

Abstract
Objective
Variations in cytochrome P450 (CYP) genes have been shown to be associated with both accelerated and delayed pharmacokinetic clearance of many psychotropic medications. Citalopram is metabolized by three CYP enzymes. CYP2C19 and CYP3A4 play a primary role in citalopram metabolism, whereas CYP2D6 plays a secondary role.
Methods
The STAR*D sample was used to examine the relationships between variations in the CYP2C19 and CYP2D6 genes and remission of depressive symptoms as well as tolerance to treatment with citalopram. The primary analyses were of the white non-Hispanic subjects adherent to the study protocol (n=1,074).
Results
Generally, subjects who had CYP2C19 genotypes associated with decreased metabolism were less likely to tolerate citalopram than those with increased metabolism, although this difference was not statistically significant (p=0.06). However, subjects with the inactive 2C19*2 allele had significantly lower odds of tolerance (p=0.02). Patients with the poor metabolism CYP2C19 genotype based category who were classified as citalopram tolerant were more likely to experience remission (p=0.03). No relationship between 2D6 genotype based categories and either remission or tolerance was identified, although exploratory analyses identified a potential interaction between CYP2C19 and CYP2D6 effects.
Conclusions
Despite several limitations including the lack of serum drug levels, this study demonstrated that variations in CYP2C19 were associated with tolerance and remission in a large sample of white non-Hispanic patients treated with citalopram.
Keywords: pharmacogenomics, cytochrome P450 2C19, cytochrome P450 2D6, citalopram, remission, tolerance
Introduction
Antidepressant medications are widely prescribed [1]. However, meta-analyses of outcome studies of antidepressant treatment have concluded that fewer than 50% of depressed subjects treated in clinical trials using selective serotonin reuptake inhibitors experience a complete remission of their symptoms [2]. Pharmacogenomic testing to determine metabolic capacity is a novel strategy to identify individuals who may be better able to tolerate medications as well as those who are more likely to achieve a therapeutic response [3].
Citalopram is a chiral drug composed of an S-enantiomer and an R-enantiomer. The S-enantiomer is the pharmacologically active component and is available in a pure form that is referred to as escitalopram. Citalopram is a highly selective serotonin reuptake inhibitor metabolized by the cytochrome P450 2C19 enzyme and the cytochrome P450 3A4 enzyme. The cytochrome P450 2D6 enzyme also plays a secondary role in metabolizing citalopram [4; 5]. An early study described the roles of the cytochrome P450 2C19 enzyme and cytochrome P450 2D6 enzyme on citalopram metabolism [6]. Subsequent reports have demonstrated the involvement of cytochrome P450 3A4 enzyme [7; 8]. Quite recently, the homozygous CYP2C19*17 genotype has been demonstrated to be associated with somewhat increased escitalopram metabolism [9; 10]. Previous dose adjustments estimated for poor 2C19 metabolizers have suggested using 61% of the standard dose of citalopram, whereas minimal downward dose adjustment has been suggested for poor 2D6 metabolizers [11].
“The Sequenced Treatment Alternatives to Relieve Depression” (STAR*D) study is a multisite clinical trial designed to determine the effectiveness of a sequential pharmacological treatment approach for patients with major depressive disorder. Pharmacogenomic studies of remission and response following citalopram treatment based on subjects in the STAR*D Level 1 sample included a genome-wide association analysis [12] and analyses of candidate genes expected to influence the pharmacokinetics and pharmacodynamics of citalopram [13-17]. Initial analysis of the relationship between CYP2C19 or CYP2D6 genotypes and treatment response or tolerance to citalopram did not detect an association between variants of either gene and these primary outcome variables [17]. As per a recent discussion of levels of evidence by Swen and colleagues, this report presents an independent analysis of the relationships between genotype based categories derived from genotyping the CYP2C19 and CPP2D6 genes and the clinical endpoints of drug tolerance and remission of depressive symptoms in the white non-Hispanic (WNH) subjects who were systematically treated with citalopram in the STAR*D sample [18].
Methods and Materials
Study Design and Participants
The rationale and design of the STAR*D study have been previously described [19-21]. All participants provided written informed consent. Subjects were between ages 18 and 75 and met DSM-IV criteria for non-psychotic major depressive disorder. Symptomatic status was measured using the 16-item Quick Inventory of Depressive Symptomatology—Clinician Rating (QIDS-C16), which rates all nine criterion symptom domains of major depressive disorder [22-24]. Our analyses of the STAR*D study data were approved by the Mayo Clinic Institutional Review Board.
Three standardized outcomes were utilized in previous pharmacogenomics analyses of the STAR*D trial: (1) remission of depression, (2) response to treatment, and (3) tolerance of citalopram treatment. Definitions for all three outcomes were established in advance of these analyses. The primary analyses presented in this report focus on tolerance and remission. Tolerance was defined based on study exit data, as described previously [15]. Patients who continued citalopram treatment after the completion of Level 1 of the STAR*D trial were considered tolerant. In contrast, patients who reported side effects and subsequently either switched to another medication or left the study were considered intolerant. Patients who did not indicate side effects at the exit time point, but chose to switch to another medication were classified as probably tolerant or probably intolerant based on their last Global Rating of Side Effect Burden score. Remission was defined as a score of ≤5 on the QIDS-C16 scale at the last clinic visit [22-24].
Blood samples were collected from 1,953 of the 4,041 subjects enrolled in the STAR*D study. Documented differences between subjects who contributed DNA and those for whom DNA was not obtained have been reported [16]. Subjects who provided DNA were more likely to be white, married, older, more educated, and treated in primary care clinics. These subjects also had more depressive episodes with a longer duration of illness. 1,926 DNA samples were genotyped at the Mayo Clinic. After removing twelve subjects because of missing or discrepant genetic data, the valid genotyped sample consisted of 1,914 subjects, as shown by the Consolidated Standards of Reporting Trials (CONSORT) diagram in Figure 1.
Figure 1.
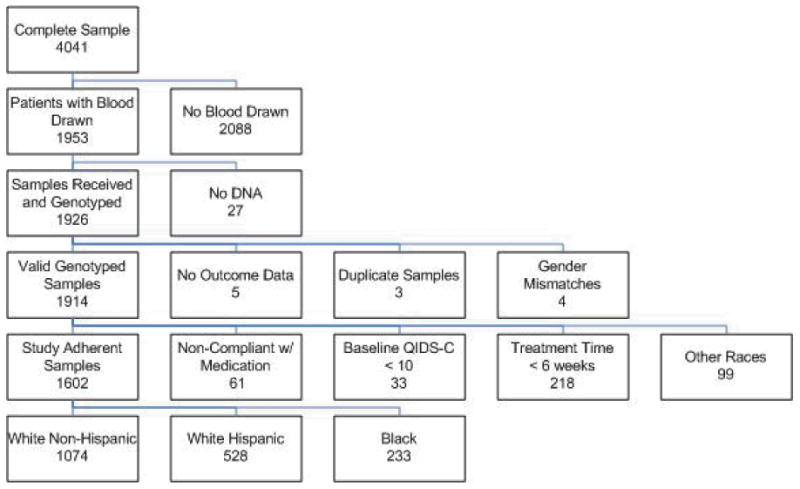
CONSORT Diagram of Study Sample. Initial samples dropped from the genotype analysis included (1) five subjects missing clinical data, (2) three subjects who were duplicates, and (3) four subjects' whose DNA results did not match reported gender. To define the “remission analysis sample,” 411 additional subjects were excluded because (1) 61 were medication non-compliant, (2) 33 had a baseline QIDS-C16 <10, (3) 218 dropped out within the first six weeks, and (4) 99 were part of an ethnically diverse group. From reference [16] with permission.
Inclusion and exclusion criteria for the primary analysis were similar to those used in previous publications [14-16]. Specifically, 312 subjects were excluded because they did not meet the criteria necessary to assess remission (33 did not have sufficient depressive symptoms at baseline to meet the severity criteria for inclusion in the study; 218 discontinued citalopram treatment within the first six weeks and thus did not have an adequate trial of citalopram; 61 were determined to be non-compliant with taking the medication based on a global rating of compliance). Additionally, 99 ethnically diverse subjects consisting of Asian, multi-racial, Pacific Islander, and unspecified racial subgroups were excluded from these genetic association analyses. The remaining study adherent sample (n = 1,503) was analyzed in ethnically homogeneous categories based on subject self-report: white non-Hispanic (n=1074), white Hispanic (n=196), and black (n=233). Sensitivity analyses that included the non-compliant subjects and those that discontinued treatment prior to 6 weeks were also completed.
Study sample demographic characteristics have been previously described [16]. Comparisons revealed significant differences between ethnic groups in terms of age, education, household income, gender, employment status, medical insurance, and marital status. Demographic characteristics of the 1,503 subjects in the study adherent sample and the 411 subjects excluded from the primary analysis have also been compared [16]. The excluded subjects had a significantly lower number of years of education (p < 0.0001), lower household income (p = 0.0004), and were more likely to be unemployed (p = 0.005).
Genotyping
CYP2C19 genotyping (other than *17) was performed on genomic DNA extracted from whole blood using the xTAG Assay for P450-2C19 v2, which incorporates multiplex polymerase chain reaction (PCR) and multiplex allele-specific primer extension (ASPE) with Luminex Molecular Diagnostics' proprietary Universal Tag sorting system on the Luminex 100 xMAP platform. Direct polymorphism analysis for 681G>A (*2), 636G>A (*3), 1A>G (*4), 1297C>T (*5), 395G>A (*6), IVS5+2T>A (*7), and 358T>C (*8) was performed for CYP2C19. The CYP2C19 assay has been validated by comparison with DNA sequencing (about 200 samples in a different study) to assess accuracy.
Similarly, CYP2D6 genotyping (other than *41) was performed on genomic DNA extracted from whole blood using the xTAG assay for CYP2D6, which incorporates multiplex PCR and multiplex ASPE with Luminex Molecular Diagnostics' proprietary Universal Tag sorting system on the Luminex 100 xMAP platform. Direct polymorphism analysis for -1584C>G (*2A), 100C>T (*4, *10), 124G>A (*12), 883G>C (*11), 1023C>T (*17), 1707T>del (*6), 1758G>T (*8/*14), 1846G>A (*4), 2549A>del (*3), 2613delAGA (*9), 2850C>T (*2/*17), 2935A>C (*7), gene deletion, and tandem gene duplication was performed for CYP2D6 following PCR amplification. Polymorphisms were detected by multiplex allele-specific primer extension. This methodology has been validated for clinical use in the Department of Laboratory Medicine at the Mayo Clinic (data now shown). In addition, the duplicated allele was determined [19] using PCR amplification of each gene present followed by genotyping using the xTAG Kit for CYP2D6 as described in Kramer and colleagues [25]. In earlier studies, the CYP2D6 assay was verified for accuracy by concordance with sequencing of each allele.
Genotyping for CYP2C19 *17 (rs11188072 and rs12248560) and CYP2D6 *41 (rs28371725) was performed as part of a custom 96 plex Illumina VeraCode panel according to the standard operating procedure, in accordance with the manufacturer's protocol [26]. The concordance rate for *17 calls based on rs11188072 and rs12248560 was 100%. For quality control, a CEPH family trio (parent, parent, child) was included in duplicate within each 96 well plate. In addition, two samples per plate were duplicated (blinded to the genotyping laboratory). There was complete concordance for genotypes in the control CEPH DNA samples.
Variant star (*) alleles were identified using a commercial genotyping assay protocol and proprietary software (now Luminex Molecular Diagnostics). Naming of these various alleles followed the format developed by the Cytochrome P450 Allele Nomenclature Committee (www.cypalleles.ki.se/).
Statistical Analysis
All association analyses were performed within subgroups based on self-reported race/ethnicity with a primary emphasis on the WNH group. Prior to analysis of treatment outcomes, chi-square tests of Hardy-Weinberg Equilibrium were performed for all genetic variants within the ethnically homogeneous sub-groups. Associations of CYP2C19 and CYP2D6 genotype based categories with tolerance and remission rates were assessed in each of the ethnic groups using logistic regression. Logistic regression models were also used to assess the effects of common CYP2C19 single nucleotide polymorphisms (SNPs) on tolerance and remission. For the SNP analyses, log-additive allele effects were assessed by coding the genotypes based on the number of copies of the minor allele as 0, 1, or 2. As tolerance is a potentially important contributor to remission, analyses of the remission outcome were repeated with tolerance included as a covariate (i.e., adjusted for tolerance), as well as within the tolerant subset of patients. For the analysis of the tolerance outcome, the small groups of “probably intolerant” and “probably tolerant” individuals were excluded, resulting in a dichotomous outcome variable. The probability of tolerance (among tolerant or intolerant patients) was then modeled in terms of genetic predictors using logistic regression. This approach was previously used for other STAR*D analyses [17]. For each predictor variable in the logistic regression analyses, the p-value, odds ratio (OR), and the corresponding 95% confidence interval (CI) were calculated.
For each of the two investigated genes, the combination of SNP variants observed for each individual was used to determine their two haplotypes, which are often referred to as alleles in the literature, using the standard nomenclature. For the primary pharmacogenomic analyses, individuals were assigned to genotype based categories on the basis of the metabolic activity of their two alleles. The genotype based categories were derived as a quantitative measure, by adding the activity scores for the two alleles as has been described [3]. The activity scores are based on expected metabolic activity of the alleles (Table 1). For example, for CYP2C19, for a *1/*2 genotype the activity score would be 1 (the activity score of the *1 allele + the activity score of the *2 allele = 1 + 0 = 1). The quantitative scores were then labeled using the traditionally used categories: poor metabolizer (PM), intermediate metabolizer (IM), extensive metabolizer (EM) and ultra-rapid metabolizer (URM). Because our classification system takes the increased activity of the *17 allele into account [10], there are 6 CYP2C19 genotype activity levels. We therefore utilize the additional categories IM+ (intermediate to IM and EM) and EM+ (intermediate to EM and URM) and thus achieve a more precise grouping. The genotype based categories for CYP2D6 were PM, IM, EM, URM. Alleles were assigned scores based on their level of activity (Table 1). The scores of duplicated active alleles were doubled. The total score for the combined alleles was then used to assign a genotype based category to each subject that quantifies their CYP2D6 metabolic capacity. Subjects with a total score of ≤0.5 were classified as a PM, those with a total score of 1.0 or 1.5 as IM, those with a total score of 2.0 or 2.5 as EM, and those with a total score of ≥3.0 as UM. For example, a subject with a CYP2D6 *4/*9 would receive a score of 0.5 (0 for *4 and 0.5 for *9), placing her in the PM genotype based category group. In several subjects the duplicated allele was unknown. These subjects were excluded from analyses of CYP2D6. The genotype based categories were used as either a categorical or quantitative predictor variable in logistic regression models for tolerance and remission outcomes.
Table 1.
CYP2C19 and CYP2D6 allele activity scores.
| Activity | Score | CYP2D6 Alleles | CYP2C19 Alleles |
|---|---|---|---|
| None | 0 | *3, *4, *5, *6, *7 | *2, *3, *4, *5, *6, *8 |
| Reduced | 0.5 | *2 (not 2A), *9, *10, *17, *41 | |
| Normal | 1 | *1 | *1 |
| Increased | 1.5 | *2A | *17 |
As in previous STAR*D pharmacogenomics analyses, subjects who discontinued treatment prior to the sixth week, or who were non-compliant, were excluded from the primary analysis as they had not been treated for a sufficient time to be able to determine whether they would have ultimately achieved remission. The exclusion of subjects who drop out early may introduce bias if early drop out is not random and is related to the predictor of interest and the study outcome. Therefore, sensitivity analyses that included the non-compliant subjects and those that discontinued treatment prior to 6 weeks were also completed.
Results
Allelic and Genotype Distributions
There were no significant deviations from Hardy Weinberg Equilibrium for any SNP in any of the ethnic subgroups. The CYP2C19 and CYP2D6 allele distributions of all three ethnic subgroups are summarized in Supplemental Table 1, Supplemental Digital Content 1, http://links.lww.com/FPC/A215. The distributions of the resulting genotype based categories are shown in Supplemental Table 2, Supplemental Digital Content 2, http://links.lww.com/FPC/A216. The data in Supplemental Table 1, Supplemental Digital Content 1, http://links.lww.com/FPC/A215 demonstrate that frequencies of certain alleles vary considerably between ethnic groups. For example, the *17 allele of CYP2D6 is rare in the WNH (<1%) and white Hispanic (2%) samples, while being relatively common (14%) in the black sample. Because the outcome and genotype distributions differ across ethnic subgroups, analyses for these groups were performed separately, with a primary focus on the large white non-Hispanic subset.
Association of CYP Genes with Tolerance
Among the entire white non-Hispanic sample (n=1,235), 72.6% (n=896) were determined to be tolerant and 12.0% (n=148) intolerant. Additionally, 2.3% (n=29) were rated as “probably intolerant” and 13.1% (n=162) were “probably tolerant.” In the study adherent white non-Hispanic sample (N=1,074), the proportions of subjects who were tolerant, probably tolerant, probably intolerant, and intolerant were 81.2% (n=872), 10.5% (n=113), 0.8% (n=9), and 7.5% (n=80) respectively. For analyses of tolerance, subjects who had been classified as either “probably intolerant” or “probably tolerant” were excluded, and “tolerant” subjects were compared with “intolerant” subjects.
Analysis of the tolerance outcome, with a quantitative classification of CYP2C19 activity as the predictor (1=PM, 2=IM, 3=IM+, 4=EM, 5=EM+, 6=URM), revealed a trend of association of higher CYP2C19 activity with increased odds of tolerance (p=0.06, OR=1.20, CI=(1.00-1.44)). Table 2 shows the results of logistic regression analyses that model the odds of tolerance in terms of log-additive effects of CYP2C19 alleles for WNH subjects. As the low numbers of the rare non-functional *4, *6, and *8 alleles were inadequate to study their effects individually, all the inactive alleles were considered together as a single allelic group in one analysis. The results suggest that in the WNH population, the inactive *2 allele of CYP2C19 is associated with lower odds of tolerance (p = 0.02, OR = 0.60, 95%CI=(0.39-0.91)). When the inactive alleles were grouped into a single category and the additive effects of “inactive alleles” were evaluated, a trend of association of inactive alleles with lower tolerance was observed (p=0.06, OR=0.66, 95% CI=(0.43-1.00)).
Table 2.
Association of CYP2C19 alleles with tolerance for white non-Hispanic subjects (n=872 tolerant, n=80 intolerant subjects). Reported results are based on logistic regression models comparing tolerant versus intolerant subjects. Analyses assume log-additive allele effects on tolerance.
| Predictor | OR (95% CI) | P |
|---|---|---|
| *2 | 0.60 (0.39,0.91) | 0.02 |
| *17 | 1.26 (0.84,1.91) | 0.26 |
| *1 | 1.08 (0.77,1.52) | 0.64 |
| Inactive (*2/*4/*6/*8) | 0.66 (0.43,1.00) | 0.06 |
There was no significant evidence of association of CYP2C19 genotypes or genotype based categories with tolerance in the white Hispanic or black subject groups. However, in both of these ethnic groups, odds ratio point estimates indicated the same direction of effects as those observed in the WNH subset, with higher activity alleles showing trends of higher tolerance (results not shown).
Sensitivity analysis based on all tolerant and intolerant WNH subjects with baseline QIDS ≥10, regardless of length of time in study or compliance, showed similar results. Again, higher activity 2C19 genotype based categories were associated with higher rates of tolerance at the level of a trend (p=0.06), and the inactive *2 allele was significantly associated with lower odds of tolerance (p=0.02, OR=0.66, 95%CI=(0.47,0.92)).
CYP2D6 genotype based categories were not found to be significantly associated with tolerance (results not shown). An exploratory analysis did not reveal significant interaction effects of CYP2C19 and CYP2D6 genotype categories on tolerance (results not shown).
Association of CYP Genes with Remission
The overall remission rate in the study adherent WNH sample was 48.5% (521/1074). Analysis of the six-category CYP2C19 genotype based groups did not show overall evidence of association with remission in the WNH sample (p=0.19). However, a trend of association of remission with the six-category CYP2C19 genotype based groups emerged when the analysis was adjusted for tolerance by including the tolerance variable as a covariate in the model (p=0.06). Moreover, the CYP2C19 genotype category variable was associated with remission when the analysis was restricted to the 872 tolerant WNH subjects (p=0.03). Figure 2 demonstrates that WNH patients who were poor metabolizers and were classified as tolerant achieved an 87% rate of remission.
Figure 2.

Percentage of tolerant white non-Hispanic subjects who remitted shown by CYP2C19 activity genotype based category. Bars represent 95% CI. The overall remission rate in the study adherent tolerant white non-Hispanic subjects was 57.8% (405/872).
Analyses of the association of CYP2C19 alleles with remission in the WNH subset are presented in Table 3. In the WNH study adherent sample, there was no significant evidence of association of common CYP2C19 alleles with remission. However, there was a trend suggesting that the number of *17 alleles is associated with lower remission rates (p=0.1, OR=0.84, CI=(0.69, 1.04)). Since tolerance was strongly associated with remission (p<0.0001), analyses of the remission outcome were repeated with tolerance included as a covariate (i.e., adjusted for tolerance), as well as within the tolerant subset of patients. This trend became more evident when the analysis was adjusted for the effects of tolerance (p=0.07, OR=0.81, CI=(0.65, 1.01)) or was restricted to patients that tolerated the medication (p=0.05, OR=0.80, CI=(0.63, 1,00)).
Table 3.
Association of CYP2C19 alleles with remission for white non-Hispanic subjects. Reported results are based on logistic regression models comparing remitters versus non-remitters. Analyses assume log-additive allele effects on remission.
| Predictor | Unadjusted (N=1074) | Adjusted for tolerancea (N=1074) | Tolerant subjects only (N=872) | |||
|---|---|---|---|---|---|---|
| Odds Ratio (CI) | P | Odds Ratio (CI) | P | Odds Ratio (CI) | P | |
| *2 | 0.90 (0.70,1.16) | 0.40 | 1.00 (0.76,1.31) | 0.98 | 0.99 (0.74,1.33) | 0.95 |
| *17 | 0.84 (0.69,1.04) | 0.11 | 0.81 (0.65,1.01) | 0.07 | 0.80 (0.63,1.00) | 0.05 |
| *1 | 1.17 (0.98,1.41) | 0.08 | 1.16 (0.96,1.41) | 0.13 | 1.19 (0.97,1.45) | 0.10 |
| Inactive (*2/*4/*6/*8) | 0.94 (0.74,1.20) | 0.64 | 1.02 (0.78,1.33) | 0.87 | 1.01 (0.77,1.35) | 0.92 |
Analysis adjusted for tolerance was performed by including an ordinal tolerance variable as a covariate in the model.
Analyses of CYP2C19 genotype based categories revealed a trend of higher remission rates in poor metabolizers in the subset of tolerant patients. Poor metabolizers are carriers of two inactive alleles. In the logistic regression analyses of either all inactive alleles or only *2 alleles, an association of inactive alleles with higher odds of remission was not observed (Table 3). In these analyses log-additive allele effects had been assumed. Consequently, the effect of carrying two inactive alleles was not tested. A post-hoc analysis of the association of inactive alleles (*2,*4, *6, and *8) with remission, under the assumption of a recessive model, demonstrated that among tolerant subjects, carriers of two inactive 2C19 alleles (i.e., 2C19 poor metabolizers) have higher odds of remission (Fisher's exact test p=0.03).
Association analyses of remission and CYP2C19 using all WNH subjects, including those that dropped out of the study before the sixth week, provided similar results to those described above for the study adherent sample. Again, among tolerant subjects, the *17 allele was associated with lower odds of remission (p=0.04), and the six phenotypic 2C19 categories showed overall association with remission (p=0.03).
CYP2D6 genotype based categories were not found to be significantly associated with remission (results not shown).
Combined CYP2C19 and CYP2D6 Variability and Remission
An exploratory analysis of the association between remission and the combined effects of CYP2C19 and CYP2D6 in the WNH study adherent sample did not detect a significant interaction of the two genes. However, when this analysis was performed for all WNH subjects, including those that dropped out of the study prior to the sixth week, a significant interaction of the genotype based categories representing activity levels of the two genes was detected (p=0.04). This logistic regression analysis tested for the effects of the quantitative activity score of CYP2C19 (ranging from 1=PM to 6=URM), the quantitative activity score of CYP2D6 (ranging from 1=PM to 4=URM), and their interaction.
Having detected evidence of interaction effects of the two genes on remission, as part of a post-hoc analysis, a “combined CYP gene” variable was created (Figure 3). The intent of this categorization was to create a variable that summarizes the overall CYP2C19 and CYP2D6 enzyme activity level based on the genotypes of both genes. Chi-square analyses of this CYP-combination variable revealed an association of this variable with remission in the study adherent WNH sample (p=0.025) as well as the entire WNH sample (p=0.013). The results demonstrate that remission rates are highest among patients with intermediate levels of enzyme activity, whereas both low and high levels of metabolic capacity appear to be associated with reduced rates of remission (Figure 4). These post-hoc analyses were exploratory and must be interpreted cautiously. The model proposed here needs to be tested in independent samples and further studies are needed to refine this model and determine the optimal method of combining CYP2C19 and CYP2D6 genotypes to predict outcomes of treatment with specific SSRIs.
Figure 3.

Methodology for defining combined CYP genotype based categories.
Subjects with reduced activity genotype based categories for both enzymes were classified as having low combined activity. The group defined as having moderate combined activity consists of subjects with an extensive genotype based category for one enzyme paired with a reduced or extensive genotype based category for the other enzyme. Subjects with at least one ultra rapid genotype based category were classified into the high activity combined CYP genotype based category group.
Figure 4.

White non-Hispanic remission rates for combined CYP genotype based categories (p = 0.01).
Discussion
These results indicate that there is an association between the CYP2C19 *2 inactive allele and tolerance of citalopram, as well as a modest association between CYP2C19 variation and remission following citalopram treatment, particularly in a subset of patients able to tolerate the medication. As expected, the inactive *2 allele of CYP2C19 was associated with lower odds of tolerance, and there was an overall trend of genotypes that correspond to higher CYP2C19 enzyme activity to be associated with higher tolerance. A small previously published study reported that patients who were poor metabolizers of 2C19 substrates were able to tolerate a 60 mg dose of citalopram [27].
There was also a trend of association of lower remission with the increased activity *17 allele, which was statistically significant in the subset of medication tolerant subjects. In the subset of subjects who were found to be tolerant of citalopram treatment, remission was highest for subjects in the “poor metabolizer” CYP2C19 activity category.
It is expected that activity of relevant drug metabolizing enzymes may impact remission rates via two mechanisms. Poor metabolizers may achieve high drug serum levels, which in some patients may be associated with lower drug tolerance. In such situations, drug dose may be reduced or treatment may be discontinued, reducing chances of remission. On the other hand, poor metabolizers who are able to tolerate the medication may have favorable treatment outcomes. In contrast, ultra-rapid metabolizers may not achieve adequate drug serum levels with standard dosing, which may also lead to lower remission rates, although via a different causal pathway than the reduced remission rates seen in poor metabolizers. Thus, in order to gain insights into such possibilities, we evaluated evidence of association of genotypes with remission without, as well as with, adjustment for the effects of tolerance on remission, as well as in the subset of subjects that tolerated citalopram treatment. The results of these analyses do in fact support the prediction that poor metabolizers have overall lower tolerance, however, poor metabolizers may also have a greater probability of achieving remission if they are able to tolerate citalopram.
The effects of other potentially important covariates (results not shown) were considered. Age was not significantly associated with tolerance or remission, thus the effects of age in the analyses of these outcomes were not assessed. Gender was not significantly associated with remission; however, it was associated with tolerance, with men being more frequently classified as intolerant than women. When gender was included as a covariate in the analysis of tolerance, results for the gene-tolerance association did not change substantially. Finally, the association of drug dose with both genotypes and treatment outcomes was evaluated. In the study adherent sample, there was no significant association between final dose and genotypes. However, in the entire WNH sample there was a trend of an association between final drug dose and the quantitative CYP2C19 genotype based enzyme activity categories, with median final dose in the PM, IM and IM+ and EM groups being 40, while in the EM+ group median final dose was 50 and in the URM group it was 55 (Spearman rho=0.06, p=0.04). Therefore, dose was considered as a covariate in the analysis of association between CYP2C19 genotype based categories and both tolerance and remission. Results of these analyses did not differ substantially from the unadjusted analyses, although the p-values were slightly higher.
Peters and others [17] analyzed data from the STAR*D study and did not find evidence of association between variations in CYP2C19 or CYP2D6 and remission of depression after treatment with citalopram. In our analyses, modest associations between CYP genotype and remission were detected. There are a number of differences between the two analyses, including different outcome definitions in the remission analyses and different groupings of genotypes for analyses. Peters and colleagues compared remitters (final QIDS-SR≤5) versus non-responders (<50% reduction in symptom severity), whereas our analysis compared remitters (final QIDS-C≤5) versus non-remitters (final QIDS-SR>5). For the predictor variables describing CYP enzyme activity genotype based category, Peters and associates considered poor metabolizers versus all other subjects, whereas we considered more categories, such as intermediate, extensive, and ultrarapid metabolizers. Most importantly, we analyzed data from all eligible subjects simultaneously, whereas Peters and coworkers performed a two-stage analysis by splitting the entire cohort into discovery and validation sets. Although this strategy allows for internal replication of results, it also leads to a reduction in power. Such differences in the analyzed sample sets and analytic approaches can lead to different estimates of effects and statistical significance. Given the modest effect sizes for the genetic effects detected in our analyses, it is not surprising that a different approach with less power could lead to failure to detect an effect and different conclusions.
In examining differences between our analyses and the analyses of Peters and colleagues [17], the distributions of the identified alleles were similar for the CYP2C19 gene, although a small number of inactive alleles were not identified by Peters and associates. In contrast, the allelic distribution of CYP2D6 was quite different. Peters and colleagues did not test for as many SNPs and did not test for duplications of active alleles (Supplemental Table 3, Supplemental Digital Content 3, http://links.lww.com/FPC/A217).
Because of the potential of false-positive associations in genetic studies of ethnically mixed populations, ancestral groups were analyzed separately. It has previously been reported that subjects of European origin generally have better outcomes when treated with selective serotonin reuptake inhibitors (SSRI) when compared to subjects from other ancestral origins [28]. This finding is replicated in the STAR*D cohort [24]. Because of the differences in allele frequencies and remission rates between groups with different geographic ancestry, separation of the sample into more homogenous ancestral subgroups is important to avoid potentially spurious association findings as a result of population stratification. Previous studies that included groups of African Americans, European Americans, and Hispanics have shown that self-reported ethnicity corresponds closely to genetic marker based classifications and that self-reported ethnicity can play an important role in controlling for population stratification [29; 30]. Consequently, analyses of self-report based ethnically homogeneous sub-groups were conducted with a primary focus on the WNH sub-sample.
These findings should be interpreted cautiously given the sample selection criteria. Specifically, the sample may have an unspecified shift toward patients who may be more likely to respond to citalopram treatment because some patients who had been unsuccessfully treated with citalopram in the past were excluded. Also, the main analyses in this paper were based on a subset of patients who completed at least six weeks in the study and for whom there was no evidence of non-compliance. Because of the study exclusion criteria and the exclusion of subjects from analysis, absolute remission rates in the analyzed subset are likely to be higher than could be achieved in the general population of patients with major depressive disorder. However, this sampling bias should not significantly bias measures of genotype-remission association unless the probability of early drop-out is related to remission and the genotype itself is related to early drop-out. To test the robustness of our findings, all analyses were repeated using data from all WNH subjects who initiated citalopram treatment and were available for genotyping, leading to similar results and conclusions.
One limitation of this study relates to the fact that only about half of the entire STAR*D full patient cohort was genotyped. Although the subset of patients who were genotyped were similar to the entire STAR*D cohort in many respects, there were some differences that reached statistical significance. McMahon and associates [15] have discussed these differences and concluded that they are unlikely to affect the genetic association results that are based on the genotyped sample. However, the differences do limit the generalizability of the findings. Other limitations of the STAR*D study include the fact that analyses did not adjust for certain potential confounders such as concomitant medication use and medication compliance. Although concomitant medications were allowed in this study as long as they were not mood stabilizing and were not contraindicated in patients taking citalopram, data on concomitant medications were not available. Serum levels were not obtained from subjects and compliance was not rigorously monitored. However, a global measure of compliance that was available for most subjects was used to exclude non-compliant patients from the primary analyses.
Given the relatively modest detected genetic effects, the results of our study do not suggest changes in the specific guidance of the use of citalopram in patients with adequate CYP2C19 metabolic capacity. However, these results do indicate that testing for deficient or completely inactive alleles could help identify individuals that may have difficulties tolerating citalopram. These findings should encourage further investigation into the role of CYP2C19 as a predictor of outcome of citalopram treatment. Identification of other factors that are associated with treatment response would allow modeling of the joint effects of relevant genomic variation and clinical factors.
Conclusion
Variations in CYP2C19 were associated with tolerance and remission in a large sample of white non-Hispanic patients treated with citalopram. Development of more sophisticated models may ultimately provide more accurate predictions of treatment response and guidance in the selection of the most appropriate treatment of individual patients.
Supplementary Material
Supplemental Table 1. CYP2C19 and CYP2D6 allele frequencies in white non-Hispanic, white Hispanic, and black groups.
Supplemental Table 2. Distributions of CYP2C19 and CYP2D6 genotype based categories in ethnic subgroups.
Supplemental Table 3. CYP2C19 and CYP2D6 allele frequency comparison for white subjects with our genotyping (Mrazek and colleagues) and the Peters analyses.
Acknowledgments
We thank Denise L. Walker and Rajeswari Avula for their technical assistance in performing the genotyping.
Financial Disclosures
The STAR*D study was funded by the National Institute of Mental Health, National Institutes of Health, under Contract N01MH90003 to UT Southwestern Medical Center at Dallas (P.I.: A. J. Rush).
Genetic analyses were funded by an NIH grant U01 GM61388 (“The Pharmacogenomics Research Network”), a PhRMA Foundation “Center of Excellence in Clinical Pharmacology Award” (P.I.: R.M. Weinshilboum), and the Cooper Family Foundation.
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Drs. Mrazek, O'Kane, and Black have an interest in intellectual property that has been licensed from the Mayo Clinic by AssureRx, which is a clinical decision support company.
Dr Rush's disclosures are:
| Financial Relationships | ||
| A. John Rush, M.D. | ||
| (Last 5 Years) | ||
| Commercial Interest | What I Received | My Role |
| Advanced Neuromodulation Systems | Consulting Fee | Consultant |
| AstraZeneca | Consulting Fee | Consultant |
| Best Practice Project Management | Consulting Fee | Consultant |
| Bristol-Myers Squibb/Otsuka | Consulting Fee | Consultant |
| Cyberonics | Consulting Fee/Honoraria | Consultant/Speaker |
| Forest Pharmaceuticals | Consulting Fee/Honoraria | Consultant/Speaker |
| Gerson Lehrman Group | Consulting Fee | Consultant |
| GlaxoSmithKline | Consulting Fee/Honoraria | Consultant/Speaker |
| Guilford Publications | Royalties | Author |
| Healthcare Technology Systems | Royalties | Author |
| Jazz Pharmaceuticals | Consulting Fee | Consultant |
| Magellan Health Services | Consulting Fee | Consultant |
| Merck & Company | Consulting Fee | Consultant |
| Neuronetics | Consulting Fee | Consultant |
| National Institute of Mental Health | Research Support | Researcher |
| Novartis Pharmaceuticals | Consulting Fee | Consultant |
| Ono Pharmaceuticals | Consulting Fee | Consultant |
| Organon | Consulting Fee | Consultant |
| Otsuka Pharmaceuticals | Consulting Fee | Consultant/Speaker |
| Pamlab | Consulting Fee | Consultant |
| Pfizer | Consulting Fee/Income | Consultant/Stock Holder |
| Society of Biological Psychiatry | Stipend | Treasurer |
| Stanley Medical Research Institute | Research Support | Researcher |
| Transcept Pharmaceuticals | Consulting Fee | Consultant |
| Urban Institute | Consulting Fee | Consultant |
| Wyeth Ayerst | Consulting Fee | Consultant |
Drs. Biernacka, Cunningham, and Weinshilboum and Mses. Drews, Snyder, and Stevens reported no biomedical financial interests or potential conflicts of interest.
Contributor Information
David A. Mrazek, Professor of Pediatrics and Psychiatry, Mayo Clinic College of Medicine, Chair, Department of Psychiatry and Psychology, Mayo Clinic, Rochester, MN.
Joanna M. Biernacka, Assistant Professor of Biostatistics and Psychiatry, Mayo Clinic College of Medicine, Division of Biomedical Statistics, Department of Health Sciences Research, Mayo Clinic, Rochester, MN
Dennis J. O'Kane, Associate Professor of Laboratory Medicine and Pathology, Mayo Clinic College of Medicine, Department of Laboratory Medicine and Pathology, Mayo Clinic, Rochester, MN
John L. Black, Professor of Psychiatry, Mayo Clinic College of Medicine, Consultant, Department of Psychiatry and Psychology, Mayo Clinic, Rochester, MN
Julie M. Cunningham, Assistant Professor of Laboratory Medicine and Pathology, Mayo Clinic College of Medicine, Department of Laboratory Medicine and Pathology, Mayo Clinic, Rochester, MN
Maureen S. Drews, Department of Psychiatry and Psychology, Mayo Clinic, Rochester, MN
Karen A. Snyder, Department of Psychiatry and Psychology, Mayo Clinic, Rochester, MN
Susanna R. Stevens, Division of Biomedical Statistics, Department of Health Sciences Research, Mayo Clinic, Rochester, MN
Augustus John Rush, Vice Dean, Clinical Sciences, Duke-NUS Graduate Medical School Singapore, Republic of Singapore
Richard M. Weinshilboum, Professor of Medicine and Pharmacology, Mayo Clinic College of Medicine, Department of Molecular Pharmacology and Experimental Therapeutics, Mayo Clinic, Rochester, MN
References
- 1.Paulose-Ram R, Safran MA, Jonas BS, Gu Q, Orwig D. Trends in psychotropic medication use among U.S. adults. Pharmacoepidemiol Drug Saf. 2007;16:560–570. doi: 10.1002/pds.1367. [DOI] [PubMed] [Google Scholar]
- 2.Thase ME, Haight BR, Richard N, Rockett CB, Mitton M, Modell JG, et al. Remission rates following antidepressant therapy with bupropion or selective serotonin reuptake inhibitors: a meta-analysis of original data from 7 randomized controlled trials. J Clin Psychiatry. 2005;66:974–981. doi: 10.4088/jcp.v66n0803. [DOI] [PubMed] [Google Scholar]
- 3.Mrazek DA. Psychiatric Pharmacogenomics. New York, NY: Oxford University Press; 2010. [Google Scholar]
- 4.Olesen OV, Linnet K. Studies on the stereoselective metabolism of citalopram by human liver microsomes and cDNA-expressed cytochrome P450 enzymes. Pharmacology. 1999;59:298–309. doi: 10.1159/000028333. [DOI] [PubMed] [Google Scholar]
- 5.von Moltke LL, Greenblatt DJ, Grassi JM, Granda BW, Venkatakrishnan K, Duan SX, et al. Citalopram and desmethylcitalopram in vitro: human cytochromes mediating transformation, and cytochrome inhibitory effects. Biol Psychiatry. 1999;46:839–849. doi: 10.1016/s0006-3223(98)00353-9. [DOI] [PubMed] [Google Scholar]
- 6.Sindrup SH, Brosen K, Hansen MGJ, Aaes-Jorgensen T, Overo KF, Gram LF. Pharmacokinetics of Citalopram in Relation to the Sparteine and the Mephenytoin Oxidation Polymorphisms. Ther Drug Monit. 1993;15:11–17. doi: 10.1097/00007691-199302000-00002. [DOI] [PubMed] [Google Scholar]
- 7.Kobayashi K, Chiba K, Yagi T, Shimada N, Taniguchi T, Horie T, et al. Identification of cytochrome P450 isoforms involved in citalopram N-demethylation by human liver microsomes. J Pharmacol Exp Ther. 1997;280:927–933. [PubMed] [Google Scholar]
- 8.Rochat B, Amey M, Gillet M, Meyer UA, Baumann P. Identification of three cytochrome P450 isozymes involved in N-demethylation of citalopram enantiomers in human liver microsomes. Pharmacogenetics. 1997;7:1–10. doi: 10.1097/00008571-199702000-00001. [DOI] [PubMed] [Google Scholar]
- 9.Ohlsson Rosenborg S, Mwinyi J, Andersson M, Baldwin RM, Pedersen RS, Sim SC, et al. Kinetics of omeprazole and escitalopram in relation to the CYP2C19*17 allele in healthy subjects. Eur J Clin Pharmacol. 2008;64:1175–1179. doi: 10.1007/s00228-008-0529-z. [DOI] [PubMed] [Google Scholar]
- 10.Rudberg I, Mohebi B, Hermann M, Refsum H, Molden E. Impact of the ultrarapid CYP2C19*17 allele on serum concentration of escitalopram in psychiatric patients. Clin Pharmacol Ther. 2008;83:322–327. doi: 10.1038/sj.clpt.6100291. [DOI] [PubMed] [Google Scholar]
- 11.Kirchheiner J, Nickchen K, Bauer M, Wong ML, Licinio J, Roots I, et al. Pharmacogenetics of antidepressants and antipsychotics: the contribution of allelic variations to the phenotype of drug response. Mol Psychiatry. 2004;9:442–473. doi: 10.1038/sj.mp.4001494. [DOI] [PubMed] [Google Scholar]
- 12.Garriock HA, Kraft JB, Shyn SI, Peters EJ, Yokoyama JS, Jenkins GD, et al. A Genomewide Association Study of Citalopram Response in Major Depressive Disorder. Biol Psychiatry. 2010;67:99–100. doi: 10.1016/j.biopsych.2009.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu XZ, Rush AJ, Charney D, Wilson AF, Sorant AJ, Papanicolaou GJ, et al. Association between a functional serotonin transporter promoter polymorphism and citalopram treatment in adult outpatients with major depression. Arch Gen Psychiatry. 2007;64:783–792. doi: 10.1001/archpsyc.64.7.783. [DOI] [PubMed] [Google Scholar]
- 14.Kraft JB, Peters EJ, Slager SL, Jenkins GD, Reinalda MS, McGrath PJ, et al. Analysis of association between the serotonin transporter and antidepressant response in a large clinical sample. Biol Psychiatry. 2007;61:734–742. doi: 10.1016/j.biopsych.2006.07.017. [DOI] [PubMed] [Google Scholar]
- 15.McMahon FJ, Buervenich S, Charney D, Lipsky R, Rush AJ, Wilson AF, et al. Variation in the Gene Encoding the Serotonin 2A Receptor Is Associated with Outcome of Antidepressant Treatment. Am J Hum Genet. 2006;78:804–814. doi: 10.1086/503820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mrazek DA, Rush AJ, Biernacka JM, O'Kane DJ, Cunningham JM, Wieben ED, et al. SLC6A4 variation and citalopram response. Am J Med Genet B Neuropsychiatr Genet. 2009;150B:341–351. doi: 10.1002/ajmg.b.30816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peters EJ, Slager SL, Kraft JB, Jenkins GD, Reinalda MS, McGrath PJ, et al. Pharmacokinetic genes do not influence response or tolerance to citalopram in the STAR*D sample. PLoS ONE [Electronic Resource] 2008;3:e1872. doi: 10.1371/journal.pone.0001872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Swen JJ, Wilting I, de Goede AL, Grandia L, Mulder H, Touw DJ, et al. Pharmacogenetics: from bench to byte. Clin Pharmacol Ther. 2008;83:781–787. doi: 10.1038/sj.clpt.6100507. [DOI] [PubMed] [Google Scholar]
- 19.Fava M, Rush AJ, Trivedi MH, Nierenberg AA, Thase ME, Sackeim HA, et al. Background and Rationale for the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) Study. Psychiatr Clin North Am. 2003;26:457–494. doi: 10.1016/s0193-953x(02)00107-7. [DOI] [PubMed] [Google Scholar]
- 20.Rush AJ, Fava M, Wisniewski SR, Lavori PW, Trivedi MH, Sackeim HA, et al. Sequenced treatment alternatives to relieve depression (STAR*D): rationale and design. Control Clin Trials. 2004;25:119–142. doi: 10.1016/s0197-2456(03)00112-0. [DOI] [PubMed] [Google Scholar]
- 21.Trivedi M, Rush A, Wisniewski S, Nierenberg A, Warden D, Ritz L, et al. Evaluation of Outcomes With Citalopram for Depression Using Measurement-Based Care in STAR*D: Implications for Clinical Practice. Am J Psychiatry. 2006;163:28. doi: 10.1176/appi.ajp.163.1.28. [DOI] [PubMed] [Google Scholar]
- 22.Rush AJ, Bernstein IH, Trivedi MH, Carmody TJ, Wisniewski S, Mundt JC, et al. An evaluation of the quick inventory of depressive symptomatology and the Hamilton Rating Scale for Depression: a Sequenced Treatment Alternatives to Relieve Depression trial report. Biol Psychiatry. 2006;59:493–501. doi: 10.1016/j.biopsych.2005.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rush AJ, Trivedi MH, Ibrahim HM, Carmody TJ, Arnow B, Klein DN, et al. The 16-Item Quick Inventory of Depressive Symptomatology (QIDS), clinician rating (QIDS-C), and self-report (QIDS-SR): a psychometric evaluation in patients with chronic major depression. Biol Psychiatry. 2003;54:573–583. doi: 10.1016/s0006-3223(02)01866-8. erratum appears in Biol Psychiatry. 2003 Sep 1;54(5):585. [DOI] [PubMed] [Google Scholar]
- 24.Trivedi MH, Rush AJ, Ibrahim HM, Carmody TJ, Biggs MM, Suppes T, et al. The Inventory of Depressive Symptomatology, Clinician Rating (IDS-C) and Self-Report (IDS-SR), and the Quick Inventory of Depressive Symptomatology, Clinician Rating (QIDS-C) and Self-Report (QIDS-SR) in public sector patients with mood disorders: a psychometric evaluation. Psychol Med. 2004;34:73–82. doi: 10.1017/s0033291703001107. [DOI] [PubMed] [Google Scholar]
- 25.Kramer WE, Walker DL, O'Kane DJ, Mrazek DA, Fisher PK, Dukek BA, et al. CYP2D6: novel genomic structures and alleles. Pharmacogenet Genomics. 2009;19:813–822. doi: 10.1097/FPC.0b013e3283317b95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oliphant A, Barker DL, Stuelpnagel JR, Chee MS. BeadArray technology: enabling an accurate, cost-effective approach to high-throughput genotyping. Biotechniques. 2002;(Suppl):56–58. 60–51. [PubMed] [Google Scholar]
- 27.Baumann P, Nil R, Souche A, Montaldi S, Baettig D, Lambert S, et al. A double-blind, placebo-controlled study of citalopram with and without lithium in the treatment of therapy-resistant depressive patients: a clinical, pharmacokinetic, and pharmacogenetic investigation. J Clin Psychopharmacol. 1996;16:307–314. doi: 10.1097/00004714-199608000-00006. [DOI] [PubMed] [Google Scholar]
- 28.Schraufnagel TJ, Wagner AW, Miranda J, Roy-Byrne PP. Treating minority patients with depression and anxiety: what does the evidence tell us? Gen Hosp Psychiatry. 2006;28:27–36. doi: 10.1016/j.genhosppsych.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 29.Liu XQ, Paterson AD, John EM, Knight JA. The role of self-defined race/ethnicity in population structure control. Ann Hum Genet. 2006;70:496–505. doi: 10.1111/j.1469-1809.2005.00255.x. [DOI] [PubMed] [Google Scholar]
- 30.Tang H, Quertermous T, Rodriguez B, Kardia SL, Zhu X, Brown A, et al. Genetic structure, self-identified race/ethnicity, and confounding in case-control association studies. Am J Hum Genet. 2005;76:268–275. doi: 10.1086/427888. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1. CYP2C19 and CYP2D6 allele frequencies in white non-Hispanic, white Hispanic, and black groups.
Supplemental Table 2. Distributions of CYP2C19 and CYP2D6 genotype based categories in ethnic subgroups.
Supplemental Table 3. CYP2C19 and CYP2D6 allele frequency comparison for white subjects with our genotyping (Mrazek and colleagues) and the Peters analyses.