

Abstract
Tau is the major microtubule associated protein (MAP) of a mature neuron. The other two neuronal MAPs are MAP1 and MAP2. An established function of MAPs is their interaction with tubulin and promotion of its assembly into microtubules and stabilization of the microtubule network. The microtubule assembly promoting activity of tau, a phosphoprotein, is regulated by its degree of phosphorylation. Normal adult human brain tau contains 2–3 moles phosphate/mole of tau protein. Hyperphosphorylation of tau depresses this biological activity of tau. In Alzheimer disease (AD) brain tau is ∼three to four-fold more hyperphosphorylated than the normal adult brain tau and in this hyperphosphorylated state it is polymerized into paired helical filaments ([PHF) admixed with straight filaments (SF) forming neurofibrillary tangles. Tau is transiently hyperphosphorylated during development and during anesthesia and hypothermia but not to the same state as in AD brain. The abnormally hyperphosphorylated tau in AD brain is distinguished from transiently hyperphosphorylated tau by its ability (1) to sequester normal tau, MAP1 and MAP2 and disrupt microtubules, and (2) to self-assemble into PHF/SF. The cytosolic abnormally hyperphosphorylated tau, because of oligomerization, unlike normal tau, is sedimentable and on self-assembly into PHF/SF, loses its ability to sequester normal MAPs. Some of the tau in AD brain is truncated which also promotes its self-assembly. Tau mutations found in frontotemporal dementia apparently promote its abnormal hyperphosphorylation. Thus, the AD abnormally hyperphosphorylated tau (1) is distinguishable from both normal and transiently hyperphosphorylated taus, and (2) is inhibitory when in a cytosolic/oligomeric state but not when it is self-assembled into PHF/SF. Inhibition of abnormal hyperphosphorylation of tau offers a promising therapeutic target for AD and related tauopathies.
Keywords: Microtubule associated proteins, hyperphosphorylation of tau, microtubule assembly, neurofibrillary tangles, paired helical filaments, tau truncation
Introduction
Tau is the major microtubule associated protein (MAP) of a normal mature neuron. The other two neuronal MAPs are MAP1 and MAP2. Tau is found as six molecular isoforms in human brain [1]. These isoforms are coded by a single gene on chromosome 17 and are generated by alternative splicing of its pre-mRNA [2]. To date, the only established function of tau, a phosphoprotein, is the promotion of the assembly of tubulin into microtubules and stabilization of their structure [3].
In Alzheimer disease (AD) and a family of related neurodegenerative diseases, called tauopathies, tau protein is abnormally hyperphosphorylated and aggregated into bundles of filaments [4]. In AD brain this tau pathology is seen as intraneuronal neurofibrillary tangles of paired helical filaments (PHF) sometimes admixed with straight filaments (SF). Aggregates of abnormally hyperphosphorylated filaments are also seen in dystrophic neurites surrounding the β-amyloid plaque core, and in the neuropil as neuropil threads [5]. Neurofibrillary degeneration of abnormally hyperphosphorylated tau is apparently required for the clinical expression of AD and related tauopathies [6-8]. Thus, understan ding the etiopathogenesis of this pivotal and hallmark lesion of AD and related tauopathies is critical to developing rational therapeutic treatments of these human CNS diseases. Studies on the role of tau in neurodegeneration and therapeutic targets based on this pathology have been the subject of several recent reviews by us and others [9-17]. This article discusses the relationship between normal and pathological taus found in AD and related tauopathies.
Structure and Function of Normal Brain Tau
In human brain the alternative splicing of the tau pre-mRNA results in six molecular isoforms of the protein [1]. These six tau isoforms differ in containing three (3R taus) or four (4R taus) microtubule binding repeats (R) of 31–32 amino acids in the carboxy terminal half and one (1N), two (2N), or zero (0N) amino terminal inserts of 29 amino acids each; the extra repeat in 4R tau is the second repeat (R2) of 4R taus. This alternative splicing of tau pre-mRNA results in the expression of three 3R taus (0N3R, 1N3R, and 2N3R) and three 4R taus (0N4R, 1N4R, and 2N4R). The 2N4R tau is the largest size human brain tau with a total of 441 amino acids (tau441) in length. The smallest size tau isoform, which lacks both the two amino terminal inserts and the extra microtubule binding repeat (0N3R; tau352), is the only form that is expressed in fetal human brain. Tau has little secondary structure; it is mostly random coil with β structure in the second and third microtubule binding repeats.
Tau interacts with tubulin and promotes its assembly into microtubules and helps stabilize their structure [3]. Like MAP1 and MAP2, tau is a phosphoprotein and its biological activity is regulated by the degree of its phosphorylation [18-20]. Normal brain tau contains 2–3 moles of phosphate per mole of the protein [19], which appears to be optimal for its interaction with tubulin and the promotion of microtubule assembly. In addition to phosphorylation, the alternative splicing also affects the biological activity of tau. Both the extra repeat (Repeat 2) in the 4R taus and the amino terminal inserts (N1 and N2) enhance the binding of tau to tubulin, which makes 2N4R tau (tau441), and 0N3Rtau (tau352, the fetal tau) the relatively most and the least effective in promoting microtubule assembly [21, 22]. All six isoforms of tau are highly hydrophilic and are, thus, soluble and heat stable.
In a normal mature neuron tubulin is present in over tenfold excess of tau. The neuronal concentration of tau is ∼2 μM [23, 24] and it binds to microtubules at a Kd of ∼100 nM [25], and thus practically all tau is likely to be microtubule bound in the cell. In cultured cells overexpression of tau can cause microtubule bundling. However, neither in AD nor in any related tauopathy such a situation has been reported.
Pathological Forms and States of Tau
Abnormal Hyperphosphorylation and Oligomerization of tau
Neurofibrillary degeneration of abnormally hyperphosphorylated tau not only occurs in AD brain but is also seen in a family of related neurodegenerative diseases, called tauopathies, such as fronto-temporal dementia with Parkinsonism linked to chromosome 17 (FTDP-17) caused by tau mutations, Pick disease, corticobasal degeneration, dementia pugilistica, and progressive supranuclear palsy. In every one of these tauopathies the neurofibrillary changes are made up of abnormally hyperphosphorylated tau and their occurrence in the neocortex is associated with dementia. In frontotemporal dementia with Parkinsonism-linked to chromosome 17 and tau pathology (FTDP-17-tau), several missense mutations in tau cosegregate with the disease [26-28]. Four of these missense mutations, G272V, P301L, V337M, and R406W, which have been most studied to date, make tau a preferable substrate for abnormal hyperphosphorylation in vitro [21]. The neurofibrillary degeneration of the Alzheimer type is primarily seen in human neurodegenerative disorders. To date, in aged and in cognitively impaired animals the neurofibrillary degeneration of abnormally hyperphosphorylated tau has been found only sparsely.
To date, not only in AD but also in every known human tauopathy, the tau pathology is made up of the abnormally hyperphosphorylated protein. In AD brain all of the six tau isoforms are hyperphosphorylated and aggregated into PHF [4, 29-33]. While conformational changes [34-36] and truncation of tau [37-39] following its hyperphosphorylation [40]have been reported in AD, the most established and the most compelling cause of dysfunctional tau in AD and related tauopathies is the abnormal hyperphosphorylation of this protein [4, 20, 31].
While in normal brain almost all tau is soluble and is recovered in 200,000 × g cytosol, from AD brain this protein is recovered in three major states, i.e. soluble, oligomeric, and fibrillized [19, 31, 41]. There is at least as much normal cytosolic tau in AD brain as in normal aged brain but the level of total tau in the former is four to eight fold higher and this increase is solely in the form of the abnormally hyperphosphorylated protein [24]. As much as 40% of the tau from AD brain is non-fibrillized but oligomeric and sediments at 200,000 × g [19]. These tau oligomers isolated from AD brain, as 27,000 × g to 200,000 × g fraction, are made up of both abnormally hyperphosphorylated and non-hyperphosphorylated taus, and the two can be separated by phosphocellulose chromatography [19, 31]. Up until recently [42] this oligomeric tau was referred to as cytosolic tau, amorphous tau, and sedimentable cytosolic abnormally hyperphosphorylated tau [19, 20, 31, 41, 43-47]. The abnormally hyperphosphorylated tau purified from the oligomers is three to four fold more hyperphosphorylated as the non-hyperphosphorylated/normal tau [19].
Neurotoxic State of Tau
Two major known functions of tau are its ability to promote assembly and to maintain structure of microtubules [3]. The tau polymerized into neurofibrillary tangles is apparently inert and neither binds to tubulin nor promotes its assembly into microtubules [45, 48, 49]. As much as 40% of the abnormally hyperphosphorylated tau in AD brain is present in the cytosol and not polymerized into paired helical filaments/neurofibrillary tangles [19, 31, 41]. The AD cytosolic abnormally hyperphosphorylated tau (AD P-tau) does not bind to tubulin and promote microtubule assembly, but instead it inhibits assembly and disrupts microtubules Fig. (1) [20, 50, 51]. This toxic property of the pathological tau involves the sequestration of normal tau by the diseased protein [20, 44]. The AD P-tau also sequesters the other two major neuronal microtubule associated proteins MAP1 A/B and MAP2 [43]. This toxic behavior of the AD P-tau appears to be solely due to its abnormal hyperphosphorylation because dephosphorylation of diseased tau converts it into a normal-like protein [20, 50-52].
Fig. (1). A schematic representation of various pathological states of tau originating from normal brain tau and associated loss of normal and gain of toxic functions.
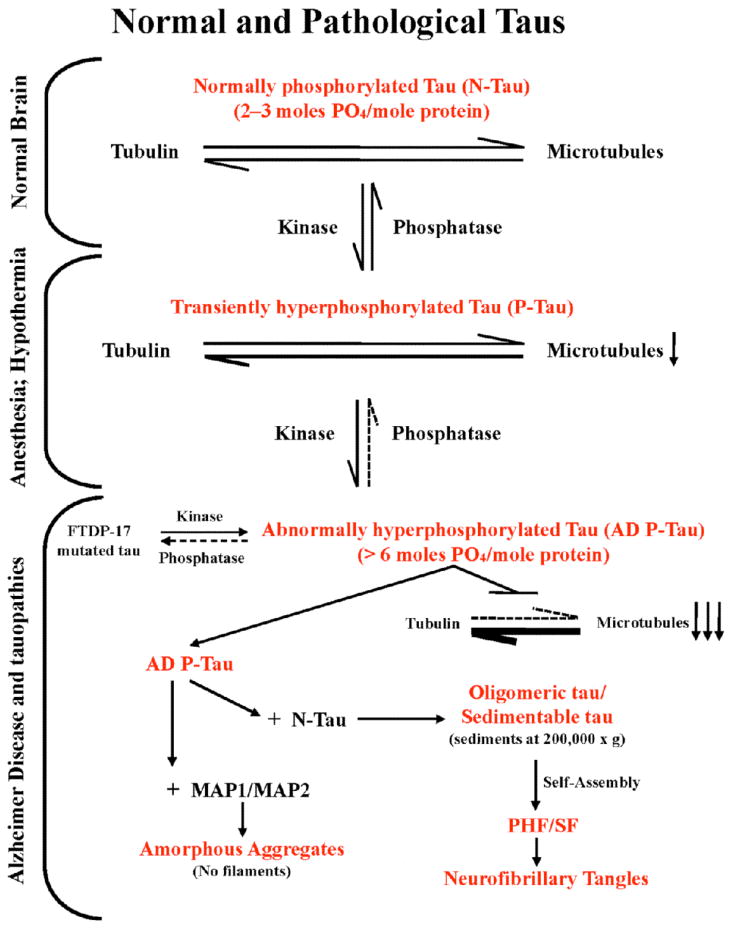
Normal brain tau, which has a stoichiometry of 2–3 moles phosphate/mole of the protein, stimulates assembly of tubulin and stabilizes the structure of microtubules produced. During development, anesthesia as well as hypothermia, such as during hibernation, tau is transiently hyperphosphorylated. During development the level of brain tubulin is >4 mg/ml, the critical concentration required for its self-assembly into microtubules and the role of tau for this function is less critical. The hyperphosphorylation of tau during anesthesia and hypothermia, however, leads to a decrease in microtubule network and the associated functions.
In AD brain a phosphorylation/dephosphorylation imbalance caused apparently by a decrease in protein phosphatase-2A activity leads to abnormal hyperphosphorylation of tau. This AD P-tau on one hand sequesters normal MAPs from microtubules and causes inhibition and disruption of microtubules. On the other hand, while the binding of AD P-tau to MAP1 or MAP2 results in amorphous aggregates, the binding to normal tau forms oligomers. Unlike normal tau, which is highly soluble, the tau oligomers formed with AD P-tau can be sedimented at 200,000 × g and self-assemble into PHF/SF in the form of neurofibrillary tangles.
The inhibitory activity of the non-fibrillized abnormally hyperphosphorylated tau has been confirmed in yeast, drosophila, and in mouse models that express human brain tau. The expression of the longest human brain tau (2N4Rtau) in yeast produces pathological phosphoepitopes, assumes a pathological conformation, and forms aggregates. These processes are modulated by yeast kinases Mds1 and Pho85, orthologues of GSK-3β and cdk5 [53, 54]. In yeast tau aggregates more when it is more phosphorylated, the mobility in SDS-PAGE is slower with increased phosphorylation, and hyperphosphorylated tau isolated from the stably transfected yeast is able to assemble into filaments, and is able to nucleate the assembly of the normal non-phosphorylated tau. These yeast studies, like those carried out previously using AD P-tau, suggest that the hyperphosphorylated tau works as a nucleation factor that initiates and promotes the aggregation of tau [44, 55].
In wild-type human tau- and mutated human tau-transgenic drosophila, the accumulation of the abnormally phosphorylated tau in the absence of its fibrillization into neurofibrillary tangles leads to neurodegeneration [56]. In a P301L tau inducible transgenic mouse model, cognitive improvement was observed when expression of human tau, which became abnormally hyperphosphorylated, was suppressed although neurofibrillary tangles continued to form, suggesting that the accumulation of the cytosolic abnormally hyperphosphorylated tau, and not its aggregation, was apparently involved in behavioral impairment in these animals [57]. Reduction of soluble Aβ and soluble abnormally hyperphosphorylated tau, but not soluble Aβ alone, was found to ameliorate cognitive decline in 3×Tg mice that express both plaque and tangle pathology [58] Furthermore, in vitro dephosphorylation of neurofibrillary tangles disaggregates filaments and, as a result, the tau released behaves like normal protein in promoting microtubule assembly [51]. Thus, two characteristics of AD abnormally hyperphosphorylated tau are (1) that it sequesters normal MAPs and disrupts microtubules and (2) that it self-assembles into PHF/SF.
In a recent study methylthioninium chloride (methylene blue dye) has been found to disaggregate PHF in vitro, reduce the number of tau aggregates in tau transgenic mice, and show significant inhibition of cognitive impairment in a PHASE II double blind clinical trial in AD patients [59, 60]. Whether disaggregation of pathological aggregates of tau with methylthioninium chloride results also in its dephosphorylation and removal remains to be studied.
Transient and Reversible Hyperphosphorylation of Tau
Hyperphosphorylation of tau, though not to the same level as in AD, is not only associated with the disease as in tauopathies, but is also employed by the neuron to down regulate its activity transiently and reversibly where required Fig. (1). For instance, during development the level of tubulin in the brain is at its highest, i.e., almost 33% of total cytosolic protein, which is almost 1.5-fold the critical concentration of 4 mg/ml tubulin required for its polymerization into microtubules [61]. Probably to avoid microtubule bundling, the fetal tau is transiently hyperphosphorylated during development. However, the level of hyperphosphorylation of tau in fetal brain is far less than that seen in AD brain. Similarly, anesthesia and hypothermia induced by hibernation in animals induces transient hyperphosphorylation of tau [62-65]. The molecular mechanism of the transient hyperphosphorylation of tau observed during development is, at present, not understood. However, during hypothermia the activity of protein phosphatase-2A (PP-2A), the major brain phosphoseryl/phosphothreonyl protein phosphatase activity is transiently and reversibly reduced and is believed to cause the hyperphosphorylation of tau [62, 63]. In AD and Down syndrome, the decrease in brain PP-2A activity apparently involves different molecular mechanisms, and occurs in a non-transient and irreversible manner [66-68]. It is the non-reversible nature of the abnormal hyperphosphorylation of tau in AD, Down syndrome, and related tauopathies which results in an involuntary slowing down of neuronal activity and a consequent chronic progressive neurodegeneration and its clinical phenotype, the dementia.
Fibrillization of Abnormally Hyperphosphorylated Tau and Neurodegeneration
Tau has long stretches of positively or negatively charged regions that are not conducive for intermolecular hydrophobic association [69]. The β-structure in tau is concentrated only in repeats R2 and R3 which can self-assemble into filaments [70] and co-assemble with heparin [71]. Both the amino terminal and the carboxy terminal flanking regions to the microtubule binding repeats in normal tau appear to inhibit its self-aggregation into filaments and on AD type abnormal hyperphosphorylation, i.e., the phosphorylation of the amino terminal and the carboxy terminal flanking regions, this inhibition is eliminated, resulting in the formation of tangles of PHF/SF [21, 46]. The co-assembly of tau with polyanions such as heparin, heparin sulfate [72-75], tRNA [76], or polyglutamate [77] appears to involve a mechanism different from what is seen in AD and in tauopathies. The polyanion-induced assembly of tau is very slow and does not result either in the lateral association of filaments into tangles nor the formation of any protofilaments seen in AD PHF. Furthermore, unlike AD and related tauopathies and transgenic animal models, the in vitro polyanions-induced assembly of tau into filaments is inhibited and not promoted by phosphorylation [78].
Dephosphorylation of PHF/neurofibrillary tangles isolated from AD brain results in their dissociation and disaggregation, and the dephosphorylated tau released behaves like normal tau in promoting microtubule assembly in vitro [51]. Similarly, dephosphorylation of AD cytosolic abnormally hyperphosphorylated tau with PP-2A inhibits its ability to self-aggregate into PHF/SF, sequester normal tau, and inhibit microtubule assembly in vitro, and rephosphorylation of the PP-2A-AD P-tau by several combinations of protein kinases restores all of its above pathological properties [47, 52].
The abnormal hyperphosphorylation of tau makes it resistant to proteolysis by the calcium activated neutral protease [51, 52] and turnover of hyperphosphorylated tau is several fold slower than the normal tau [79]. Most likely it is because of this reason that the levels of tau are several-fold increased in AD [23, 24]. Some increase in tau level in AD brain can also result from the activation of p70 S6 kinase which upregulates the translation of tau [80, 81]. It is likely that to neutralize the ability of AD P-tau to sequester normal MAPs and cause disassembly of microtubules, the affected neurons promote the self-assembly of the abnormal tau into tangles of PHF. The fact that the tangle-bearing neurons seem to survive many years [82] and that in AD brain the decrease in microtubule density was unrelated to PHFs accumulation [83] is consistent with such a self-defense role of the formation of tangles. Employing an inducible transgenic mouse model that expressed human four-repeat tau with the P301L mutation, Santacruz and colleagues [57] found that the cognitive deficiencies correlate with the appearance of soluble hyperphosphorylated tau. In this model when tau expression was turned off, there was no clearance of the polymerized tau, soluble phosphotau decreased, and there was improvement in cognition, suggesting that the polymerized tau was not sufficient to cause cognitive decline or neuronal cell death. Andorfer et al. [84] showed that in human tau transgenic mice, while there was widespread neurodegeneration, the PHF-containing neurons, however, appeared “healthy” in terms of nuclear morphology, suggesting that the polymerization of hyperphosphorylated tau into fibrils was probably neuroprotective [84]. Thus, all these studies taken together demonstrate the pivotal involvement of abnormal hyperphosphorylation in neurofibrillary degeneration and the disruptive properties to the microtubule network of the cytosolic abnormally hyperphosphorylated tau, whereas AD P-tau polymer remains inert Fig. (1).
Effect of Hyperphosphorylated Tau on Rough Endoplasmic Reticulum and Golgi
There is approximately as much tau in the somato-dendritic compartment as in the axon [23]. In the somato-dendritic compartment tau is associated with rough endoplasmic reticulum and Golgi apparatus [19, 31, 85]. The abnormal hyperphosphorylation of tau and its accumulation in the somato-dendritic compartment in AD might have been responsible for the morphological alterations of the RER and the Golgi apparatus and the abnormal N-glycosylation of tau in AD [86-88]. In AD brain abnormally hyperphosphorylated tau, in addition to forming neurofibrillary tangles, is associated with granulovacuolar changes [4, 89-91]. Overexpression of tau, which results in its hyperphosphorylation, has been found to induce fragmentation of Golgi both in neuronal cultures and in neurons in JNPL3 P301L tau transgenic mice [85]. In P301S tau transgenic mice, which show abnormal hyperphosphorylation of tau, a selective decrease in mitochondria and RER has been observed [92]. The chronic accumulation of the hyperphosphorylated tau as a misfolded protein in the ER could cause neurodegeneration due to protracted ER stress [93]. Hyperphosphorylation of tau might also be involved in neurodegeneration through alterations of RER and Golgi and a consequent reduction in RER and mitochondria.
Truncation of Tau
In addition to abnormal hyperphosphorylation, conformational changes and cleavage of tau have also been implicated in the pathogenesis of AD [34, 37, 38, 94]. The hyperphosphorylation of tau has been found to precede both conformational changes and cleavage of this protein [40]. Truncation of tau might make it a more favorable substrate for abnormal hyperphosphorylation. Transgenic rats expressing human tau truncated both N- and C-terminally tau151–391 show a marked neurofibrillary degeneration of abnormally hyperphosphorylated tau [95]. Hyperphosphorylation is known to produce conformational changes in a protein. The late appearance and low abundance of cleaved tau in neurofibrillary tangles probably represent little more than the unsuccessful attempts of the affected neuron to turn over the pathological aggregates.
Proteolysis of Abnormally Hyperphosphorylated Tau
In situ hybridization studies have revealed no significant change in the expression of tau mRNA in AD brain [96]. Thus, the increase in tau level observed in AD brain is probably mostly due to a decrease in its turnover caused by the hyperphosphorylation [51, 79, 97]. The AD abnormal hyperphosphorylation of tau (AD P-tau) makes tau resistant to both calcium activated neutral proteases, calpains, and its degradation by the ubiquitin-proteasome pathway. Unlike normal tau, the AD hyperphosphorylated tau is resistant to proteolysis by calpains [51].
Subsequent to its hyperphosphorylation in AD neurofibrillary tangles, tau becomes polyubiquitinated [19, 41, 98-105]. However, the ubiquitination of the abnormally hyper-phosphorylated tau in neurofibrillary tangles apparently does not lead to its clearance by digestion in the proteasome. This could partly be due to a faster rate of accumulation of the ubiquitinated phosphotau than the ability of the proteasomes of the degenerating neurons to digest it. Inhibition of the proteasome by its inhibitor, lactacystin, increases accumulation of both normal and hyperphosphorylated taus in rats [106]. Inhibition of the proteasome with its inhibitor, MG-132, in cultured oligodendrocytes causes ubiquitination and aggregation of tau [107]. An in vivo cause of impaired proteasome might be the occurrence, in the tangle-bearing neurons, of the one frame-shift mutation of ubiquitin (UBB+1) which inhibits the proteasome activity [108]. Another cause of the proteasome inhibition could be the increased level of BAG-1, an Hsp70/Hsc70 binding partner in the degenerating neurons. BAG-1 has been shown to inhibit degradation of tau by the 20S proteasome without affecting the ubiquitination of tau [109].
Overexpression of Hsp70 which interacts with the heat-shock cognate (Hsc) 70-interacting protein (CHIP), a ubiquitin ligase, causes a reduction of tau in transgenic mice [110]. In AD brain levels of both CHIP and Hsp70 are increased and the level of the former is inversely proportional to that of sarkosyl-insoluble tau [111]. The increase in CHIP might be protective in the early stages of AD. Interestingly, chronic administration of lithium, a known GSK-3β inhibitor, has been reported to decrease the tau lesions by promoting their ubiquitination in a tau transgenic mouse model [112]. Protein kinase B, Akt, which can hyperphosphorylate tau both directly and indirectly through GSK-3β and PAR1/MARK2, has been reported to prevent CHIP-induced tau ubiquitination and its subsequent proteolysis either by regulating Hsp90/CHIP complex directly or by competing as a client protein with tau for binding [113].
Tau Mutations Found in Frontotemporal Dementia Promote Abnormal Hyperphosphorylation
Tau mutations, which cause FTDP-17, result either in increase in 4-R:3-R tau ratio or in missense mutations in the protein. Both 4-repeat tau and the mutated protein are more easily abnormally hyperphosphorylated than the normal wild-type protein [21, 114]. Four of these missense mutations, G272V, P301L, V337M, and R406W, which have been most extensively studied to date, make tau a more favorable substrate than the wild-type protein for abnormal hyperphosphorylation by brain protein kinases in vitro [21]. These mutated taus become hyperphosphorylated at a faster rate and self-aggregate into filaments more readily, i.e., at a phosphorylation stoichiometry of 4–6 as compared to ten or more in the case of the wild-type protein. These faster kinetics of the hyperphosphorylation of the mutated tau might explain a relatively early onset, severity, and autosomal dominance of the disease in the inherited FTDP-17 cases.
The six human brain tau isoforms are differentially sequestered by AD P-tau in vitro [22]. The association of AD P-tau to normal human brain recombinant taus is 2N4Rtau>1N4Rtau>0N4Rtau and 2N3Rtau>1N3Rtau>0N-3Rtau, and 2N4Rtau>2N3Rtau. AD P-tau also inhibits the assembly and disrupts microtubules pre-assembled with each tau isoform with an efficiency which corresponds directly to the degree of interaction with these isoforms. In vitro hyperphosphorylation of recombinant tau converts it into an AD P-tau-like state in sequestering normal tau and inhibiting microtubule assembly. The preferential sequestration of 4R taus and taus with amino terminal inserts explains both (i) why fetal tau (ON3Rtau) is protected from Alzheimer neurofibrillary pathology and (ii) why intronic mutations seen in certain inherited cases of FTDP-17, which result in alternate splicing of tau mRNA and consequently an increase in 4R:3R ratio, lead to neurofibrillary degeneration and the disease. In vitro, at a phosphorylation stoichiometry of 4 and above, the hyperphosphorylated tau sequesters normal tau, whereas it requires a stoichiometry of 10 or more to self-aggregate into filaments [21, 45, 50]. On aggregation into filaments, tau loses its ability to sequester normal tau. Furthermore, AD P-tau, but not PHF, inhibits regeneration of microtubule network in detergent-extracted PC12 cells, indicating that the formation of filaments might be initiated as a self-defense response by the affected neurons [22, 45]. Opposite to FTDP-17, in Pick disease and in Down syndrome (DS) the tau 3R:4R ratio is very much increased [115-117]. Since the activity of 3R tau is lesser than of 4R tau in binding to tubulin/microtubules, the unbound 3R tau becomes abnormally hyperphosphorylated because free tau is a more favorable substrate than tau on microtubules for phosphorylation [118].
Conclusion
In conclusion, the abnormal hyperphosphorylation of tau seen in AD is different from the normal and from the transient hyperphosphorylation of this protein that occurs during development, anesthesia, or hypothermia. The cytosolic AD abnormally hyperphosphorylated tau (AD P-tau) is sedimentable/oligomeric, and probably causes neurodegeneration by sequestering normal MAPs and disrupting microtubule network. Tau mutations found in frontotemporal dementia may cause neurodegeneration through promoting abnormal hyperphosphorylation of tau. AD P-tau self-assembles into PHF/SF, forming neurofibrillary tangles. Tau truncation found in AD brain promotes its self-assembly into PHF/SF. Unlike AD P-tau, PHF/SF neither sequester normal MAPs nor disrupt microtubules. Thus, inhibition of abnormal hyperphosphorylation of tau offers a promising therapeutic target for AD and related tauopathies.
Acknowledgments
We are grateful to Ms. Janet Murphy for secretarial assistance. Dr. Ezzat El-Akkad helped prepare Fig. (1). Studies in our laboratories were supported in part by the New York State Office of Mental Retardation and Developmental Disabilities; NIH grants AG019158, AG028538, and AG27429; and Alzheimer's Association (Chicago, IL) grants IIRG-00-2002, HRG-05-13095, and NIRG-08-91126.
References
- 1.Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer's disease. Neuron. 1989;3:519–526. doi: 10.1016/0896-6273(89)90210-9. [DOI] [PubMed] [Google Scholar]
- 2.Himmler A, Drechsel D, Kirschner MW, Martin DW., Jr Tau consists of a set of proteins with repeated C-terminal microtubule-binding domains and variable N-terminal domains. Mol Cell Biol. 1989;9:1381–1388. doi: 10.1128/mcb.9.4.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW. A protein factor essential for microtubule assembly. Proc Natl Acad Sci USA. 1975;72:1858–1862. doi: 10.1073/pnas.72.5.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA. 1986b;83:4913–4917. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Braak H, Braak E, Grundke-Iqbal I, Iqbal K. Occurrence of neuropil threads in the senile human brain and in Alzheimer's disease: a third location of paired helical filaments outside of neurofibrillary tangles and neuritic plaques. Neurosci Lett. 1986;65:351–355. doi: 10.1016/0304-3940(86)90288-0. [DOI] [PubMed] [Google Scholar]
- 6.Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology. 1992;42:631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- 7.Tomlinson BE, Blessed G, Roth M. Observations on the brains of demented old people. J Neurol Sci. 1970;11:205–242. doi: 10.1016/0022-510x(70)90063-8. [DOI] [PubMed] [Google Scholar]
- 8.Alafuzoff I, Iqbal K, Friden H, Adolfsson R, Winblad B. Histopa-thological criteria for progressive dementia disorders: clinical-pathological correlation and classification by multivariate data analysis. Acta Neuropathol (Berl) 1987;74:209–225. doi: 10.1007/BF00688184. [DOI] [PubMed] [Google Scholar]
- 9.Avila J, Hernandez F. GSK-3 inhibitors for Alzheimer's disease. Expert Rev Neurother. 2007;7:1527–1533. doi: 10.1586/14737175.7.11.1527. [DOI] [PubMed] [Google Scholar]
- 10.Iqbal K, Grundke-Iqbal I. Pharmacological approaches of neurofibrillary degeneration. Curr Alzheimer Res. 2005;2:335–341. doi: 10.2174/1567205054367810. [DOI] [PubMed] [Google Scholar]
- 11.Iqbal K, Grundke-Iqbal I. Alzheimer neurofibrillary degeneration: significance, etiopathogenesis, therapeutics, and prevention. J Cell Mol Med. 2008;12:38–55. doi: 10.1111/j.1582-4934.2008.00225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iqbal K, Liu F, Gong CX, Alonso Adel C, Grundke-Iqbal I. Mechanisms of tau-induced neurodegeneration. Acta Neuropathol. 2009;118:53–69. doi: 10.1007/s00401-009-0486-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bamburg JR, Bloom GS. Cytoskeletal pathologies of Alzheimer disease. Cell Motil Cytoskeleton. 2009;66:635–649. doi: 10.1002/cm.20388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davies P, Koppel J. Mechanism-based treatments for Alzheimer's disease. Dialogues Clin Neurosci. 2009;11:159–169. doi: 10.31887/DCNS.2009.11.2/pdavies. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gotz J, Ittner LM, Lim YA. Common features between diabetes mellitus and Alzheimer's disease. Cell Mol Life Sci. 2009;66:1321–1325. doi: 10.1007/s00018-009-9070-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martinez A, Perez DI. GSK-3 inhibitors: a ray of hope for the treatment of Alzheimer's disease? J Alzheimers Dis. 2008;15:181–191. doi: 10.3233/jad-2008-15204. [DOI] [PubMed] [Google Scholar]
- 17.Pei JJ, Sjogren M, Winblad B. Neurofibrillary degeneration in Alzheimer's disease: from molecular mechanisms to identification of drug targets. Curr Opin Psychiatry. 2008;21:555–561. doi: 10.1097/YCO.0b013e328314b78b. [DOI] [PubMed] [Google Scholar]
- 18.Lindwall G, Cole RD. Phosphorylation affects the ability of tau protein to promote microtubule assembly. J Biol Chem. 1984;259:5301–5305. [PubMed] [Google Scholar]
- 19.Kopke E, Tung YC, Shaikh S, Alonso AC, Iqbal K, Grundke-Iqbal I. Microtubule-associated protein tau. Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J Biol Chem. 1993;268:24374–24384. [PubMed] [Google Scholar]
- 20.Alonso AD, Zaidi T, Grundke-Iqbal I, Iqbal K. Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc Natl Acad Sci USA. 1994;91:5562–5566. doi: 10.1073/pnas.91.12.5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alonso AD, Mederlyova A, Novak M, Grundke-Iqbal I, Iqbal K. Promotion of hyperphosphorylation by frontotemporal dementia tau mutations. J Biol Chem. 2004;279:34873–34881. doi: 10.1074/jbc.M405131200. [DOI] [PubMed] [Google Scholar]
- 22.Alonso AD, Zaidi T, Novak M, Barra HS, Grundke-Iqbal I, Iqbal K. Interaction of tau isoforms with Alzheimer's disease abnormally hyperphosphorylated tau and in vitro phosphorylation into the disease-like protein. J Biol Chem. 2001;276:37967–37973. doi: 10.1074/jbc.M105365200. [DOI] [PubMed] [Google Scholar]
- 23.Butner KA, Kirschner MW. Tau protein binds to microtubules through a flexible array of distributed weak sites. J Cell Biol. 1991;115:717–730. doi: 10.1083/jcb.115.3.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khatoon S, Grundke-Iqbal I, Iqbal K. Brain levels of microtubule-associated protein tau are elevated in Alzheimer's disease: a radioimmuno-slot-blot assay for nanograms of the protein. J Neurochem. 1992;59:750–753. doi: 10.1111/j.1471-4159.1992.tb09432.x. [DOI] [PubMed] [Google Scholar]
- 25.Ackmann M, Wiech H, Mandelkow E. Nonsaturable binding indicates clustering of tau on the microtubule surface in a paired helical filament-like conformation. J Biol Chem. 2000;275:30335–30343. doi: 10.1074/jbc.M002590200. [DOI] [PubMed] [Google Scholar]
- 26.Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, et al. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol. 1998;43:815–825. doi: 10.1002/ana.410430617. [DOI] [PubMed] [Google Scholar]
- 27.Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 28.Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci USA. 1998;95:7737–7741. doi: 10.1073/pnas.95.13.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grundke-Iqbal I, Iqbal K, Quinlan M, Tung YC, Zaidi MS, Wisniewski HM. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J Biol Chem. 1986a;261:6084–6089. [PubMed] [Google Scholar]
- 30.Iqbal K, Grundke-Iqbal I, Smith AJ, George L, Tung YC, Zaidi T. Identification and localization of a tau peptide to paired helical filaments of Alzheimer disease. Proc Natl Acad Sci USA. 1989;86:5646–5650. doi: 10.1073/pnas.86.14.5646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Iqbal K, Grundke-Iqbal I, Zaidi T, Merz PA, Wen GY, Shaikh SS, et al. Defective brain microtubule assembly in Alzheimer's disease. Lancet. 1986;2:421–426. doi: 10.1016/s0140-6736(86)92134-3. [DOI] [PubMed] [Google Scholar]
- 32.Lee VM, Balin BJ, Otvos L, Jr, Trojanowski JQ. A68: a major subunit of paired helical filaments and derivatized forms of normal Tau. Science. 1991;251:675–678. doi: 10.1126/science.1899488. [DOI] [PubMed] [Google Scholar]
- 33.Goedert M, Spillantini MG, Cairns NJ, Crowther RA. Tau proteins of Alzheimer paired helical filaments: abnormal phosphorylation of all six brain isoforms. Neuron. 1992;8:159–168. doi: 10.1016/0896-6273(92)90117-v. [DOI] [PubMed] [Google Scholar]
- 34.Jicha GA, Lane E, Vincent I, Otvos L, Jr, Hoffmann R, Davies P. A conformation- and phosphorylation-dependent antibody recognizing the paired helical filaments of Alzheimer's disease. J Neurochem. 1997;69:2087–2095. doi: 10.1046/j.1471-4159.1997.69052087.x. [DOI] [PubMed] [Google Scholar]
- 35.Jicha GA, Berenfeld B, Davies P. Sequence requirements for formation of conformational variants of tau similar to those found in Alzheimer's disease. J Neurosci Res. 1999;55:713–723. doi: 10.1002/(SICI)1097-4547(19990315)55:6<713::AID-JNR6>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 36.Jicha GA, Rockwood JM, Berenfeld B, Hutton M, Davies P. Altered conformation of recombinant frontotemporal dementia-17 mutant tau proteins. Neurosci Lett. 1999;260:153–156. doi: 10.1016/s0304-3940(98)00980-x. [DOI] [PubMed] [Google Scholar]
- 37.Novak M, Jakes R, Edwards PC, Milstein C, Wischik CM. Difference between the tau protein of Alzheimer paired helical filament core and normal tau revealed by epitope analysis of monoclonal antibodies 423 and 7.51. Proc Natl Acad Sci USA. 1991;88:5837–5841. doi: 10.1073/pnas.88.13.5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gamblin TC, Chen F, Zambrano A, Abraha A, Lagalwar S, Guillozet AL, et al. Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer's disease. Proc Natl Acad Sci USA. 2003;100:10032–10037. doi: 10.1073/pnas.1630428100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cotman CW, Poon WW, Rissman RA, Blurton-Jones M. The role of caspase cleavage of tau in Alzheimer disease neuropathology. J Neuropathol Exp Neurol. 2005;64:104–112. doi: 10.1093/jnen/64.2.104. [DOI] [PubMed] [Google Scholar]
- 40.Delobel P, Lavenir I, Fraser G, Ingram E, Holzer M, Ghetti B, et al. Analysis of tau phosphorylation and truncation in a mouse model of human tauopathy. Am J Pathol. 2008;172:123–131. doi: 10.2353/ajpath.2008.070627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bancher C, Brunner C, Lassmann H, Budka H, Jellinger K, Wiche G, et al. Accumulation of abnormally phosphorylated tau precedes the formation of neurofibrillary tangles in Alzheimer's disease. Brain Res. 1989;477:90–99. doi: 10.1016/0006-8993(89)91396-6. [DOI] [PubMed] [Google Scholar]
- 42.Takashima A. Amyloid-beta, Tau, and Dementia. J Alzheimers Dis. 2009 doi: 10.3233/JAD-2009-1090. [DOI] [PubMed] [Google Scholar]
- 43.Alonso AD, Grundke-Iqbal I, Barra HS, Iqbal K. Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc Natl Acad Sci USA. 1997;94:298–303. doi: 10.1073/pnas.94.1.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alonso AD, Grundke-Iqbal I, Iqbal K. Alzheimer's disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat Med. 1996;2:783–787. doi: 10.1038/nm0796-783. [DOI] [PubMed] [Google Scholar]
- 45.Alonso AD, Li B, Grundke-Iqbal I, Iqbal K. Polymerization of hyperphosphorylated tau into filaments eliminates its inhibitory activity. Proc Natl Acad Sci USA. 2006;23:8864–8869. doi: 10.1073/pnas.0603214103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alonso AD, Zaidi T, Novak M, Grundke-Iqbal I, Iqbal K. Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc Natl Acad Sci USA. 2001;98:6923–6928. doi: 10.1073/pnas.121119298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang JZ, Grundke-Iqbal I, Iqbal K. Kinases and phosphatases and tau sites involved in Alzheimer neurofibrillary degeneration. Eur J Neurosci. 2007;25:59–68. doi: 10.1111/j.1460-9568.2006.05226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Iqbal K, Zaidi T, Bancher C, Grundke-Iqbal I. Alzheimer paired helical filaments. Restoration of the biological activity by dephosphorylation. FEBS Lett. 1994;349:104–108. doi: 10.1016/0014-5793(94)00650-4. [DOI] [PubMed] [Google Scholar]
- 49.Khatoon S, Grundke-Iqbal I, Iqbal K. Guanosine triphosphate binding to beta-subunit of tubulin in Alzheimer's disease brain: role of microtubule-associated protein tau. J Neurochem. 1995;64:777–787. doi: 10.1046/j.1471-4159.1995.64020777.x. [DOI] [PubMed] [Google Scholar]
- 50.Li B, Chohan MO, Grundke-Iqbal I, Iqbal K. Disruption of microtubule network by Alzheimer abnormally hyperphosphorylated tau. Acta Neuropathol (Berl) 2007;113:501–511. doi: 10.1007/s00401-007-0207-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang JZ, Gong CX, Zaidi T, Grundke-Iqbal I, Iqbal K. Dephosphorylation of Alzheimer paired helical filaments by protein phosphatase-2A and -2B. J Biol Chem. 1995;270:4854–4860. doi: 10.1074/jbc.270.9.4854. [DOI] [PubMed] [Google Scholar]
- 52.Wang JZ, Grundke-Iqbal I, Iqbal K. Restoration of biological activity of Alzheimer abnormally phosphorylated tau by dephosphorylation with protein phosphatase-2A, -2B and -1. Brain Res Mol Brain Res. 1996;38:200–208. doi: 10.1016/0169-328x(95)00316-k. [DOI] [PubMed] [Google Scholar]
- 53.Vandebroek T, Terwel D, Vanhelmont T, Gysemans M, Van Haesendonck C, Engelborghs Y, et al. Microtubule binding and clustering of human Tau-4R and Tau-P301L proteins isolated from yeast deficient in orthologues of glycogen synthase kinase-3beta or cdk5. J Biol Chem. 2006;281:25388–25397. doi: 10.1074/jbc.M602792200. [DOI] [PubMed] [Google Scholar]
- 54.Vandebroek T, Vanhelmont T, Terwel D, Borghgraef P, Lemaire K, Snauwaert J, et al. Identification and isolation of a hyperphosphorylated, conformationally changed intermediate of human protein tau expressed in yeast. Biochemistry. 2005;44:11466–11475. doi: 10.1021/bi0506775. [DOI] [PubMed] [Google Scholar]
- 55.Maeda S, Sahara N, Saito Y, Murayama S, Ikai A, Takashima A. Increased levels of granular tau oligomers: an early sign of brain aging and Alzheimer's disease. Neurosci Res. 2006;54:197–201. doi: 10.1016/j.neures.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 56.Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, Hutton M, et al. Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science. 2001;293:711–714. doi: 10.1126/science.1062382. [DOI] [PubMed] [Google Scholar]
- 57.Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Oddo S, Vasilevko V, Caccamo A, Kitazawa M, Cribbs DH, LaFerla FM. Reduction of soluble Abeta and tau, but not soluble Abeta alone, ameliorates cognitive decline in transgenic mice with plaques and tangles. J Biol Chem. 2006;281:39413–39423. doi: 10.1074/jbc.M608485200. [DOI] [PubMed] [Google Scholar]
- 59.Wischik CM, Bentham P, Wischik DJ, Seng KM. Tau aggregation inhibitor (TAI) therapy with rember arrests disease progression in mild and moderate Alzheimer's disease over 50 weeks. Alzheimer's & Dementia. 2008;4:167. [Google Scholar]
- 60.Harrington C, Rickard JE, Horsley D, Harrington KA, Hindley KP, Riedel G, et al. Methylthioninium chloride (MTC) acts as a tau aggregation inhibitor (TAI) in a cellular model and reverses tau pathology in transgenic mouse model of Alzheimer's disease. Alzheimer's & Dementia. 2008;4:120. [Google Scholar]
- 61.Schmitt H, Gozes I, Littauer UZ. Decrease in levels and rates of synthesis of tubulin and actin in developing rat brain. Brain Res. 1977;121:327–342. doi: 10.1016/0006-8993(77)90155-x. [DOI] [PubMed] [Google Scholar]
- 62.Su B, Wang X, Drew KL, Perry G, Smith MA, Zhu X. Physiological regulation of tau phosphorylation during hibernation. J Neurochem. 2008;105:2098–2108. doi: 10.1111/j.1471-4159.2008.05294.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Planel E, Richter KE, Nolan CE, Finley JE, Liu L, Wen Y, et al. Anesthesia leads to tau hyperphosphorylation through inhibition of phosphatase activity by hypothermia. J Neurosci. 2007;27:3090–3097. doi: 10.1523/JNEUROSCI.4854-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Arendt T, Stieler J, Strijkstra AM, Hut RA, Rudiger J, Van der Zee EA, et al. Reversible paired helical filament-like phosphorylation of tau is an adaptive process associated with neuronal plasticity in hibernating animals. J Neurosci. 2003;23:6972–6981. doi: 10.1523/JNEUROSCI.23-18-06972.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Run X, Liang Z, Zhang L, Iqbal K, Grundke-Iqbal I, Gong C. Anesthesia induces phosphorylation of tau. J Alzheimers Dis. 2008 doi: 10.3233/JAD-2009-1003. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gong CX, Shaikh S, Wang JZ, Zaidi T, Grundke-Iqbal I, Iqbal K. Phosphatase activity toward abnormally phosphorylated tau: decrease in Alzheimer disease brain. J Neurochem. 1995;65:732–738. doi: 10.1046/j.1471-4159.1995.65020732.x. [DOI] [PubMed] [Google Scholar]
- 67.Gong CX, Singh TJ, Grundke-Iqbal I, Iqbal K. Phosphoprotein phosphatase activities in Alzheimer disease brain. J Neurochem. 1993;61:921–927. doi: 10.1111/j.1471-4159.1993.tb03603.x. [DOI] [PubMed] [Google Scholar]
- 68.Liang Z, Liu F, Iqbal K, Grundke-Iqbal I, Wegiel J, Gong CX. Decrease of protein phosphatase 2A and its association with accumulation and hyperphosphorylation of tau in Down syndrome. J Alzheimers Dis. 2008;13:295–302. doi: 10.3233/jad-2008-13307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ruben GC, Iqbal K, Grundke-Iqbal I, Wisniewski HM, Ciardelli TL, Johnson JE., Jr The microtubule-associated protein tau forms a triple-stranded left-hand helical polymer. J Biol Chem. 1991;266:22019–22027. [PubMed] [Google Scholar]
- 70.von Bergen M, Friedhoff P, Biernat J, Heberle J, Mandelkow EM, Mandelkow E. Assembly of tau protein into Alzheimer paired helical filaments depends on a local sequence motif ((306)VQIVYK(311)) forming beta structure. Proc Natl Acad Sci USA. 2000;97:5129–5134. doi: 10.1073/pnas.97.10.5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Arrasate M, Perez M, Armas-Portela R, Avila J. Polymerization of tau peptides into fibrillar structures. The effect of FTDP-17 mutations. FEBS Lett. 1999;446:199–202. doi: 10.1016/s0014-5793(99)00210-0. [DOI] [PubMed] [Google Scholar]
- 72.Hasegawa M, Crowther RA, Jakes R, Goedert M. Alzheimer-like changes in microtubule-associated protein Tau induced by sulfated glycosaminoglycans. Inhibition of microtubule binding, stimulation of phosphorylation, and filament assembly depend on the degree of sulfation. J Biol Chem. 1997;272:33118–33124. doi: 10.1074/jbc.272.52.33118. [DOI] [PubMed] [Google Scholar]
- 73.Goedert M, Jakes R, Spillantini MG, Hasegawa M, Smith MJ, Crowther RA. Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature. 1996;383:550–553. doi: 10.1038/383550a0. [DOI] [PubMed] [Google Scholar]
- 74.Sibille N, Sillen A, Leroy A, Wieruszeski JM, Mulloy B, Landrieu I, et al. Structural impact of heparin binding to full-length Tau as studied by NMR spectroscopy. Biochemistry. 2006;45:12560–12572. doi: 10.1021/bi060964o. [DOI] [PubMed] [Google Scholar]
- 75.Perez M, Valpuesta JM, Medina M, Montejo de Garcini E, Avila J. Polymerization of tau into filaments in the presence of heparin: the minimal sequence required for tau-tau interaction. J Neurochem. 1996;67:1183–1190. doi: 10.1046/j.1471-4159.1996.67031183.x. [DOI] [PubMed] [Google Scholar]
- 76.Kampers T, Friedhoff P, Biernat J, Mandelkow EM, Mandelkow E. RNA stimulates aggregation of microtubule-associated protein tau into Alzheimer-like paired helical filaments. FEBS Lett. 1996;399:344–349. doi: 10.1016/s0014-5793(96)01386-5. [DOI] [PubMed] [Google Scholar]
- 77.Kampers T, Pangalos M, Geerts H, Wiech H, Mandelkow E. Assembly of paired helical filaments from mouse tau: implications for the neurofibrillary pathology in transgenic mouse models for Alzheimer's disease. FEBS Lett. 1999;451:39–44. doi: 10.1016/s0014-5793(99)00522-0. [DOI] [PubMed] [Google Scholar]
- 78.Schneider A, Biernat J, von Bergen M, Mandelkow E, Mandelkow EM. Phosphorylation that detaches tau protein from microtubules (Ser262, Ser214) also protects it against aggregation into Alzheimer paired helical filaments. Biochemistry. 1999;38:3549–3558. doi: 10.1021/bi981874p. [DOI] [PubMed] [Google Scholar]
- 79.Poppek D, Keck S, Ermak G, Jung T, Stolzing A, Ullrich O, et al. Phosphorylation inhibits turnover of the tau protein by the proteasome: influence of RCAN1 and oxidative stress. Biochem J. 2006;400:511–520. doi: 10.1042/BJ20060463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.An WL, Cowburn RF, Li L, Braak H, Alafuzoff I, Iqbal K, et al. Up-regulation of phosphorylated/activated p70 S6 kinase and its relationship to neurofibrillary pathology in Alzheimer's disease. Am J Pathol. 2003;163:591–607. doi: 10.1016/S0002-9440(10)63687-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pei JJ, An WL, Zhou XW, Nishimura T, Norberg J, Benedikz E, et al. P70 S6 kinase mediates tau phosphorylation and synthesis. FEBS Lett. 2006;580:107–114. doi: 10.1016/j.febslet.2005.11.059. [DOI] [PubMed] [Google Scholar]
- 82.Morsch R, Simon W, Coleman PD. Neurons may live for decades with neurofibrillary tangles. J Neuropathol Exp Neurol. 1999;58:188–197. doi: 10.1097/00005072-199902000-00008. [DOI] [PubMed] [Google Scholar]
- 83.Cash AD, Aliev G, Siedlak SL, Nunomura A, Fujioka H, Zhu X, et al. Microtubule reduction in Alzheimer's disease and aging is independent of tau filament formation. Am J Pathol. 2003;162:1623–1627. doi: 10.1016/s0002-9440(10)64296-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Andorfer C, Acker CM, Kress Y, Hof PR, Duff K, Davies P. Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J Neurosci. 2005;25:5446–5454. doi: 10.1523/JNEUROSCI.4637-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liazoghli D, Perreault S, Micheva KD, Desjardins M, Leclerc N. Fragmentation of the Golgi apparatus induced by the overexpression of wild-type and mutant human tau forms in neurons. Am J Pathol. 2005;166:1499–1514. doi: 10.1016/S0002-9440(10)62366-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lin WL, Lewis J, Yen SH, Hutton M, Dickson DW. Ultrastructural neuronal pathology in transgenic mice expressing mutant (P301L) human tau. J Neurocytol. 2003;32:1091–1105. doi: 10.1023/B:NEUR.0000021904.61387.95. [DOI] [PubMed] [Google Scholar]
- 87.Gray EG, Paula-Barbosa M, Roher A. Alzheimer's disease: paired helical filaments and cytomembranes. Neuropathol Appl Neurobiol. 1987;13:91–110. doi: 10.1111/j.1365-2990.1987.tb00174.x. [DOI] [PubMed] [Google Scholar]
- 88.Wang JZ, Grundke-Iqbal I, Iqbal K. Glycosylation of microtubule-associated protein tau: an abnormal posttranslational modification in Alzheimer's disease. Nat Med. 1996;2:871–875. doi: 10.1038/nm0896-871. [DOI] [PubMed] [Google Scholar]
- 89.Ikegami K, Kimura T, Katsuragi S, Ono T, Yamamoto H, Miyamoto E, et al. Immunohistochemical examination of phosphorylated tau in granulovacuolar degeneration granules. Psychiatry Clin Neurosci. 1996;50:137–140. doi: 10.1111/j.1440-1819.1996.tb01678.x. [DOI] [PubMed] [Google Scholar]
- 90.Lagalwar S, Berry RW, Binder LI. Relation of hippocampal phospho-SAPK/JNK granules in Alzheimer's disease and tauopathies to granulovacuolar degeneration bodies. Acta Neuropathol. 2007;113:63–73. doi: 10.1007/s00401-006-0159-4. [DOI] [PubMed] [Google Scholar]
- 91.Ghoshal N, Smiley JF, DeMaggio AJ, Hoekstra MF, Cochran EJ, Binder LI, et al. A new molecular link between the fibrillar and granulovacuolar lesions of Alzheimer's disease. Am J Pathol. 1999;155:1163–1172. doi: 10.1016/S0002-9440(10)65219-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 93.Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2008;7:1013–1030. doi: 10.1038/nrd2755. [DOI] [PubMed] [Google Scholar]
- 94.Luna-Munoz J, Garcia-Sierra F, Falcon V, Menendez I, Chavez-Macias L, Mena R. Regional conformational change involving phosphorylation of tau protein at the Thr231, precedes the structural change detected by Alz-50 antibody in Alzheimer's disease. J Alzheimers Dis. 2005;8:29–41. doi: 10.3233/jad-2005-8104. [DOI] [PubMed] [Google Scholar]
- 95.Zilka N, Filipcik P, Koson P, Fialova L, Skrabana R, Zilkova M, et al. Truncated tau from sporadic Alzheimer's disease suffices to drive neurofibrillary degeneration in vivo. FEBS Lett. 2006;580:3582–3588. doi: 10.1016/j.febslet.2006.05.029. [DOI] [PubMed] [Google Scholar]
- 96.Mah VH, Eskin TA, Kazee AM, Lapham L, Higgins GA. In situ hybridization of calcium/calmodulin dependent protein kinase II and tau mRNAs; species differences and relative preservation in Alzheimer's disease. Brain Res Mol Brain Res. 1992;12:85–94. doi: 10.1016/0169-328x(92)90071-i. [DOI] [PubMed] [Google Scholar]
- 97.Yang LS, Ksiezak-Reding H. Calpain-induced proteolysis of normal human tau and tau associated with paired helical filaments. Eur J Biochem. 1995;233:9–17. doi: 10.1111/j.1432-1033.1995.009_1.x. [DOI] [PubMed] [Google Scholar]
- 98.Mori H, Kondo J, Ihara Y. Ubiquitin is a component of paired helical filaments in Alzheimer's disease. Science. 1987;235:1641–1644. doi: 10.1126/science.3029875. [DOI] [PubMed] [Google Scholar]
- 99.Perry G, Friedman R, Shaw G, Chau V. Ubiquitin is detected in neurofibrillary tangles and senile plaque neurites of Alzheimer disease brains. Proc Natl Acad Sci USA. 1987;84:3033–3036. doi: 10.1073/pnas.84.9.3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Perry G, Mulvihill P, Fried VA, Smith HT, Grundke-Iqbal I, Iqbal K. Immunochemical properties of ubiquitin conjugates in the paired helical filaments of Alzheimer disease. J Neurochem. 1989;52:1523–1528. doi: 10.1111/j.1471-4159.1989.tb09203.x. [DOI] [PubMed] [Google Scholar]
- 101.Grundke-Iqbal I, Vorbrodt AW, Iqbal K, Tung YC, Wang GP, Wisniewski HM. Microtubule-associated polypeptides tau are altered in Alzheimer paired helical filaments. Brain Res. 1988;464:43–52. doi: 10.1016/0169-328x(88)90017-4. [DOI] [PubMed] [Google Scholar]
- 102.Bancher C, Grundke-Iqbal I, Iqbal K, Fried VA, Smith HT, Wisniewski HM. Abnormal phosphorylation of tau precedes ubiquitination in neurofibrillary pathology of Alzheimer disease. Brain Res. 1991;539:11–18. doi: 10.1016/0006-8993(91)90681-k. [DOI] [PubMed] [Google Scholar]
- 103.Morishima-Kawashima M, Hasegawa M, Takio K, Suzuki M, Titani K, Ihara Y. Ubiquitin is conjugated with amino-terminally processed tau in paired helical filaments. Neuron. 1993;10:1151–1160. doi: 10.1016/0896-6273(93)90063-w. [DOI] [PubMed] [Google Scholar]
- 104.Cripps D, Thomas SN, Jeng Y, Yang F, Davies P, Yang AJ. Alzheimer disease-specific conformation of hyperphosphorylated paired helical filament-Tau is polyubiquitinated through Lys-48, Lys-11, and Lys-6 ubiquitin conjugation. J Biol Chem. 2006;281:10825–10838. doi: 10.1074/jbc.M512786200. [DOI] [PubMed] [Google Scholar]
- 105.Yang L, Ksiezak-Reding H. Ubiquitin immunoreactivity of paired helical filaments differs in Alzheimer's disease and corticobasal degeneration. Acta Neuropathol. 1998;96:520–526. doi: 10.1007/s004010050928. [DOI] [PubMed] [Google Scholar]
- 106.Liu YH, Wei W, Yin J, Liu GP, Wang Q, Cao FY, et al. Proteasome inhibition increases tau accumulation independent of phosphorylation. Neurobiol Aging. 2008 doi: 10.1016/j.neurobiolaging.2008.02.012. [DOI] [PubMed] [Google Scholar]
- 107.Goldbaum O, Richter-Landsberg C. Proteolytic stress causes heat shock protein induction, tau ubiquitination, and the recruitment of ubiquitin to tau-positive aggregates in oligodendrocytes in culture. J Neurosci. 2004;24:5748–5757. doi: 10.1523/JNEUROSCI.1307-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.van Leeuwen FW, Hol EM, Fischer DF. Frameshift proteins in Alzheimer's disease and in other conformational disorders: time for the ubiquitin-proteasome system. J Alzheimers Dis. 2006;9:319–325. doi: 10.3233/jad-2006-9s336. [DOI] [PubMed] [Google Scholar]
- 109.Elliott E, Tsvetkov P, Ginzburg I. BAG-1 associates with Hsc70.Tau complex and regulates the proteasomal degradation of Tau protein. J Biol Chem. 2007;282:37276–37284. doi: 10.1074/jbc.M706379200. [DOI] [PubMed] [Google Scholar]
- 110.Petrucelli L, Dickson D, Kehoe K, Taylor J, Snyder H, Grover A, et al. CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum Mol Genet. 2004;13:703–714. doi: 10.1093/hmg/ddh083. [DOI] [PubMed] [Google Scholar]
- 111.Sahara N, Murayama M, Mizoroki T, Urushitani M, Imai Y, Takahashi R, et al. In vivo evidence of CHIP up-regulation attenuating tau aggregation. J Neurochem. 2005;94:1254–1263. doi: 10.1111/j.1471-4159.2005.03272.x. [DOI] [PubMed] [Google Scholar]
- 112.Nakashima H, Ishihara T, Suguimoto P, Yokota O, Oshima E, Kugo A, et al. Chronic lithium treatment decreases tau lesions by promoting ubiquitination in a mouse model of tauopathies. Acta Neuropathol. 2005;110:547–556. doi: 10.1007/s00401-005-1087-4. [DOI] [PubMed] [Google Scholar]
- 113.Dickey CA, Koren J, Zhang YJ, Xu YF, Jinwal UK, Birnbaum MJ, et al. Akt and CHIP coregulate tau degradation through coordinated interactions. Proc Natl Acad Sci USA. 2008;105:3622–3627. doi: 10.1073/pnas.0709180105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bhaskar K, Yen SH, Lee G. Disease-related modifications in tau affect the interaction between Fyn and Tau. J Biol Chem. 2005;280:35119–35125. doi: 10.1074/jbc.M505895200. [DOI] [PubMed] [Google Scholar]
- 115.Yoshida M. Cellular tau pathology and immunohistochemical study of tau isoforms in sporadic tauopathies. Neuropathology. 2006;26:457–470. doi: 10.1111/j.1440-1789.2006.00743.x. [DOI] [PubMed] [Google Scholar]
- 116.Oyama F, Cairns NJ, Shimada H, Oyama R, Titani K, Ihara Y. Down's syndrome: up-regulation of beta-amyloid protein precursor and tau mRNAs and their defective coordination. J Neurochem. 1994;62:1062–1066. doi: 10.1046/j.1471-4159.1994.62031062.x. [DOI] [PubMed] [Google Scholar]
- 117.Shi J, Zhang T, Zhou C, Chohan MO, Gu X, Wegiel J, et al. Increased dosage of Dyrk1A alters alternative splicing factor (ASF)-regulated alternative splicing of tau in Down syndrome. J Biol Chem. 2008;283:28660–28669. doi: 10.1074/jbc.M802645200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sengupta A, Grundke-Iqbal I, Iqbal K. Regulation of phosphorylation of tau by protein kinases in rat brain. Neurochem Res. 2006;31:1473–1480. doi: 10.1007/s11064-006-9205-9. [DOI] [PubMed] [Google Scholar]