

Abstract
Nucleophilic mixed chromium(II) and chromium(III) acetylides are generated from the smooth reduction of primary 1,1,1-trichloroalkanes with chromium(II) chloride in the presence of an excess amount of triethylamine at room temperature. These species arise from chromium(III) vinylidene carbenoids. It has been demonstrated that uncommon low-valent CrII acetylides are formed by C–H insertion of CrIICl2 into terminal alkynes, formed in situ through the Fritsch– Buttenberg–Wiechell (FBW) rearrangement, whereas CrIII acetylides are concomitantly generated by HCl elimination from the chromium(III) vinylidene carbenoid. Both divergent pathways result, overall, in the formation of nucleophilic acetylides. In situ trapping with electrophilic aldehydes afforded propargyl alcohols. Furthermore, deuteration experiments and the use of deuterium labeled 1,1,1-trichloroalkane substrates demonstrated the prevalence of low-valent CrII acetylides, potentially useful, yet highly elusive synthetic intermediates.
Keywords: Chromium, Carbenoids, Rearrangement, Alkynes, Alcohols
Introduction
Since their discovery in 1957, organochromium reagents have been the focus of constant development and innovations.[1] Due to their unique combination of chemical features and remarkable compatibility with a wide range of functional groups, these reagents have become indispensable tools for advanced organic synthesis and for natural product synthesis.[2] The last 10 years, in particular, have witnessed an enormous growth in terms of new reagents and reaction modalities.[3–11] For instance, our laboratories and others have described several new chromium intermediates including chromium vinylidene carbenoids,[4] halogenated chromium enolates,[6] chromium Fischer halocarbenes,[5,7,11] and carbynes.[9] In continuation of our harvest of novel intermediates with unusual reactivities formed through the chromium-mediated reduction of 1,1,1-trihaloalkanes, we report herein that the reduction of 1,1,1-trichloroalkanes by chromium(II) chloride in the presence of triethylamine (TEA) induces the smooth formation of alkynylchromium reagents in high yields under exceptionally mild reaction conditions. Interception of the alkynylchromium intermediates with electrophilic aldehydes provides convenient access to functionalized propargyl alcohols (Scheme 1).[12]
Scheme 1.

Generation of mixed chromium acetylides from 1,1,1-trichloroalkanes and in situ reaction with aldehydes.
Heretofore, the generation of metal acetylides under mild conditions compatible with various functional groups has long been an unresolved synthetic challenge.[13] Classical methods have mainly exploited the relatively high acidity of terminal acetylenic C–H bonds to form metal alkynylides, either by direct metalation using strong bases, such as n-butyllithium or lithium diisopropylamide at low temperature (−100 to −80 °C),[14] or upon treatment with tertiary amines in the presence of a stoichiometric or catalytic amount of the metal salt of interest (Figure 1, route a).[15] Lithium and silver acetylides prepared by this approach are also utilized for the preparation of other acetylides by transmetalation with magnesium, zinc, cerium, and other metals (Figure 1, route b).[16–20] Alternatively, lithium acetylides can also be prepared through the Fritsch– Buttenberg–Wiechell (FBW) rearrangement/metalation of 1,1-dibromoolefins when treated with an excess amount of n-butyllithium (Figure 1, route d).[20,21] An in situ metalation/desilylation strategy has recently been applied successfully to the preparation of highly stable ruthenium acetylides of interest for their electronic properties (Figure 1, route c).[22]
Figure 1.

General approaches for the generation of metalated acetylides.
Chromium(III) acetylides, that are mainly generated by reduction of alkynyl halides with chromium(II) chloride (Figure 1, route e),[23] or more recently by transmetalation of lithium acetylides (Figure 1, route b),[24] have received scant attention, in spite of their demonstrated synthetic utility in a great number of natural product total syntheses[25] and for their interesting electronic properties.[26]
Results and Discussion
The generation of alkynylchromium(III) reagents by the most widely used reductive route (Figure 1, route e) is, however, plagued by drawbacks, inter alia, dependency upon nickel(II) additives, polar solvents, and/or high reaction temperatures.[1c] In sharp contrast, we have discovered that alkynylchromiums can be easily synthesized from the reduction of gem-1,1,1-trihaloalkanes by using 6 equiv. of CrCl2 and 10 equiv. of TEA (Figure 1, route f; Scheme 2).
Scheme 2.

Postulated mechanism for the formation of mixed chromium acetylides.
The postulated mechanism for the formation of chromium acetylides likely proceeds through a comparatively stable chromium(III) vinylidene carbenoid 6, generated through the syn-β-elimination of chromium hydride from the unstable 1-chloro-1,1-bis-chromium alkane carbenoid 5, initially formed by the reduction of two C-Cl bonds in 1,1,1-trichloralkane 4 (Scheme 2).[4a] Subsequent β-elimination of hydrogen chloride induced by Et3N abstraction of the vinylic proton of 6 gives rise to chromium(III) acetylide 7 (Scheme 2, path a).
Surprisingly, whereas DBU, pyridine, 1,5,7-triazabicyclo-[4.4.0]dec-5-ene (TBD), and DABCO were completely inactive and even inhibited the reduction of trichloroalkane 4, TEA was unique amongst the common organic bases and did not interfere with the overall transformations of gem-1,1,1-trichloroalkanes. The role of TEA is not known yet, although it is assumed that the dramatic decrease in the pKa of the carbenoid vinylic proton can be ascribed to the concerted metal-assisted ionization phenomenon (MAI),[27] which triggers the formation of chromium(III) acetylide 7 as shown in postulated binuclear complex 13 (Scheme 3).
Scheme 3.

Possible intermediate for the abstraction of HCl and FBW rearrangement of dinuclear chromium vinylidene carbenoid 13.
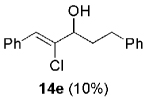
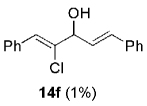
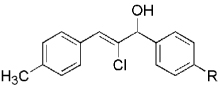
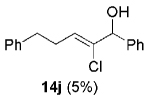
Hydrogen abstraction from 13 (Scheme 2, path a) is assumed to be kinetically competitive with the FBW rearrangement that leads to terminal alkyne 9 (Scheme 2, path b).[28] We postulate that this unprecedented reactivity of chromium vinylidene carbenoids is a result of coordination of the σ-donor lone pair of the basic nitrogen to chromium(III), yielding chromium(III) acetylides 7. This mechanistic pathway was partially corroborated by the fact that (Z)-2-chloroalk-2-en-l-ols 14 were formed in low amounts (4–10%) as byproducts when the reaction was performed under Barbier conditions in the presence of various aldehydes (Scheme 4).
Scheme 4.

Application to the synthesis of propargyl alcohols.
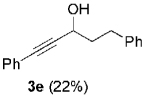
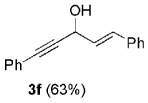
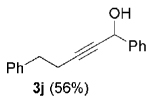
The high yield of propargyl alcohol 3, combined with the ready availability of 1,1,1-trichloroalkanes la–d, make this methodology very attractive for the preparation of a large panel of propargyl alcohols (Table 1).[29] Aromatic aldehydes 12a–d and 12g–i bearing diverse, sensitive functional groups such as bromo, cyano, acetoxy, methoxy, and fluoro, were well tolerated and afforded expected adducts 3a– d[30,31] and 3g–i[32] in good to excellent yields (Table 1, Entries 1–4, 7–9). Similarly, an α,β-unsaturated aldehyde like (E)-cinnamaldehyde (12f) reacted smoothly and delivered cleanly the corresponding propargyl allyl alcohol 3f[30] in a good isolated yield of 63% (Table 1, Entry 6). In sharp contrast, the use of an enolizable aliphatic aldehyde like dihydrocinnamaldehyde (12e) under the same conditions was problematic, and the yield of addition product 3e[33] was drastically decreased to 22% because of competitive cross-aldol as well as elimination. (Table 1, Entry 5).[34] Primary 1,1,1-trichloroalkanes lc reacted moderately and resulted in the formation of 3j[35] in reasonable isolated yield (56%) when benzaldehyde (12j) was used as the electrophile (Table 1, Entry 10). It is worth mentioning that the use of allylic 1,1,1-trichloroalkane 1d offered the opportunity to extend the scope of this transformation in generating useful enyn–alcohol 3k[36] in excellent yield (Table 1, Entry 11). Attempts to reduce the amount of chromium reagent by using multicomponent redox system for chromium recycling [CrCl2 (10 mol-%)/Mn0/TMSCl][37] and TEA (10 equiv.) in THF at room temperature for 12 h were not satisfactory as a result of limited conversion of the starting 1,1,1-trichloroalkanes (<10%). This result suggests, most likely, that the initial generation of key vinylidene carbenoid 6 (Scheme 2) might be the rate-limiting step of the process under these specific conditions.
Table 1.
Synthesis of different propargyl alcohols.
| Entry | Trichloroalkane | Aldehyde | Propargyl alcohol | Vinyl chloride |
|---|---|---|---|---|
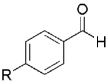 |
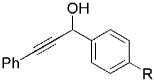 |
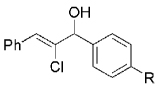 |
||
| 1 | 1a | R = CH312a | 3a (79%) | 14a (4%) |
| 2 | 1a | R = Br 12b | 3b (86%) | 14b (8%) |
| 3 | 1a | R = CN 12c | 3c (63%) | 14c (4%) |
| 4 | 1a | R = OAc 12d | 3d (72%) | 14d(10%) |
| 5 | 1a | 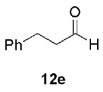 |
 |
 |
| 6 | 1a | 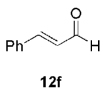 |
 |
 |
| 7 | 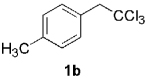 |
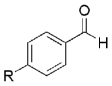 |
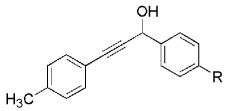 |
 |
| 8 | 1b | R = OAc 12h | 3h (79%) | 14h(10%) |
| 9 | 1b | R = F 12i | 3i(81%) | 14i (10%) |
| 10 | 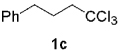 |
PhCHO 12j |
 |
 |
| 11 | 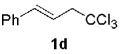 |
12j | 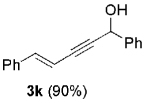 |
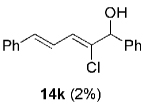 |
Mechanistically, to determine whether terminal alkyne 9, generated by FBW rearrangement from 6, could eventually be an intermediate in the overall transformation (Scheme 2, path b), we examined the reactivity of terminal alkynes with chromium(III) and chromium(II) chloride in the presence or absence of TEA. Notably, as a control experiment, 1-phenylacetylene (11) does not react with chromium(III) chloride in the presence or in the absence of TEA in THF under the same conditions as 1, thus excluding pathway a in the formation of chromium(III) acetylide 7a (Scheme 5).
Scheme 5.

Generation of alkynylating chromium agents from phenylacetylene (11).
However, we were surprised by the fact that CrIICl2 reacted smoothly with 11 in the presence of TEA (Scheme 5, path b), as evidenced by the formation of a minimum of 60% of adduct 16 when the organometallic species was trapped with benzaldehyde under Barbier conditions. Because the direct insertion of chromium(III) into the C–H bond of terminal alkynes is excluded for the formation of chromium(III) acetylide 7a, these results suggest strongly that the nucleophilic metalated acetylide is the uncommon low-valent chromium(II) acetylide 15. This result might be explained by ligand exchange of CrII, allowing nucleophilic substitution of labile ligands (e.g., Cl) to give nucleophilic chromium(II) acetylide 15. Indeed, like ZnII, CuI, or AuI acetylides that are generated in situ from terminal alkynes at room temperature upon treatment with an organic base (TEA, iPr2NnPr, or NH4OH)[13,15c,15d,29] by ligand exchange, this substitution reaction occurs for CrII. This mechanism is supported by kinetic studies reported by Merbach, who showed that this ligand exchange is kinetically very fast and favored for CrII, whereas CrIII is known to be extremely resistant to this process.[38] Indeed, the exchange ligand rates are ca. 15 orders of magnitude higher for CrII than CrIII.[39] The synthesis of end-bound acetylide ligands with monovalent and divalent octahedral chromium(II) has been recently reported; however, to the best of our knowledge, there is no report that accounts for their reactivity towards C–C bond-forming reactions.[26] At this point in our investigation, we provided evidence that the reduction of 1,1,1-trichloroalkanes with chromium(II) chloride in the presence of TEA affords unprecedented mixed chromium(II) and chromium(III) acetylides through two divergent pathways from chromium(III) vinylidene carbenoid 6 (Scheme 2). To further distinguish the prevalence of one pathway with respect to the other, we examined the reduction of deuterated substrate 17 and performed additional deuteration experiments (Scheme 6). Substrate 17 was first treated with 4 equiv. of CrCl2 under the same experimental conditions as those outlined for compounds 1. Quenching the reaction with H2O afforded a 2:3 mixture of protonated terminal alkyne 18 and its deuterated analogue 19 (FBW product) as determined by quantitative GC–MS analysis.
Scheme 6.

Mechanistic deuteration experiments.
Interestingly, this result was corroborated by the reduction of 1a with 4 equiv. of CrCl2,[4a] in the presence of an excess amount of TEA, and subsequent deuteration with DCl in D2O, which yielded 30–35% of [D1]phenylacetylene (20), along with 70–65% of 11. These observations demonstrated unambiguously the propensity of vinylidene carbenoids 6, prepared in the presence of TEA, to undergo FBW rearrangement predominantly and, therefore, the prevalence of pathway b that leads to CrII acetylides 10 (Scheme 2). The smooth generation of low-valent chromium(II) acetylides from 1,1,1-trihaloalkanes and CrCl2 as well as the reactivity of such organometallic species as new alkynylating agents have not been studied earlier. Although their nucleophilic behavior has been shown by trapping with electrophiles such as aldehydes, these reagents could be eventually used either in one-pot, metal (Pd, Ni, Fe) cross-coupling reactions or engaged in situ in [3+2]-Huisgen dipolar cycloaddition with azides.[40] The optimization as well as the scope and the potential applications of these synthetically useful reagents are underway in our laboratories and will be disclose elsewhere.
Conclusions
In summary, we have found that the reduction of 1,1,1-trichloroalkanes with an excess amount of chromium(II) provides direct access to mixed nucleophilic chromium(II) and chromium(III) acetylides. Both species are formed through divergent pathways, from chromium(III) vinylidene carbenoids, which favor the formation of uncommon low-valent chromium(II) acetylides, generated through the FBW route. We have demonstrated that in situ generated mixed chromium acetylides react smoothly with electrophilic aldehydes, providing access to propargyl alcohols, complementing other known strategies. Further promising developments using this new reductive/oxidation reaction of 1,1,1-trichloroalkanes by CrCl2 are expected to emerge and will be disseminated shortly.
Experimental Section
General
All reactions were performed under an argon atmosphere. The solvent (THF) was distilled from Na and benzophenone. All commercially available reagents were used without further purification. Analytical thin-layer chromatography (TLC) was performed on glass-backed silica gel plates. Visualization of the developed chromatogram was performed by using UV absorbance and staining with a vanillin, phosphomolybdic acid, or cerium sulfate solution. Flash column chromatography was performed with silica gel (40–63 µm) according to a standard technique. Nuclear magnetic resonance spectra (1H, 13C, and 19F) were recorded with a Bruker 400 MHz spectrometer equipped with a BBI or a DUAL probe. Chemical shifts for 1H and 13C NMR spectra are recorded in parts per million by using the residual chloroform as an internal standard (1H, δ = 7.26 ppm; 13C, δ = 77.16 ppm). Multiplicities are indicated by s (singlet), br. s (broad singlet), d (doublet), t (triplet), and m (multiplet). Mass spectra were recorded with a MS–MS high-resolution Micromass ZABSpecTOF spectrometry. Infrared spectra were recorded with an FTIR spectrometer equipped with KRS-5.
General Procedure for the Generation of Chromium Acetylides and Their Reaction with Aldehydes
All the reactions were performed on 1-mmol scale of trichloroalkanes 1a–d. To a solution of trichloroalkane 1 (1 equiv.) in THF (15 mL) was added aldehyde 12 (1 equiv.), CrCl2 (6 equiv.), and TEA (10 equiv.) under an inert atmosphere. The whole mixture was allowed to stir overnight at room temperature (10 h). After completion of the reaction (TLC analysis), the mixture was quenched with 1 n HCl (5 mL) and extracted with EtOAc (2 × 10 mL). The organic layer was washed with water and brine and dried with Na2SO4. Evaporation of the solvent under reduced pressure gave the crude product, which was purified by silica gel column chromatography. Elution with EtOAc/cyclohexane gave desired propargyl alcohol 3 and (Z)-2-chloroalk-2-en-1-ol (14) as an inseparable mixture. [32]
1-[4-(1-hydroxy-3-phenylprop-2-ynyl)phenyl]ethanone (3d)
Yield: 192 mg (72%), colorless sticky solid. 1H NMR (400 MHz, CDCl3): δ = 7.54 (d, J = 8.4 Hz, 2 H), 7.37–7.39 (m, 2 H), 7.22–7.24 (m, 3 H), 7.03 (d, J = 8.8 Hz, 2 H), 5.5 (br. s, 1 H), 2.41 (br. s, 1 H), 2.20 (s, 3 H) ppm. 13C NMR (100 MHz, CDCl3): δ = 169.5, 169.4, 150.6, 138.2, 131.7, 128.3, 127.9, 122.3, 121.7, 88.5, 86.8, 64.5, 21.1 ppm. IR (film): ṽ = 3414, 3056, 2183, 1753, 1504, 1195, 1164, 1064, 733 cm−1. HRMS (EI): calcd. for C17H14NaO3 [M + Na]+ 289.0835; found 289.0845.
1-(4-Methoxyphenyl)-3-(p-tolyl)prop-2-yn-1-ol (3g)
Yield: 164 mg (65%), light-yellow-colored viscous solid. 1H NMR (400 MHz, CDCl3): δ = 7.48–7.45 (m, 2 H), 7.29 (d, J = 8.4 Hz, 2 H), 7.05 (d, J = 8 Hz, 2 H), 6.84–6.86 (m, 2 H), 5.56 (d, J = 5.6 Hz, 1 H), 3.75 (s, 3 H), 2.27 (s, 3 H), 2.12 (d, J = 6 Hz, 1 H) ppm. 13CNMR (100 MHz, CDCl3): δ = 159.7, 138.7, 131.6, 133.1, 129.2, 129.0, 128.9, 128.1, 128.0, 119.4, 114.0, 88.2, 86.6, 64.8, 55.3, 21.5 ppm. IR (film): ṽ= 3400, 2921, 2835, 2197, 1608, 1508, 1245, 1170, 1029, 814 cm−1. HRMS (EI): calcd. for C17H16NaO2 [M + Na]+ 275.1042; found 275.1053.
1-{4-[1-hydroxy-3-(4-methylphenyl)prop-2-ynyl]phenyl}ethanone (3h)
Yield: 239 mg (79%), colorless sticky solid. 1H NMR (400 MHz, CDCl3): δ = 7.53–7.51 (m, 2 H), 7.27–7.25 (m, 2 H), 7.03–7.00 (m, 4 H), 5.56 (br. s, 1 H), 2.51 (br. s, 1 H), 2.25 (s, 3 H), 2.20 (s, 3 H) ppm. 13C NMR (100 MHz, CDCl3): δ = 169.5, 150.5, 138.8, 138.4, 131.6, 129.1, 127.9, 121.7, 119.2, 87.9, 86.9, 64.5, 21.5, 21.3, 21.1 ppm. IR (film): ṽ = 3432, 3043, 2931, 2195, 1754, 1505, 1194, 1163, 1012, 815, 734 cm−1. HRMS(EI): calcd. for C18H16NaO3 [M + Na]+ 303.0991; found 303.1000.
1-(4-Fluorophenyl)-3-(p-tolyl)prop-2-yn-1-ol (3i)
Yield: 195 mg (81%), colorless sticky solid. 1H NMR (400 MHz, CDCl3): δ = 7.52–7.48 (m, 2 H), 7.28–7.26 (m, 2 H), 7.05–6.96 (m, 2 H), 7.04 (d, J = 8 Hz, 2 H), 5.57 (d, J = 5.2 Hz, 1 H), 2.30 (d, J = 6 Hz, 1 H), 2.26 (s, 3 H) ppm. 13C NMR (100 MHz, CDCl3): δ = 163.9 (d, 1 J= 246 Hz, C), 161.4, 138.9, 136.6 (d, 4J= 3 Hz, C), 131.6, 129.1, 128.6 (d, 3J = 8 Hz, C), 128.5, 125.5, 119.1, 115.5 (d, 2J = 22 Hz, C), 115.3, 115.2, 87.8, 87.0, 64.4, 21.5, 21.3 ppm. 19F NMR (376.49 MHz, CDCl3): δ = −113.8 (s, 1 F) ppm. IR (film): ṽ = 3338, 2921, 2229, 1604, 1506, 1221, 1156, 1013, 836, 814 cm−1. HRMS (EI): calcd. for C16H14FO [M + H]+ 241.1023; found 241.1036.
Supplementary Material
Acknowledgments
Les Laboratoires Pierre-Fabre, le Centre National pour la Recherche Scientifique (CNRS), and l’Agence National pour la Recherche (ANR) Proteasome are warmly acknowledged for grants to S. T. and D. K. J. R. F. and N. P. were supported by the National Institutes of Health (NIH) (GM31278) and the Robert A. Welch Foundation. The authors are grateful to Cyril Antheaume from the University of Strasbourg for helpful scientific discussions.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/ejoc.200901476.
Supporting Information (see footnote on the first page of this article): 1H, 13C, and 19F (for 3i) NMR spectra of the original compounds.
Contributor Information
John R. Falck, Email: j.falck@utsouthwestern.edu.
Rachid Baati, Email: baati@bioorga.u-strasbg.fr.
References
- 1.a) Anet FAL, Leblanc E. J. Am. Chem. Soc. 1957;79:2649–2650. [Google Scholar]; b) Baati R. Synlett. 2001;5:722. [Google Scholar]; c) Fürstner A. Chem. Rev. 1999;99:991–1045. doi: 10.1021/cr9703360. [DOI] [PubMed] [Google Scholar]
- 2.a) Nilewski C, Geisser RW, Carreira EM. Nature. 2009;457:573–576. doi: 10.1038/nature07734. [DOI] [PubMed] [Google Scholar]; b) Baati R, Mioskowski C, Falck JR. Actual. Chim. 2009;326:25–30. [Google Scholar]
- 3.Toratsu C, Fujii T, Suzuki T, Takai K. Angew. Chem. Int. Ed. 2000;39:2725–2727. [PubMed] [Google Scholar]
- 4.a) Baati R, Barma DK, Falck JR, Mioskowski C. J. Am. Chem. Soc. 2001;123:9196–9197. doi: 10.1021/ja016515n. [DOI] [PubMed] [Google Scholar]; b) Falck JR, Bandyopadhyay A, N Puli, Kundu A, Reddy LM, Barma DK, He A, Zhang H, Dhurke K, Baati R. Org. Lett. 2009;11:4764–4766. doi: 10.1021/ol901985c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takai K, Toshikawa S, Inoue A, Kokumai R. J. Am. Chem. Soc. 2003;125:12990–12991. doi: 10.1021/ja0373061. [DOI] [PubMed] [Google Scholar]
- 6.a) Baati R, Mioskowski C, Dhurke K, Kodepelly S, Biao L, Falck JR. Tetrahedron Lett. 2009;50:402–405. [Google Scholar]; b) Barma DK, Kundu A, Zhang H, Mioskowski C, Falck JR. J. Am. Chem. Soc. 2003;125:3218–3219. doi: 10.1021/ja029938d. [DOI] [PubMed] [Google Scholar]
- 7.Bejot R, Tisserand S, Reddy LM, Barma DK, Baati R, Falck JR, Mioskowski C. Angew. Chem. Int. Ed. 2005;44:2008–2011. doi: 10.1002/anie.200461884. [DOI] [PubMed] [Google Scholar]
- 8.Baati R, Mioskowski C, Barma DK, Kache R, Falck JR. Org. Lett. 2006;8:2949–2951. doi: 10.1021/ol0607140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bejot R, He A, Falck JR, Mioskowski C. Angew. Chem. Int. Ed. 2007;46:1719–1722. doi: 10.1002/anie.200604015. [DOI] [PubMed] [Google Scholar]
- 10.Concellon JM, Rodrigez-Solla H, Mejica C, Blanco EG. Org. Lett. 2007;9:2981–2984. doi: 10.1021/ol070896d. [DOI] [PubMed] [Google Scholar]
- 11.Dhurke K, Mioskowski C, Falck JR, Goli M, Meunier S, Baati R, Wagner A. Org. Biomol. Chem. 2009;7:1771–1774. doi: 10.1039/b903244b. [DOI] [PubMed] [Google Scholar]
- 12.a) Vourloumis D, Kim KD, Petersen JL, Magriotis PA. J. Org. Chem. 1996;61:4848–4852. doi: 10.1021/jo9520506. [DOI] [PubMed] [Google Scholar]; b) Trost B, Krische MJ. J. Am. Chem. Soc. 1999;121:6131–6141. [Google Scholar]; c) Duffey MO, Tiran AL, Morken JP. J. Am. Chem. Soc. 2003;125:1458–1459. doi: 10.1021/ja028941u. [DOI] [PubMed] [Google Scholar]; d) Noyori R, Tomino I, Yamada M, Nishizawa MJ. J. Am. Chem. Soc. 1984;106:6717–6725. [Google Scholar]; e) Micalizio GC, Schreiber SL. Angew. Chem. Int. Ed. 2002;41:152–154. doi: 10.1002/1521-3773(20020104)41:1<152::aid-anie152>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 13.a) Coste A, Karthikeyan G, Couty F, Evano G. Angew. Chem. Int. Ed. 2009;48:1–6. doi: 10.1002/anie.200901099. [DOI] [PubMed] [Google Scholar]; b) Tejedor D, Lopez-Tosco S, Cruz-Acosta F, Mendez-Abt G, Garcia-Tellado F. Angew. Chem. Int. Ed. 2008;48:2–11. doi: 10.1002/anie.200801987. [DOI] [PubMed] [Google Scholar]
- 14.a) Midland MM, Tramontano A, Cable JR. J. Org. Chem. 1980;45:28–29. [Google Scholar]; b) Yamada K, Miyaura N, Itoh M, Suzuki A. Synthesis. 1977:679–682. [Google Scholar]
- 15.a) Pu L. Tetrahedron. 2003;59:9873–9886. [Google Scholar]; b) Aschwanden P, Carreira in EM. In: Acetylene Chemistry: Chemistry, Biology, and Material Science. Diederich F, Stang PJ, RR Tykwinski, editors. Wiley-VCH; Weinheim: 2005. pp. 101–138. [Google Scholar]; c) Frantz DE, Fassler R, Tomoka CS, Carreira EM. Acc. Chem. Res. 2001;34:373–381. doi: 10.1021/ar990078o. [DOI] [PubMed] [Google Scholar]; d) Frantz DE, Fassler R, Carreira EM. J. Am. Chem. Soc. 2000;122:1806–1807. [Google Scholar]; e) Anand NK, Carreira EM. J. Am. Chem. Soc. 2001;123:9687–9688. doi: 10.1021/ja016378u. [DOI] [PubMed] [Google Scholar]; f) Anaya de Parrodi C, Walsh PJ. Angew. Chem. Int. Ed. 2009;26:4679–4682. doi: 10.1002/anie.200900900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clive DLJ, Tao Y, Bo Y, Hu Y-Z, Selvakumar N, Sun S, Daigneault S, Wu Y-J. Chem. Commun. 2000:1341–1350. [Google Scholar]
- 17.a) Anastasia L, Negishi E. Org. Lett. 2001;3:3111–3113. doi: 10.1021/ol010145q. [DOI] [PubMed] [Google Scholar]; b) Hiroya K, Matsumoto S, Sakamoto T. Org. Lett. 2004;6:2953. doi: 10.1021/ol0489548. [DOI] [PubMed] [Google Scholar]
- 18.a) Hanessian S, Yun H, Hou Y, Tintelnot-Blomley M. J. Org. Chem. 2005;70:6746–6756. doi: 10.1021/jo050740w. [DOI] [PubMed] [Google Scholar]; b) Vedejs E, Piotrowski DW, Tucci FC. J. Org. Chem. 2000;65:5498–5505. doi: 10.1021/jo0001277. [DOI] [PubMed] [Google Scholar]
- 19.a) Donner CD. Tetrahedron Lett. 2007;48:8888–8890. [Google Scholar]; b) Kauffmann GS, Watson PS, Nugent WA. J. Org. Chem. 2006;71:8975–8977. doi: 10.1021/jo0616963. [DOI] [PubMed] [Google Scholar]; c) Ramana CV, Srinivas B, Puranick VG, Gurjar MK. J. Org. Chem. 2005;70:8216–8219. doi: 10.1021/jo050972v. [DOI] [PubMed] [Google Scholar]
- 20.Luu T, Morisaki Y, Cunningham N, Tywinski RR. J. Org. Chem. 2007;72:9622–9629. doi: 10.1021/jo701810g. [DOI] [PubMed] [Google Scholar]
- 21.Luu T, Morisaki Y, Tywinski RR. Synthesis. 2008;7:1158–1162. [Google Scholar]
- 22.a) Fox MA, Roberts RL, Baines TE, Le Guennec B, Halet JF. J. Am. Chem. Soc. 2008;130:3566–3578. doi: 10.1021/ja0779755. [DOI] [PubMed] [Google Scholar]; b) Bruce MI, Hall BC, Kelly BD, Low PJ, Skelton BW, White AH. J. Chem. Soc., Dalton Trans. 1999:3719–3722. [Google Scholar]
- 23.Takai K, Kuroda T, Nakatsukasa S, Oshima K, Nozaki H. Tetrahedron Lett. 1985;26:5585–5587. [Google Scholar]
- 24.Berben LA, Kozimor SA. Inorg. Chem. 2008;47:4639–4649. doi: 10.1021/ic702275g. [DOI] [PubMed] [Google Scholar]
- 25.Takai K. In: Organic Reactions. Overmann LA, editor. vol. 64. Wiley; 2004. pp. 253–612. [Google Scholar]
- 26.Berry JF, Cotton FA, Murillo CA, Roberts BK. Inorg. Chem. 2004;43:2277–2283. doi: 10.1021/ic0354320. [DOI] [PubMed] [Google Scholar]
- 27.Duraisamy M, Walborsky HM. J. Am. Chem. Soc. 1984;106:5035–5037. [Google Scholar]
- 28.a) Bejot R, Tisserand S, De Run L, Falck JR, Mioskowski C. Tetrahedron Lett. 2007;48:3855–3858. [Google Scholar]; b) Fritsch P. Justus Liebigs Ann. Chem. 1894;279:319. [Google Scholar]; c) Buttenberg WP. Justus Liebigs Ann. Chem. 1894;279:324. [Google Scholar]; d) Wiechel H. Justus Liebigs Ann. Chem. 1894;279:337. [Google Scholar]
- 29.Wong KM-C, Hung L-L, Lam WH, Zhu N, Yam VW-W. J. Am. Chem. Soc. 2007;129:4350–4365. doi: 10.1021/ja068264u. [DOI] [PubMed] [Google Scholar]
- 30.For the analytical data of 3a, 3b, and 3f see: Koyuncu H, Dogan z. Org. Lett. 2007;9:3477–3479. doi: 10.1021/ol701535y.
- 31.For the analytical data of 3c see: Ahn JH, Joung MJ, Min Yoon N. J. Org. Chem. 1995;60:6173–6175.
- 32.All the resulting propargyl alcohols were characterized by 1H NMR, 13C NMR, IR spectroscopy and HRMS. The 1H and 13C NMR spectra of known compounds were compared with data reported in the literature. See Supporting Information for the 1H NMR and 13C NMR spectroscopic data for original products 3d, 3g, 3h, and 3i.
- 33.For the analytical data of 3e see: Fang T, Du D-M, Lu S-F, Xu J. Org. Lett. 2005;7:2081–2084. doi: 10.1021/ol050047v. Gao G, Moore D, Xie RG, Pu L. Org. Lett. 2002;4:1855–1857. doi: 10.1021/ol025825n.
- 34.We have also noticed that ketones reacted sluggishly to furnish propargyl alcohols (<10%), and enolizable ketones afforded cross-aldol and elimination compounds as byproducts.
- 35.For the analytical data of 3j see: Takita R, Yakura K, Ohshima T, Shibasaki M. J. Am. Chem. Soc. 2005;127:13760–13761. doi: 10.1021/ja053946n.
- 36.For the analytical data of 3e see: Karatholuvhu MS, Fuchs PL. J. Am. Chem. Soc. 2004;126:14314–14315. doi: 10.1021/ja045112v.
- 37.Fürstner A. J. Am. Chem. Soc. 1996;126:12349–12357. [Google Scholar]; Fürstner A. J. Am. Chem. Soc. 1996;118:2533–2534. [Google Scholar]
- 38.Wessjohann LA, Scheid G. Synthesis. 1999;1:1–36. [Google Scholar]
- 39.a) Merbach AE. Pure Appl. Chem. 1987;59:161–172. [Google Scholar]; b) Merbach AE. Pure Appl. Chem. 1982;54:1479–1796. [Google Scholar]
- 40.Krasinski A, Fokin VV, Sharpless KB. Org. Lett. 2004;6:1237–1240. doi: 10.1021/ol0499203. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.