

Summary
Messenger RNA decay plays a central role in the regulation and surveillance of eukaryotic gene expression. The conserved multi-domain exoribonuclease Xrn1 targets cytoplasmic RNA substrates marked by a 5′ monophosphate for processive 5′-to-3′ degradation by an unknown mechanism. Here we report the crystal structure of an Xrn1-substrate complex. The single-stranded substrate is held in place by stacking of the 5′-terminal trinucleotide between aromatic side chains while a highly basic pocket specifically recognizes the 5′ phosphate. Mutations of residues involved in binding the 5′-terminal nucleotide impair Xrn1 processivity. The substrate recognition mechanism allows Xrn1 to couple processive hydrolysis to duplex melting in RNA substrates with sufficiently long single-stranded 5′ overhangs. The Xrn1-substrate complex structure thus rationalizes the exclusive specificity of Xrn1 for 5′-monophosphorylated substrates, ensuring fidelity of mRNA turnover, and posits a model for translocation-coupled unwinding of structured RNA substrates.
Introduction
During constitutive mRNA turnover in eukaryotic cells, or in response to regulatory pathways such miRNA-mediated silencing, mRNA decay is triggered by a gradual shortening of the mRNA 3′ poly(A) tail by enzymatic deadenylation (Fabian et al., 2010; Garneau et al., 2007). Deadenylated mRNAs are substrates for exosome-catalyzed 3′–5′ degradation (Conti et al., 2009; Lebreton and Seraphin, 2008), or the removal of the 5′ 7-methylguanosine (m7GpppN) cap by the decapping Dcp1/2 complex (Coller and Parker, 2004; Franks and Lykke-Andersen, 2008). Decapping irreversibly commits the mRNA for degradation, as it generates a 5′-monosphorylated RNA intermediate that is recognized and rapidly hydrolyzed by the cytoplasmic 5′-3′ exonuclease Xrn1 (Houseley and Tollervey, 2009; Parker and Song, 2004).
In addition to its role in degrading the products of deadenylation-dependent decapping, Xrn1 also participates in degrading RNA intermediates generated by endonucleolytic mRNA cleavage in quality-control pathways such as nonsense-mediated decay and No-Go decay (Doma and Parker, 2006; Eberle et al., 2009; Gatfield and Izaurralde, 2004; Huntzinger et al., 2008). Furthermore, 5′-monophosphorylated RNAs are generated by Argonaute-catalyzed endonucleolytic cleavage during RNA interference (Liu et al., 2004; Martinez and Tuschl, 2004; Schwarz et al., 2004), and these are likewise degraded by Xrn1 (Orban and Izaurralde, 2005).
Xrn1 is a ∼175 kDa processive exoribonuclease that is conserved from yeast to mammals (Johnson and Kolodner, 1991; Stevens, 2001). Xrn1 activity requires divalent metal ions and generates nucleoside monophosphates as products (Stevens, 1980). The enzyme is highly specific for RNAs bearing a 5′-monophosphate group; the in vitro activity of Xrn1 towards RNA substrates bearing a 5′ triphosphate or an m7GpppN cap is at least two orders of magnitude lower than for RNA substrates carrying a 5′ monophosphate (Pellegrini et al., 2008; Stevens, 2001; Stevens and Poole, 1995). Xrn1 interacts genetically and physically with the Dcp1/2 complex and the decapping activators Lsm1-7 and Pat1, and co-localizes with these proteins to discrete cytoplasmic foci known as P-bodies (Eulalio et al., 2007; Parker and Sheth, 2007; Sheth and Parker, 2003)
A related nuclear exonuclease Xrn2 (known as Rat1 in yeast) is involved in the processive degradation of nuclear RNAs as well as ribosomal RNA maturation, transcriptional termination and quality control of mRNA capping (Fang et al., 2005; Jiao et al., 2010; Kim et al., 2004; West et al., 2004). In yeast, Rat1 exists in a complex with Rai1, a co-activator that stabilizes Rat1 and also possesses pyrophosphatase and decapping activities (Jiao et al., 2010; Xiang et al., 2009). A recent structure of the Rat1-Rai1 complex defined the active center of Xrn-family exonucleases but did not offer further insights into substrate recognition or catalytic mechanism (Xiang et al., 2009).
Both the molecular basis of 5′ monophosphate specificity and the mechanism of processive RNA degradation are critical to the physiological function of Xrn-family enzymes. In Xrn1, these two features ensure that the enzyme targets only mRNAs that have been committed to decay, and that once engaged, the enzyme completely hydrolyzes its substrate without releasing partially degraded intermediates. As the sole cytoplasmic enzyme responsible for removing mRNAs marked for destruction by means of a signature 5′-monophosphate group, Xrn1 plays a central role in all eukaryotic cells in executing the controlled turnover of the mRNA transcriptome. To gain insights into the mechanisms underlying Xrn1-mediated RNA decay, we determined the crystal structure of Drosophila melanogaster Xrn1 (DmXrn1) in complex with a nucleic acid substrate. The DmXrn1-substrate co-crystal structure reveals a conserved binding site that engages the 5′-terminal trinucleotide of a single-stranded substrate and specifically recognizes the 5′ phosphate group. Recognition by Xrn1 of the 5′-terminal nucleotide is intimately coupled to processivity, explaining not only the specificity of Xrn1 but also its catalytic efficiency. Furthermore, this mode of substrate binding allows Xrn1 to combine substrate hydrolysis with duplex unwinding to overcome secondary structures in RNA substrates.
Results and Discussion
Structure Determination of the Xrn1-substrate Complex
To elucidate the molecular mechanism of 5′ phosphate recognition and RNA degradation by Xrn1, we sought to trap DmXrn1 in a complex with a nucleic acid substrate. Xrn enzymes are distantly related to other Mg2+-dependent nucleases, such as the flap endonuclease FEN-1 and bacteriophage T4 RNase H, which utilize a cluster of seven acidic residues to bind two Mg2+ ions in their active sites (Hwang et al., 1998; Mueser et al., 1996). Alanine substitution of the invariant residue Asp207 (Figure S1A) in an otherwise catalytically competent, C-terminally truncated form of DmXrn1 (residues 1-1141, hereafter referred to as DmXrn1ΔC) resulted in loss of activity in vitro (data not shown). The D207A mutation is analogous to the D181A mutation in human FEN-1 that abolishes catalytic activity without compromising substrate binding (Shen et al., 1997). We co-crystallized DmXrn1ΔC-D207A with a 5′-phosphorylated 11-mer DNA oligonucleotide and determined the structure of this complex at 3.2 Å resolution. The final model has been refined to an Rfree of 27.1% and Rwork of 22.5%, and has good stereochemistry (Table 1). The structure represents a bona fide substrate complex, as Xrn1 is capable of degrading single-stranded DNA, albeit more slowly than RNA substrates (Stevens, 2001).
Table 1. Data collection and refinement statistics.
| Native | SeMet SAD | |
|---|---|---|
| Data collection | ||
| X-ray source | ALS 8.2.2 | ALS 8.2.2 |
| Space group | P42212 | P42212 |
| Cell dimensions | ||
| a, b, c (Å) | 150.0, 150.0, 154.9 | 149.7, 149.7, 155.1 |
| α, β, γ (°) | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 |
| Wavelength (Å) | 1.016246 | 0.979023 |
| Resolution (Å)* | 100–3.20 (3.28–3.20) | 100–3.8 (3.9–3.8) |
| Rsym (%)* | 10.1 (83.3) | 17.1 (55.2) |
| I/σI* | 20.0 (2.52) | 19.7 (6.3) |
| Completeness (%)* | 99.1 (95.7) | 99.9 (99.9) |
| Redundancy* | 7.2 (7.0) | 15.4 (15.3) |
| Refinement | ||
| Resolution (Å) | 75.0–3.2 | |
| No. reflections | 29498 | |
| Rwork/ Rfree | 0.225 / 0.271 | |
| No. atoms | ||
| Protein | 8104 | |
| Nucleic Acid | 65 | |
| Ion (Mg2+) | 1 | |
| Water | 17 | |
| B-factors | ||
| Protein | 78.8 | |
| Nucleic Acid | 131.5 | |
| Ion | 69.0 | |
| Water | 57.4 | |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.011 | |
| Bond angles (°) | 1.02 |
Values in parentheses denote highest resolution shell
Xrn1 Adopts a Distinct Domain Architecture
The structure of DmXrn1ΔC reveals a complex domain arrangement that was previously undetectable by sequence analysis (Figure 1A). Overall, the protein adopts a bipartite architecture. The N-terminal globular core of the protein is formed by the catalytic domain (residues 1-674) and a β-barrel domain (residues 674-799) that displays structural similarity to PAZ and Tudor domains in a DALI search (Figure 1B) (Holm and Sander, 1993). The C-terminal region of DmXrn1ΔC arches over the N-terminal core and is composed of three domains (Figure 1A, B). The base of the arch is formed by a β-barrel domain (consisting of two discontinuous stretches of sequence, residues 800-850 and 1031-1045) that aligns most closely with SH3 and KOW (Kyrpides-Ouzonis-Woese) domains. At the apex of the arch, closing over the active site, is a mostly α-helical domain (residues 851-1026) that contains a winged-helix (WH) motif. The extreme C-terminus of DmXrn1ΔC (residues 1046-1140) adopts an SH3-like fold and is located on the concave face of the C-terminal arch. The domain arrangement observed in DmXrn1 is absent from Xrn2 proteins, which only contain the N-terminal catalytic domain (Figure S1B and S1C). In Rat1, a non-conserved C-terminal loop located on the opposite face of the catalytic domain from the active site mediates the interaction with Rai1 (Xiang et al., 2009). In DmXrn1, this region is occupied by a long loop connecting the catalytic and PAZ/Tudor domains.
Figure 1. Domain architecture of Drosophila melanogaster Xrn1 (DmXrn1).
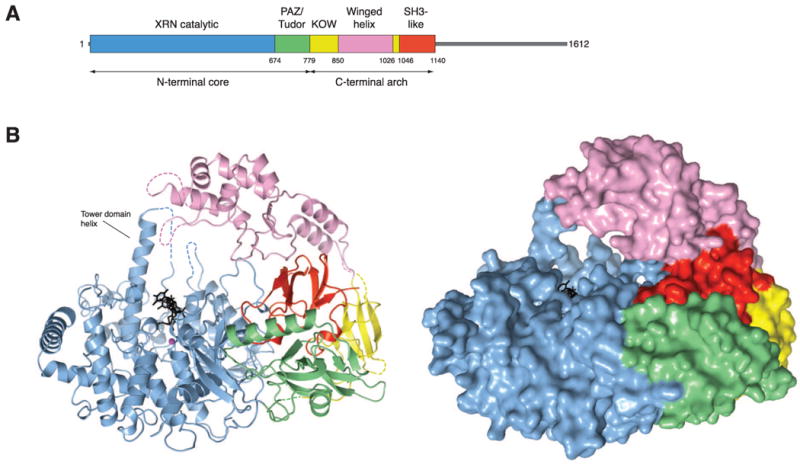
(A) Schematic representation of DmXrn1 domains. The unstructured C-terminal region of DmXrn1 that was excluded from the crystallized construct is depicted in grey. (B) Overall view of DmXrn1ΔC-substrate complex, shown as ribbon (left) and surface (right) representations, with domains colored according to the schematic diagram in A. The nucleic acid substrate is colored black and shown in stick format. A Mg2+ ion is depicted as a purple sphere. Dashed lines indicate loops not resolved in electron density maps. Illustrations were generated using Pymol (Delano Scientific).
The C-terminal SH3-like domain in DmXrn1ΔC lacks the canonical SH3 residues normally involved in binding proline-rich peptide motifs and instead engages in non-canonical interactions with the catalytic domain (Figure S2A). The SH3-like domain is essential in maintaining the structural integrity of Xrn1 since truncation of yeast Xrn1 after residue 1206 (corresponding to residue 1112 in DmXrn1) yields an inactive protein that is unable to rescue the growth defect of an xrn1 null mutant (Page et al., 1998). A long loop projecting from the SH3-like domain contacts the PAZ/Tudor domain, occluding the functional surface that binds RNA or peptide motifs containing methylated arginines, respectively, in canonical PAZ and Tudor domains (Figure S2B). The extensive interface between the PAZ/Tudor and the catalytic domains suggests that the PAZ/Tudor domain plays a primarily structural role in DmXrn1, possibly stabilizing the structure of the catalytic domain, analogous to the function of Rai1 in stabilizing Rat1 (Xiang et al., 2009).
The three dimensional architecture of DmXrn1 places the WH domain in close proximity of the catalytic center. We speculate that the domain might function as a flexible lid, closing over the active site and providing a shielded environment for RNA degradation. Interestingly, the WH domain features a conserved, positively charged surface patch located approximately 30 Å away from the active site that might aid in RNA binding and recruitment (Figure S2C). Alternatively, the WH domain might function in Xrn1-specific modulation of catalytic activity or in mediating protein-protein interactions.
Xrn1 Binds the 5′-terminal Trinucleotide of the Substrate
The catalytic domain of DmXrn1 is composed of two highly conserved regions separated by a variable insertion that is disordered in the DmXrn1ΔC structure (Figure S1A). The domain shares 42% sequence identity with the catalytic domain of Rat1 and superposes with a root mean square deviation (rmsd) of 2.2 Å over 539 Ca atoms (Figure S1B-C). A prominent feature of the domain is a long helix α4, termed the tower domain helix in the Rat1 structure (Xiang et al., 2009), that protrudes from the catalytic domain and contacts the WH domain in DmXrn1 (Figure 1B). The catalytic center of Xrn1 is located at the base of the tower domain helix. Experimentally-phased electron density maps showed clear density in the catalytic site, which we interpret as the first three nucleotides of the substrate, counted from the 5′ end, plus an additional 3′-terminal phosphate group (Figure 2A). The remainder of the 11-mer substrate is disordered, although it is present in the crystals (data not shown). The bases of substrate nucleotides 1–3 form a continuous π-π stack. The stack is capped by the strictly conserved residues His41 and Trp540 at the 5′ and 3′ ends, respectively (Figure 2B). Besides these stacking interactions, there are no other contacts with nucleotide bases, allowing the active site of Xrn1 to accommodate both pyrimidine and purine bases indiscriminately. The phosphate and ribose moieties of the third nucleotide are positioned directly over the amino-terminal helix α1. The 3′-terminal phosphate group is located near the positively charged rim of the active site, in the vicinity of Lys5 and Arg8 (Figure 2B, C). The trajectory of the phosphate backbone cannot be traced beyond the phosphate group of the fourth nucleotide, suggesting that Xrn1 does not make stable interactions with the substrate beyond the fourth nucleotide. Consistent with this, the DmXrn1ΔC-D207A protein protected up to four 5′-terminal nucleotides of a 5′-phosphorylated RNA oligonucleotide substrate in an RNase protection assay (Figure 2D).
Figure 2. Xrn1 binds the N-terminal trinucleotide of the nucleic acid substrate.
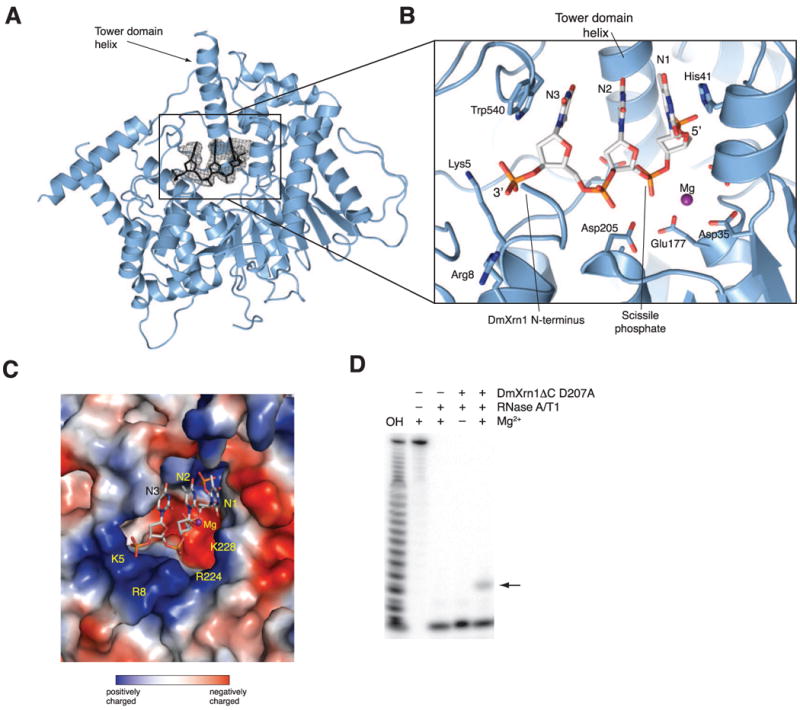
(A) The catalytic domain of DmXrn1 is shown in blue. The nucleic acid substrate is shown in black. The electron density map corresponds to the experimental SAD-phased map obtained after solvent flipping and phase extension to 3.2 Å, and is contoured at 1.0 σ. (B) Close-up view of substrate binding. The oligodeoxythymidine substrate is shown in stick format with carbon atoms colored white. The nucleotides are labeled N1–N3 in the 5′-3′ direction. Strictly conserved residues contacting the substrate are shown in stick format. (C) Surface representation of the active site of Xrn1, colored according to electrostatic potential. (D) A 5′-end-labeled G(CU)10C RNA oligonucleotide (200 nM) was pre-incubated in the presence or absence of DmXrn1ΔC (740 nM) and subsequently digested with RNase T1 and RNase A. Products were resolved by electrophoresis on a 16% polyacrylamide and 8 M urea gel, and visualized by phosphorimaging. The Xrn1-protected fragment is indicated with an arrow.
Mg2+ Binding and Catalytic Mechanism of Xrn1
The scissile phosphate of the substrate lies directly over the conserved heptad of Mg2+-binding acidic residues located on the floor of the active site (Figure 2B). One magnesium ion is evident in electron density maps, surrounded by Asp35, Asp86 and Glu177 (Figure S3A). The distances between the carboxyl groups and the metal center are such that Glu177 most likely coordinates the Mg2+ ion directly while Asp35 and Asp86 interact via inner-sphere-coordinated water molecules. In the context of the wild-type enzyme, a second solvated Mg2+ ion is predicted to bind to Glu177, Asp205, Asp207 and Asp288, based on similarities to FEN-1 and T4 RNase H (Hwang et al., 1998; Mueser et al., 1996) (Figure S3A-D). This Mg2+ ion likely coordinates and activates a water molecule for an in-line nucleophilic attack at the scissile phosphate (Figure S3B). Of note, the distance between the scissile phosphate oxygens and the observed magnesium ion is too large to permit direct inner-sphere coordination, as seen in the structure of T4 RNase H in a magnesium-free complex with a DNA substrate (Figure S3E) (Devos et al., 2007). In the absence of active site or substrate rearrangements, the catalytic mechanism of Xrn1 thus may be distinct from the canonical two-metal-ion mechanism employed by many nucleases, in which both magnesium ions directly coordinate the scissile phosphate group (Yang et al., 2006).
A Conserved Basic Pocket Mediates Specific 5′ Phosphate Recognition
With the base of the first nucleotide stacked onto the invariant His41, the 5′ end of the substrate oligonucleotide packs orthogonally against the tower domain helix (Figure 3A). The closure of the active site by the tower helix thus ensures specific exonuclease activity in Xrn-family enzymes, as noted previously (Xiang et al., 2009). The 5′-phosphate group is inserted in a highly basic pocket lined with the side chains of the strictly conserved residues Lys93, Gln97, Arg100 and Arg101 (Figure 3A). The guanidinium groups of Arg100 and Arg101 make hydrogen-bonding interactions with the 5′ phosphate oxygens. The observed mode of 5′ phosphate binding readily explains why m7GpppN-capped or 5′-triphosphorylated RNAs are refractory to hydrolysis by Xrn1. The 5′ phosphate is not only specifically recognized by Xrn-family enzymes, but the 5′-phosphate-binding pocket also sterically excludes larger 5′-terminal groups. The specific recognition of the 5′ phosphate likely makes a significant contribution to substrate binding affinity since RNAs with a free 5′-hydroxyl group make poor Xrn1 substrates under multiple-turnover conditions (Mathy et al., 2007; Pellegrini et al., 2008; Stevens and Poole, 1995).
Figure 3. A conserved basic pocket in Xrn1 specifically recognizes the 5′ phosphate moiety of the substrate.
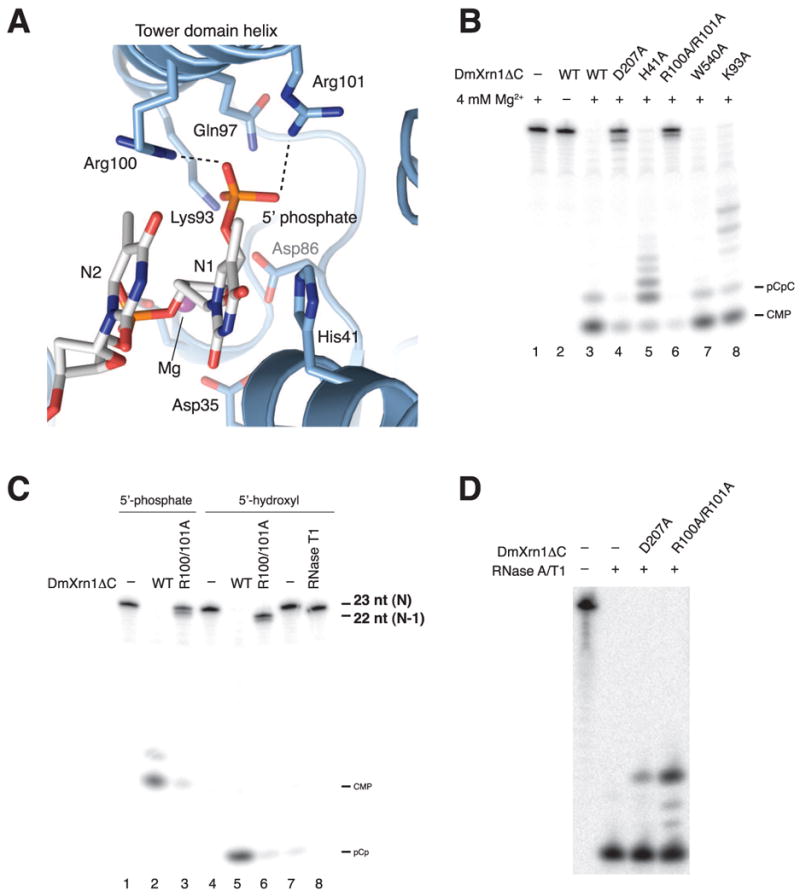
(A) Close-up view of the 5′-phosphate-binding pocket. Strictly conserved residues are shown in stick format. (B) Single-turnover exonuclease activity assay of wild-type and mutant DmXrn1ΔC proteins. DmXrn1ΔC constructs (370 nM) were incubated with a 3′-[32P]-pCp labeled, 5′-phosphorylated G(CU)10C RNA oligonucleotide (200 nM) for 2 min at 20°C. Reactions were analyzed on a 16% denaturing gel, followed by phosphorimaging. (C) Wild-type (WT) and R100A/R101A DmXrn1ΔC proteins were incubated with 3′ end-labeled G(CU)10C oligonucleotides bearing either 5′-terminal phosphate or hydroxyl groups. Note that 5′-phosphorylation of the 3′-end-labeled substrate by polynucleotide kinase removes the 3′ phosphate group, resulting in different terminal products (CMP vs pCp). RNase T1 served as positive control for removal of a single nucleotide from the 5′ end. The slightly lower mobility of the RNase T1 digested fragment is due to the absence of a 5′ phosphate. (D) RNase protection assay of WT and R100A/R101A DmXrn1ΔC. The assay was performed as in Figure 2D.
To confirm the functional roles of residues implicated in substrate recognition, we generated point mutant proteins and tested their exonucleolytic activity towards a 5′-phosphorylated 23-mer RNA oligonucleotide under single turnover conditions. Alanine substitutions of Arg100 and Arg101 effectively abolished Xrn1 activity to an extent comparable with the inactivating D207A mutation (Figure 3B, lane 6). The K93A mutation also slightly impaired activity as stalled intermediates could be observed (Figure 3B, lane 8). Similarly, alanine substitution of the invariant His41 also impaired activity and processivity (Figure 3B, lane 5), Overall, these findings indicate that both monophosphate recognition by the 5′-phosphate binding pocket and stacking between the base of the 5′-terminal nucleotide and His41 are required for efficient substrate hydrolysis. Our observations are consistent with a previous screen for non-complementing missense mutations in yeast Xrn1, which identified His41, Gln97 and Arg101 among the residues essential for Xrn1 function in vivo (Page et al., 1998).
5′-terminal Nucleotide Recognition Contributes to Processivity
Xrn1 is a highly processive enzyme that degrades its substrates without releasing partially hydrolyzed intermediates (Stevens, 2001). Therefore, the catalytic mechanism of Xrn1 must include a translocation step in which the nucleic acid product advances by one nucleotide following the release of a nucleoside monophosphate. His41 appears to be directly involved in processivity since the H41A DmXrn1ΔC mutant produced stalled intermediates in vitro (Figure 3B). This processivity defect was more evident at earlier time points (Figure S4A, lanes 11-14) and occurred independently of substrate length (Figure S4B, compare lanes 5-7 and 15-17). Moreover, the H41A mutant yielded a dinucleotide as the predominant terminal hydrolytic product (Figures 3B and S4A-B). As the substrate is unable to form a continuous π-π stack between His41 and Trp540 once it is shortened to two nucleotides, this observation suggests that in the absence of His41, the dinucleotide cannot advance into the active site, possibly due to base-stacking with Trp540. Together, these results strongly suggest that the stacking interaction between His41 and the base of the 5′ terminal nucleotide of the substrate promotes substrate translocation, thereby enhancing the processivity of degradation. In contrast, Trp540 is not required for processive degradation as the W540A mutant protein retained near wild-type activity and no stalling could be observed (Figures 3B and S4A-B).
In addition to His41, mutation of Lys93 also led to a marked processivity defect that was independent of substrate length (Figure S7), suggesting that 5′ monophosphate recognition also contributes to processivity. In light of this result, we reasoned that the positive electrostatic surface potential of the 5′ phosphate-binding pocket might also promote substrate translocation. To test this hypothesis, we compared the activities of wild-type (WT) DmXrn1ΔC and the R100A/R101A double mutant with an RNA substrate bearing a 5′ hydroxyl group. Under single-turnover conditions at a saturating substrate concentration, WT DmXrn1ΔC efficiently degraded both the 5′-monophosphorylated and 5′-hydroxylated substrates. While the 5′-monophosphorylated RNA was refractory to hydrolysis by the R100A/R101A mutant, the 5′-hydroxylated substrate underwent a single round of hydrolysis to yield a stalled 5′-monophosphorylated intermediate (Figure 3C, compare lanes 3 and 6). The failure of the 5′-monophosphorylated species to undergo hydrolysis could be explained by stalling in a product-like state due to a translocation block. Alternatively, the substrate could be fully inserted in the active site, but distorted such that hydrolysis is prevented. To distinguish between these possibilities, we examined the R100A/R101A mutant using the RNase protection assay described in Figure 2D. The double mutant was capable of protecting a 4-nucleotide fragment, as for the D207A mutant, but shorter species (3- and 2-nt fragments) were also present (Figure 3D). Thus, rather than being stalled in a product-like state, the 5′-monophosphorylated substrate appears to be mostly fully inserted in a non-productive conformation and might be dynamically sampling multiple states in the absence of 5′-phosphate recognition. Altogether, the activities of the K93A and R100A/R101A mutants and the RNase protection data are consistent with a role of the 5′ phosphate binding pocket in anchoring the 5′-terminal nucleotide of the substrate in a productive conformation to permit cleavage once the substrate has advanced forward.
Based on these results, we propose that Xrn1 achieves processive degradation by a Brownian ratchet mechanism in which His41, in concert with the 5′ phosphate-binding pocket, imparts directionality to the process (Figure S4C). Subsequent to hydrolysis at the scissile phosphate and nucleoside monophosphate dissociation, π-π stacking between the base of the 5′-terminal nucleotide of the product and His41 drives substrate translocation. The translocation is reinforced by electrostatic contacts between the 5′ phosphate of the substrate and the basic phosphate-binding pocket, which ensures correct orientation of the 5′-terminal nucleotide for the next round of cleavage.
Hydrolysis-coupled Duplex Unwinding by Xrn1
Xrn1 is capable of processively degrading RNA substrates containing stem-loop secondary structures; stalling Xrn1-mediated RNA degradation in yeast usually requires insertion of a G-quartet-forming tract into the substrate RNA (Decker and Parker, 1993; Poole and Stevens, 1997). The structure of the DmXrn1-substrate complex reveals that the active site cannot accommodate an A-form RNA duplex since the complementary duplex strand would clash with the tower domain helix, a loop harboring the conserved Trp540 (residues 533-542), and a disordered loop (residues 896-910) projecting from the WH domain. Moreover, stacking of the third base onto Trp540 might introduce a kink in the single-stranded substrate, potentially disrupting helical structures.
To examine the ability of Xrn1 to couple processive single-strand hydrolysis and secondary structure unwinding, we assayed the activity of DmXrn1ΔC towards RNA-DNA hybrid substrates with variable-length oligo(A) 5′-overhangs in the RNA strand. The substrates shared a common 17 base pair duplex sequence; the RNA strands were 5′-monophosphorylated while the DNA strand carried a 5′ hydroxyl group. DmXrn1ΔC processively degraded RNA strands in duplexes containing 5′-overhangs of 5 or 8 nucleotides but failed to degrade a duplex containing a 2-nucleotide overhang (Figure 4A). These results suggest that in order to initiate duplex unwinding, Xrn1 requires a single-stranded 5′-overhang that is sufficiently long to reach the active site. This is consistent with the mode of single-stranded substrate binding observed in the DmXrn1-substrate complex and points to a mechanism whereby duplex unwinding is brought about by substrate translocation past a steric barrier that excludes double-stranded regions (Figure 4B). The narrow entrance into the catalytic site located between the N-terminus of helix α1 and the Trp540-containing loop could serve as the steric barrier since it is approximately 9 Å wide and thus can only accommodate a single strand of a nucleic acid substrate. In the Xrn1-substrate complex, this site is occupied by the phosphate group of the fourth nucleotide (Figure 2B). Although substrate translocation could actively promote base pair separation in a duplex substrate, it is also plausible that the mechanism simply exploits thermal breathing of the duplex, while concomitant translocation of the substrate and subsequent hydrolysis ensure irreversibility of the process (Figure 4B). As such, the mechanism of duplex unwinding by Xrn1 is an extension of a Brownian ratchet mechanism of processive hydrolysis afforded by the steric barrier at the entrance to the active site.
Figure 4. Xrn1 couples hydrolysis to duplex unwinding in substrates with sufficiently long single-stranded 5′ extensions.
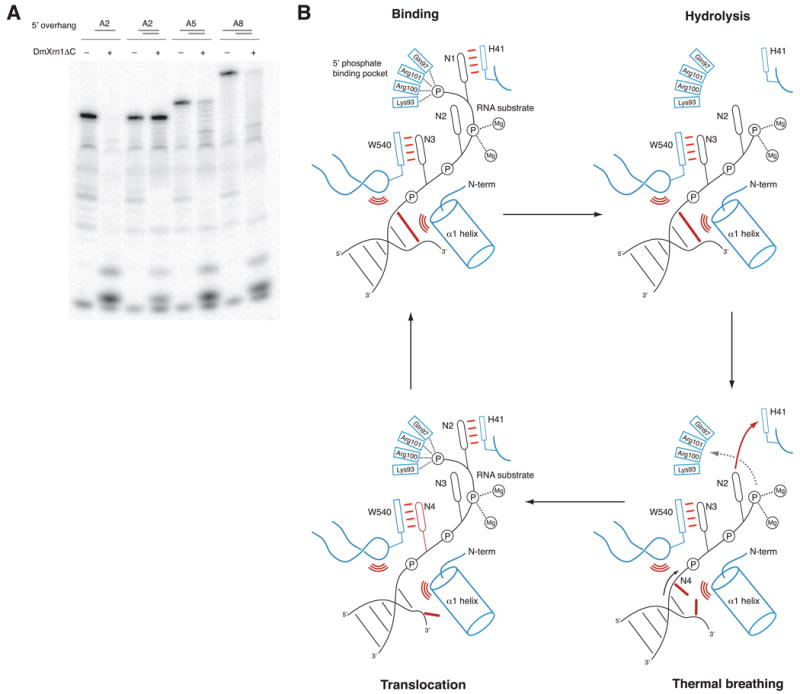
(A) Hydrolysis-coupled duplex unwinding by DmXrn1ΔC. The substrates contain a common 17 base-pair GC-rich RNA/DNA duplex and a 2-, 5- or 8-nt oligo(A) overhang at the 5′ end of the RNA strand. The RNA strands were 3′-end labeled and 5′-phosphorylated. The two leftmost lanes represent control degradation of the A2 overhang RNA oligonucleotide in the absence of the complementary DNA strand. (B) Model of hydrolysis-coupled duplex unwinding by Xrn1. Following cleavage and release of the nucleotide monophosphate product, Xrn1 remains bound to substrate. Transient opening of the terminal base pair of the duplex by thermal breathing allows the substrate to advance through the narrow channel between the N-terminal helix α1 and the Trp540-containing loop. Substrate translocation occurs by a Brownian ratchet mechanism in which interactions of the 5′-terminal nucleotide with His41 and the 5′ phosphate binding pocket and subsequent hydrolysis impart directionality.
Our findings draw unexpected parallels between the mechanism of Xrn enzymes and degradation-coupled substrate unwinding observed for the RNase II-family of processive 3′-5′ exonucleases such as bacterial RNase R and Rrp44/Dis3, the hydrolytic subunit of the eukaryotic exosome complex (Lorentzen et al., 2008; Vincent and Deutscher, 2006). Thus, Xrn1-mediated 5′-3′ exonucleolytic mRNA decay and exosome-catalyzed 3′-5′ degradation both appear to require initial recognition of a single-stranded terminal segment in order to commence degradation of structured RNAs and employ an unwinding mechanism based on steric exclusion (Bonneau et al., 2009). This is likely to have implications not only for Xrn1-mediated mRNA decay but also for Xrn2-specific functions such a transcriptional termination. Recent studies have shown that Xrn1 is involved in 18S nonfunctional rRNA decay and, together with Rat1, in tRNA quality control in yeast, further underscoring the role of Xrn-family enzymes in degrading structured RNA substrates (Chernyakov et al., 2008; Cole et al., 2009).
Conclusions
The structure of the Xrn1-substrate complex presented here reveals that Xrn1 specifically recognizes 5′-monophosphated substrates through electrostatic interactions mediated by a conserved electropositive pocket. This ensures that Xrn1 is essentially inactive towards m7GpppN-capped RNA and thus targets only mRNAs that have been irreversibly committed to decay by decapping or through endonucleolytic cleavage. Specific recognition of the 5′-terminal nucleotide of the substrate promotes substrate translocation after a round of hydrolysis, thereby contributing to a processive mode of RNA degradation. This not only leads to rapid clearance of targeted RNAs but also allows Xrn1 to couple processive hydrolysis to ATP-independent melting of structured substrates through a mechanism involving steric occlusion.
Experimental Procedures
Structure determination of the DmXrn1-substrate complex
Full details of experimental procedures are given in the Supplemental Information. Briefly, a C-terminally truncated construct of Drosophila melanogaster Xrn1 (residues 1-1141), carrying a D207A point mutation and a C-terminal hexahistidine tag, was expressed using the insect cell/baculovirus expression system as per standard procedures. The protein was purified by affinity and anion exchange chromatographic steps, followed by size-exclusion chromatography in 20 mM HEPES pH 7.5, 150 mM KCl, 4 mM MgCl2 and 1 mM TCEP. The Xrn1-substrate complex was reconstituted by incubating the protein (4 mg·ml-1) with 100 μM 5′-phosphorylated dT11 oligonucleotide, and crystallized using the hanging drop vapor diffusion method from 100 mM MES pH 6.2, 1% (v/v) PEG 300. X-ray diffraction data were measured at beamline 8.2.2 of the Advanced Light Source (Lawrence Berkeley National Laboratory) and processed with XDS (Kabsch, 2010). The structure was solved by a single-wavelength anomalous dispersion (SAD) experiment using crystals containing selenomethionine-substituted protein. Phases were calculated using AutoSHARP (Vonrhein et al., 2007) and the model was refined with Phenix.refine (Adams et al., 2010). Data measurement and refinement statistics are summarized in Table 1.
RNase protection assays
2.0 μg DmXrn1ΔC (740 nM final concentration) was incubated with 4 pmol [32P] 5′-end-labeled RNA oligonucleotide (sequence G(CU)10C) in 20 mM HEPES pH 7.5, 100 mM KCl, 4 mM MgCl2, 5% (v/v) glycerol, 1 mM DTT in a total volume of 20 μl for 30 min at 0°C. The mixtures were subsequently digested with 2.5 U RNase T1 and 1 mg RNase A (Fermentas) for 5 min at 20 °C. The samples were resolved by electrophoresis on a 16% polyacrylamide and 8 M urea gel and visualized by phosphorimaging (Storm, GE Life Sciences).
Nuclease activity assays
3′-end labelled oligonucleotide substrate was prepared by labeling a G(CU)10C synthetic RNA with [5′-32P] pCp and T4 RNA ligase and subsequently 5′-phosphorylated using T4 polynucleotide kinase. Assays were carried out in 20 mM HEPES pH 7.5, 100 mM KCl, 4 mM MgCl2, 5% (v/v) glycerol, 1 mM DTT. 1 μg of wild type or mutant Xrn1 (370 μM final concentration) was incubated with 5 pmol RNA substrate in a volume of 20 μl at 20 °C for 2 minutes. Duplex substrates were generated by annealing 3′ end-labeled, 5′-phosphorylated RNA oligonucleotides (2 μM) with a 2-fold molar excess of a complementary 17-mer DNA oligonucleotide (see Supplemental Information for full details). Assays with duplex substrates were carried out as above using 200 nM duplex, 37 nM enzyme and 5 minute incubation time.
Supplementary Material
Acknowledgments
We thank the staff at beamline 8.2.2 of the Advanced Light Source (Lawrence Berkeley National Laboratory) for support during X-ray data measurement. We thank D. King for mass spectrometry and S. Lorenz for dynamic light scattering analysis. We are grateful to J. Cate and E. Conti for discussions and members of the J.A.D. laboratory for critical reading of the manuscript. M.J. was supported by a Long-Term Fellowship from the Human Frontier Science Program. J.A.D. is a Howard Hughes Medical Institute Investigator. This work was funded in part by the National Institutes of Health.
Footnotes
Accession Numbers: Coordinates and structure factor amplitudes for the DmXrn1-substrate complex crystal structure have been deposited in the Protein Data Bank under ID code 2Y35.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonneau F, Basquin J, Ebert J, Lorentzen E, Conti E. The yeast exosome functions as a macromolecular cage to channel RNA substrates for degradation. Cell. 2009;139:547–559. doi: 10.1016/j.cell.2009.08.042. [DOI] [PubMed] [Google Scholar]
- Chernyakov I, Whipple JM, Kotelawala L, Grayhack EJ, Phizicky EM. Degradation of several hypomodified mature tRNA species in Saccharomyces cerevisiae is mediated by Met22 and the 5′-3′ exonucleases Rat1 and Xrn1. Genes Dev. 2008;22:1369–1380. doi: 10.1101/gad.1654308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole SE, LaRiviere FJ, Merrikh CN, Moore MJ. A convergence of rRNA and mRNA quality control pathways revealed by mechanistic analysis of nonfunctional rRNA decay. Mol Cell. 2009;34:440–450. doi: 10.1016/j.molcel.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coller J, Parker R. Eukaryotic mRNA decapping. Annu Rev Biochem. 2004;73:861–890. doi: 10.1146/annurev.biochem.73.011303.074032. [DOI] [PubMed] [Google Scholar]
- Conti E, Bonneau F, Ebert J, Basquin J, Lorentzen E. Molecular mechanisms of RNA degradation by the exosome. Febs J. 2009;276:37–38. [Google Scholar]
- Decker CJ, Parker R. A turnover pathway for both stable and unstable mRNAs in yeast: evidence for a requirement for deadenylation. Genes Dev. 1993;7:1632–1643. doi: 10.1101/gad.7.8.1632. [DOI] [PubMed] [Google Scholar]
- Devos JM, Tomanicek SJ, Jones CE, Nossal NG, Mueser TC. Crystal structure of bacteriophage T4 5′ nuclease in complex with a branched DNA reveals how flap endonuclease-1 family nucleases bind their substrates. J Biol Chem. 2007;282:31713–31724. doi: 10.1074/jbc.M703209200. [DOI] [PubMed] [Google Scholar]
- Doma MK, Parker R. Endonucleolytic cleavage of eukaryotic mRNAs with stalls in translation elongation. Nature. 2006;440:561–564. doi: 10.1038/nature04530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberle AB, Lykke-Andersen S, Muhlemann O, Jensen TH. SMG6 promotes endonucleolytic cleavage of nonsense mRNA in human cells. Nat Struct Mol Biol. 2009;16:49–55. doi: 10.1038/nsmb.1530. [DOI] [PubMed] [Google Scholar]
- Eulalio A, Behm-Ansmant I, Izaurralde E. P bodies: at the crossroads of post-transcriptional pathways. Nat Rev Mol Cell Biol. 2007;8:9–22. doi: 10.1038/nrm2080. [DOI] [PubMed] [Google Scholar]
- Fabian MR, Sonenberg N, Filipowicz W. Regulation of mRNA translation and stability by microRNAs. Annu Rev Biochem. 2010;79:351–379. doi: 10.1146/annurev-biochem-060308-103103. [DOI] [PubMed] [Google Scholar]
- Fang F, Phillips S, Butler JS. Rat1p and Rai1p function with the nuclear exosome in the processing and degradation of rRNA precursors. RNA. 2005;11:1571–1578. doi: 10.1261/rna.2900205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franks TM, Lykke-Andersen J. The control of mRNA decapping and P-body formation. Mol Cell. 2008;32:605–615. doi: 10.1016/j.molcel.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garneau NL, Wilusz J, Wilusz CJ. The highways and byways of mRNA decay. Nat Rev Mol Cell Biol. 2007;8:113–126. doi: 10.1038/nrm2104. [DOI] [PubMed] [Google Scholar]
- Gatfield D, Izaurralde E. Nonsense-mediated messenger RNA decay is initiated by endonucleolytic cleavage in Drosophila. Nature. 2004;429:575–578. doi: 10.1038/nature02559. [DOI] [PubMed] [Google Scholar]
- Holm L, Sander C. Protein structure comparison by alignment of distance matrices. J Mol Biol. 1993;233:123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- Houseley J, Tollervey D. The many pathways of RNA degradation. Cell. 2009;136:763–776. doi: 10.1016/j.cell.2009.01.019. [DOI] [PubMed] [Google Scholar]
- Huntzinger E, Kashima I, Fauser M, Sauliere J, Izaurralde E. SMG6 is the catalytic endonuclease that cleaves mRNAs containing nonsense codons in metazoan. RNA. 2008;14:2609–2617. doi: 10.1261/rna.1386208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang KY, Baek K, Kim HY, Cho Y. The crystal structure of flap endonuclease-1 from Methanococcus jannaschii. Nat Struct Biol. 1998;5:707–713. doi: 10.1038/1406. [DOI] [PubMed] [Google Scholar]
- Jiao X, Xiang S, Oh C, Martin CE, Tong L, Kiledjian M. Identification of a quality-control mechanism for mRNA 5′-end capping. Nature. 2010 doi: 10.1038/nature09338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson AW, Kolodner RD. Strand exchange protein 1 from Saccharomyces cerevisiae. A novel multifunctional protein that contains DNA strand exchange and exonuclease activities. J Biol Chem. 1991;266:14046–14054. [PubMed] [Google Scholar]
- Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Krogan NJ, Vasiljeva L, Rando OJ, Nedea E, Greenblatt JF, Buratowski S. The yeast Rat1 exonuclease promotes transcription termination by RNA polymerase II. Nature. 2004;432:517–522. doi: 10.1038/nature03041. [DOI] [PubMed] [Google Scholar]
- Lebreton A, Seraphin B. Exosome-mediated quality control: substrate recruitment and molecular activity. Biochim Biophys Acta. 2008;1779:558–565. doi: 10.1016/j.bbagrm.2008.02.003. [DOI] [PubMed] [Google Scholar]
- Liu J, Carmell MA, Rivas FV, Marsden CG, Thomson JM, Song JJ, Hammond SM, Joshua-Tor L, Hannon GJ. Argonaute2 is the catalytic engine of mammalian RNAi. Science. 2004;305:1437–1441. doi: 10.1126/science.1102513. [DOI] [PubMed] [Google Scholar]
- Lorentzen E, Basquin J, Tomecki R, Dziembowski A, Conti E. Structure of the active subunit of the yeast exosome core, Rrp44: diverse modes of substrate recruitment in the RNase II nuclease family. Mol Cell. 2008;29:717–728. doi: 10.1016/j.molcel.2008.02.018. [DOI] [PubMed] [Google Scholar]
- Martinez J, Tuschl T. RISC is a 5′ phosphomonoester-producing RNA endonuclease. Genes Dev. 2004;18:975–980. doi: 10.1101/gad.1187904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathy N, Benard L, Pellegrini O, Daou R, Wen T, Condon C. 5′-to-3′ exoribonuclease activity in bacteria: role of RNase J1 in rRNA maturation and 5′ stability of mRNA. Cell. 2007;129:681–692. doi: 10.1016/j.cell.2007.02.051. [DOI] [PubMed] [Google Scholar]
- Mueser TC, Nossal NG, Hyde CC. Structure of bacteriophage T4 RNase H, a 5′ to 3′ RNA-DNA and DNA-DNA exonuclease with sequence similarity to the RAD2 family of eukaryotic proteins. Cell. 1996;85:1101–1112. doi: 10.1016/s0092-8674(00)81310-0. [DOI] [PubMed] [Google Scholar]
- Orban TI, Izaurralde E. Decay of mRNAs targeted by RISC requires XRN1, the Ski complex, and the exosome. RNA. 2005;11:459–469. doi: 10.1261/rna.7231505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page AM, Davis K, Molineux C, Kolodner RD, Johnson AW. Mutational analysis of exoribonuclease I from Saccharomyces cerevisiae. Nucleic Acids Res. 1998;26:3707–3716. doi: 10.1093/nar/26.16.3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker R, Sheth U. P bodies and the control of mRNA translation and degradation. Mol Cell. 2007;25:635–646. doi: 10.1016/j.molcel.2007.02.011. [DOI] [PubMed] [Google Scholar]
- Parker R, Song H. The enzymes and control of eukaryotic mRNA turnover. Nat Struct Mol Biol. 2004;11:121–127. doi: 10.1038/nsmb724. [DOI] [PubMed] [Google Scholar]
- Pellegrini O, Mathy N, Condon C, Benard L. In vitro assays of 5′ to 3′-exoribonuclease activity. Methods Enzymol. 2008;448:167–183. doi: 10.1016/S0076-6879(08)02609-8. [DOI] [PubMed] [Google Scholar]
- Poole TL, Stevens A. Structural modifications of RNA influence the 5′ exoribonucleolytic hydrolysis by XRN1 and HKE1 of Saccharomyces cerevisiae. Biochem Biophys Res Commun. 1997;235:799–805. doi: 10.1006/bbrc.1997.6877. [DOI] [PubMed] [Google Scholar]
- Schwarz DS, Tomari Y, Zamore PD. The RNA-induced silencing complex is a Mg2+-dependent endonuclease. Curr Biol. 2004;14:787–791. doi: 10.1016/j.cub.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Shen B, Nolan JP, Sklar LA, Park MS. Functional analysis of point mutations in human flap endonuclease-1 active site. Nucleic Acids Res. 1997;25:3332–3338. doi: 10.1093/nar/25.16.3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheth U, Parker R. Decapping and decay of messenger RNA occur in cytoplasmic processing bodies. Science. 2003;300:805–808. doi: 10.1126/science.1082320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens A. Purification and characterization of a Saccharomyces cerevisiae exoribonuclease which yields 5′-mononucleotides by a 5′ leads to 3′ mode of hydrolysis. J Biol Chem. 1980;255:3080–3085. [PubMed] [Google Scholar]
- Stevens A. 5′-exoribonuclease 1: Xrn1. Meth Enzymol. 2001;342:251–259. doi: 10.1016/s0076-6879(01)42549-3. [DOI] [PubMed] [Google Scholar]
- Stevens A, Poole TL. 5′-exonuclease-2 of Saccharomyces cerevisiae. Purification and features of ribonuclease activity with comparison to 5′-exonuclease-1. J Biol Chem. 1995;270:16063–16069. doi: 10.1074/jbc.270.27.16063. [DOI] [PubMed] [Google Scholar]
- Vincent HA, Deutscher MP. Substrate recognition and catalysis by the exoribonuclease RNase R. J Biol Chem. 2006;281:29769–29775. doi: 10.1074/jbc.M606744200. [DOI] [PubMed] [Google Scholar]
- Vonrhein C, Blanc E, Roversi P, Bricogne G. Automated structure solution with autoSHARP. Methods Mol Biol. 2007;364:215–230. doi: 10.1385/1-59745-266-1:215. [DOI] [PubMed] [Google Scholar]
- West S, Gromak N, Proudfoot NJ. Human 5′ --> 3′ exonuclease Xrn2 promotes transcription termination at co-transcriptional cleavage sites. Nature. 2004;432:522–525. doi: 10.1038/nature03035. [DOI] [PubMed] [Google Scholar]
- Xiang S, Cooper-Morgan A, Jiao X, Kiledjian M, Manley JL, Tong L. Structure and function of the 5′-->3′ exoribonuclease Rat1 and its activating partner Rai1. Nature. 2009;458:784–788. doi: 10.1038/nature07731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Lee JY, Nowotny M. Making and breaking nucleic acids: two-Mg2+-ion catalysis and substrate specificity. Mol Cell. 2006;22:5–13. doi: 10.1016/j.molcel.2006.03.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.