)02546-0. [DOI] [PubMed] [Google Scholar]
- 46.Wada E, Wada K, Boulter J, Deneris E, Heinemann S, Patrick J, et al. Distribution of alpha 2, alpha 3, alpha 4, and beta 2 neuronal nicotinic receptor subunit mRNAs in the central nervous system: a hybridization histochemical study in the rat. J Comp Neurol. 1989;284:314–35. doi: 10.1002/cne.902840212. [DOI] [PubMed] [Google Scholar]
- 47.Azam L, McIntosh JM. Characterization of nicotinic acetylcholine receptors that modulate nicotine-evoked [3H]norepinephrine release from mouse hippocampal synaptosomes. Mol Pharmacol. 2006;70:967–76. doi: 10.1124/mol.106.024513. [DOI] [PubMed] [Google Scholar]
- 48.Luo S, Kulak JM, Cartier GE, Jacobsen RB, Yoshikami D, Olivera BM, et al. alpha-conotoxin AuIB selectively blocks alpha3 beta4 nicotinic acetylcholine receptors and nicotine-evoked norepinephrine release. J Neurosci. 1998;18:8571–9. doi: 10.1523/JNEUROSCI.18-21-08571.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kulak JM, Nguyen TA, Olivera BM, McIntosh JM. Alpha-conotoxin MII blocks nicotine-stimulated dopamine release in rat striatal synaptosomes. J Neurosci. 1997;17:5263–70. doi: 10.1523/JNEUROSCI.17-14-05263.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Le Novere N, Changeux JP. Molecular evolution of the nicotinic acetylcholine receptor: an example of multigene family in excitable cells. J Mol Evol. 1995;40:155–72. doi: 10.1007/BF00167110. [DOI] [PubMed] [Google Scholar]
- 51.Westphalen RI, Gomez RS, Hemmings HC., Jr Nicotinic receptor-evoked hippocampal norepinephrine release is highly sensitive to inhibition by isoflurane. Br J Anaesth. 2009;102:355–60. doi: 10.1093/bja/aen387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fu Y, Matta SG, James TJ, Sharp BM. Nicotine-induced norepinephrine release in the rat amygdala and hippocampus is mediated through brainstem nicotinic cholinergic receptors. J Pharmacol Exp Ther. 1998;284:1188–96. [PubMed] [Google Scholar]
- 53.Barik J, Wonnacott S. Indirect modulation by alpha7 nicotinic acetylcholine receptors of noradrenaline release in rat hippocampal slices: interaction with glutamate and GABA systems and effect of nicotine withdrawal. Mol Pharmacol. 2006;69:618–28. doi: 10.1124/mol.105.018184. [DOI] [PubMed] [Google Scholar]
- 54.Ji D, Dani JA. Inhibition and disinhibition of pyramidal neurons by activation of nicotinic receptors on hippocampal interneurons. J Neurophysiol. 2000;83:2682–90. doi: 10.1152/jn.2000.83.5.2682. [DOI] [PubMed] [Google Scholar]
- 55.Rahman S, Zhang Z, Papke RL, Crooks PA, Dwoskin LP, Bardo MT. Region-specific effects of N,N′-dodecane-1,12-diyl-bis-3-picolinium dibromide on nicotine-induced increase in extracellular dopamine in vivo. Br J Pharmacol. 2008;153:792–804. doi: 10.1038/sj.bjp.0707612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cao YJ, Surowy CS, Puttfarcken PS. Different nicotinic acetylcholine receptor subtypes mediating striatal and prefrontal cortical [3H]dopamine release. Neuropharmacology. 2005;48:72–9. doi: 10.1016/j.neuropharm.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 57.Mogg AJ, Whiteaker P, McIntosh JM, Marks M, Collins AC, Wonnacott S. Methyllycaconitine is a potent antagonist of alpha-conotoxin-MII-sensitive presynaptic nicotinic acetylcholine receptors in rat striatum. J Pharmacol Exp Ther. 2002;302:197–204. doi: 10.1124/jpet.302.1.197. [DOI] [PubMed] [Google Scholar]
- 58.Albayati ZA, Dwoskin LP, Crooks PA. Pharmacokinetics of the novel nicotinic receptor antagonist N,N′-dodecane-1,12-diyl-bis-3-picolinium dibromide in the rat. Drug Metab Dispos. 2008;36:2024–9. doi: 10.1124/dmd.108.020354. [DOI] [PMC free article] [PubMed] [Google Scholar]