

Abstract
Terpenoid synthases are ubiquitous enzymes that catalyze the formation of structurally and stereochemically diverse isoprenoid natural products. Many isoprenoid coupling enzymes and terpenoid cyclases from bacteria, fungi, protists, plants, and animals share the class I terpenoid synthase fold. Despite generally low amino acid sequence identity among these examples, class I terpenoid synthases contain conserved metal binding motifs that coordinate to a trinuclear metal cluster. This cluster not only serves to bind and orient the flexible isoprenoid substrate in the precatalytic Michaelis complex, but it also triggers the departure of the diphosphate leaving group to generate a carbocation that initiates catalysis. Additional conserved hydrogen bond donors assist the metal cluster in this function. Crystal structure analysis reveals that the constellation of three metal ions required for terpenoid synthase catalysis is generally identical among all class I terpenoid synthases of known structure.
Keywords: enzyme catalysis, inorganic pyrophosphate, geranyl diphosphate, farnesyl diphosphate, Mg2+
INTRODUCTION
The terpenome is comprised of a family of more than 55,000 structurally and stereochemically diverse natural products, all of which ultimately derive from the universal 5 carbon precursors dimethylallyl diphosphate (DMAPP) and isopentenyl diphosphate (IPP) (Fig. 1) [1]. Terpenoids are ubiquitous throughout nature and serve a multitude of specific functions in plants, animals, insects, bacteria and fungi. For example, terpenoids are critical for plant survival and account for a large number of primary metabolites, including molecules involved in photosynthesis, respiration, and membrane structure. Terpenoids also account for a wide range of secondary metabolites in plants, where they bestow unique flavors and fragrances, provide chemical defense against pests, and facilitate interactions between plants and other organisms [2,3]. From a medicinal perspective, terpenoids are of great interest because many of these natural products exhibit anti-cancer, anti-malarial, and anti-microbial activities [2]. In humans, the 15 carbon linear isoprenoid farnesyl diphosphate (FPP) is a precursor in the biosynthesis of steroids and is also utilized for posttranslational prenylation of Ras in GTPase signaling [4,5]. Recently, human farnesyl diphosphate synthase has been identified as the target of nitrogen-containing bisphosphonate drugs used for the treatment of bone diseases such as osteoporosis, hypercalcemia, and metastatic bone disease [6,7].
Fig. 1.

General scheme of terpenoid nomenclature and biosynthesis (OPP = diphosphate, PPi = inorganic diphosphate).
Terpenoid synthases can be divided into two categories: class I enzymes adopt the FPP synthase α-helical fold and initiate catalysis by metal triggered ionization of the substrate diphosphate group, and class II enzymes adopt an unrelated double α-barrel fold and initiate catalysis by protonation of an epoxide ring or carbon-carbon double bond (Fig. 2). Class I terpenoid synthases can be further subdivided into three categories: coupling enzymes that catalyze chain elongation reactions to form increasingly longer polyisoprenoid diphosphates, coupling enzymes that catalyze irregular (i.e., non-head-to-tail) isoprenoid condensation reactions such as cyclopropanation, cyclobutanation, or branching reactions [8], and cyclization enzymes that catalyze the conversion of linear isoprenoid substrates into single and multi-ringed hydrocarbon products [9-12]. The potential diversity of carbon-carbon bond formation afforded by the flexible linear isoprenoid substrate, and the chemical potential for subsequent biosynthetic functionalization of cyclic terpenoids by cytochromes P450, monooxygenases, etc., make terpenoid biosynthesis an attractive system for engineering novel compounds [2,13,14]
Fig. 2.

Structural similarities among various terpenoid synthases define the core class I terpenoid cyclase fold (blue). Conserved metal binding motifs are the aspartate-rich motifs (red) and “NSE/DTE” motifs (orange) highlighted in (a) E. coli FPP synthase (PDB code 1RQI), (b) epi-isozizaene synthase (PDB code T.B.A.), and (c) (+)-bornyl diphosphate synthase (PDB code 1N22), which contains an additional N-terminal domain (cyan). This α-helical domain is topologically similar to the α-barrel fold of the class II terpenoid cyclases, which occurs in a double domain architecture in the triterpene cyclase (d) oxidosqualene cyclase (PDB code 1W6K).
To date, the crystal structures of several class I terpenoid coupling and cyclization enzymes have been solved, revealing a conserved α-helical terpenoid synthase fold across all domains of life. Structures of enzyme complexes with substrates, inhibitors, and/or products have also revealed the universal conservation of a trinuclear metal cluster implicated in the molecular recognition of the substrate diphosphate group as well as the initiation of catalysis. Metal ions are coordinated by metal binding motifs on opposing helices near the mouth of the active site. The metal binding motifs are generally described as either “aspartate-rich” [DDXX(XX)D/E] or “NSE/DTE” [(N,D)D(L,I,V)X(S,T)XXXE], in which boldface residues typically coordinate to catalytically obligatory Mg2+ or Mn2+ ions (throughout this review, metal ligands are indicated in boldface) [15]. X-ray crystal structures have been instrumental in understanding the catalytic mechanisms of terpenoid synthases: the active site of each synthase provides a template that binds the flexible substrate(s) in the proper orientation and conformation so that, upon the departure of the diphosphate leaving group and resultant generation of a reactive carbocation, the active site template ensures a specific trajectory of intermolecular and intramolecular carbon-carbon bond formation in the ensuing cyclization cascade [16]. Here, we review the available crystal structures of class I terpenoid synthases complexed with trinuclear metal clusters and isoprenoid diphosphates or inorganic pyrophosphate (PPi) to highlight conserved structural aspects of 3-metal ion catalysis in terpenoid biosynthesis.
ISOPRENOID COUPLING ENZYMES
Farnesyl diphosphate synthase
Farnesyl disphosphate synthase, the archetypical prenyltransferase, catalyzes the formation of farnesyl diphosphate (FPP), the linear isoprenoid precursor of sesquiterpene natural products. Chain elongation to form FPP proceeds in two distinct steps (Fig. 1): first, isopentenyl disphosphate (IPP) and dimethylallyl diphosphate (DMAPP) are coupled to form geranyl diphosphate (GPP), and then a second molecule of IPP is coupled to GPP to form FPP. The first crystal structure of FPP synthase was that of the avian enzyme [17], which revealed a novel α-helical fold. The structure revealed two conserved aspartate-rich (DDXXD) sequences [18] on helices D and H, which flank the mouth of the active site cavity. Additionally, a single Sm3+ ion, used for heavy metal derivatization for MIR phasing, was bound by each DDXXD motif.
The crystal structure of E. coli FPP synthase was the first to reveal the binding of a trinuclear magnesium cluster in the active site of an isoprenoid coupling enzyme [19], similar to the trinuclear magnesium clusters previously observed in fungal and plant terpenoid cyclases [20,21]. The structure of E. coli FPP synthase was solved as the enzyme-substrate ternary complex with the noncleavable DMAPP analogue, dimethylallyl S-thiolodiphosphate (DMSPP), and a molecule of IPP. Applying the Mg2+A, Mg2+B, and Mg2+C nomenclature first established for the trinuclear magnesium cluster of trichodiene synthase [20], the crystal structure of the E. coli FPP synthase-Mg2+3-DMSPP-IPP complex reveals octahedral coordination of all three metal ions (Fig. 3a): Mg2+A is coordinated by D105 and D111 of the first aspartate-rich motif on helix D, two diphosphate oxygen atoms, and two water molecules; Mg2+C is coordinated by the side chains of D105, and D111, as well as one diphosphate oxygen and three water molecules; and Mg2+B is coordinated by D244 of the second aspartate-rich motif, two diphosphate oxygen atoms, and three water molecules. The diphosphate group of DMSPP also accepts hydrogen bonds from R116, K202, and K258.
Fig. 3.

Conservation of Mg2+3-PPi and -diphosphate binding motifs among isoprenoid coupling enzymes. Metal coordination (black) and hydrogen bond (red) interactions with phosphate(s) are indicated. (a) E. coli FPP synthase-Mg2+3-DMSPP-IPP complex (PDB code 2EGW); (b) human FPP synthase-Mg2+3-zoledronate complex (PDB 2F8Z); (c) T. cruzi FPP synthase-Mg2+3-risedronate complex (PDB code 1YHL); (d) T. brucei FPP synthase-Mg2+3-BPH-721 complex (PDB code 3DYH); (e) S. cerevisae GGPP synthase-Mg2+2-BPH-252 complex (PDB code 2Z4X); (f) C. parvum nonspecific prenyl synthase-Mg2+3-zoledronate complex (PDB code 2Q58).
More recently, the structure of the human FPP synthase-Mg2+3-zoledronate-IPP complex [4] reveals complete conservation of Mg2+3-diphosphate recognition between E. coli and human FPP synthases (Fig. 3b). In human FPP synthase, two DDXXD motifs coordinate to the Mg2+3 cluster: the first aspartate of the D103DXXD107 motif coordinates to Mg2+A and Mg2+C with syn,syn-bidentate geometry, and one oxygen atom of D107 bridges Mg2+A and Mg2+C with syn,anti-coordination stereochemistry; the first aspartate of the second D243DXXD motif coordinates to Mg2+B. The diphosphate moiety additionally accepts hydrogen bonds from R112, K200, and K257.
Interestingly, the closed active site conformation is also stabilized by newly formed hydrogen bonds between K266 and D107 and D174. The r.m.s. deviation between the unliganded enzyme and the closed conformation of the Mg2+3-zoledronate-IPP complex is 1.3 Å (341 Cα atoms). Analysis of X-ray crystal structures of several human FPP synthase-Mg2+3-bisphosphonate complexes suggests a two-step mechanism for substrate binding [4]. First, the binding of DMAPP and 3 Mg2+ ions brings together the two DDXXD motifs, and loops D-E and H-I come together to form a hydrogen bond between T111 and the backbone of I258. These structural changes close the entrance to the allylic binding site and complete the formation of the IPP binding site. Secondly, as IPP binds, and as the basic C-terminal tail of the enzyme becomes ordered and closes the IPP binding site, IPP and DMAPP are properly oriented for catalysis.
The binding of a trinuclear magnesium cluster is similarly conserved in FPP synthases from parasitic organisms. The flagellated protozoan T. cruzi causes Chagas disease, primarily in Latin America [22]. Bisphosphonates have emerged as a potential treatment for Chagas disease by inhibiting T. cruzi FPP synthase [23]. The crystal structures of T. cruzi FPP synthase-Mg2+3-inhibitor complexes [24] suggest conservation of the trinuclear magnesium cluster for substrate binding and catalysis. In the complex with risedronate (Fig. 3c), the first carboxylate of the D98DXXD102 motif on helix D coordinates to Mg2+A and Mg2+C with syn,syn-bidentate geometry, and one oxygen atom of D102 bridges Mg2+A and Mg2+C with syn,anti-coordination stereochemistry. The first carboxylate of the D250DXXD motif on helix H is the only residue that directly coordinates to Mg2+B; however, D251 and D254 indirectly interact with Mg2+B via bridging water molecules. Each Mg2+ ion is coordinated with octahedral geometry, with non-protein coordination sites occupied by oxygen atoms of the inhibitor phosphonate groups and water molecules. Oxygen atoms of the two risedronate phosphonate groups accept hydrogen bonds from the side chains of R107, K207, and K264.
Trypanosoma brucei is an African parasitic protist, and its FPP synthase is related to that of T. cruzi by 70 % amino acid sequence identity. The crystal structure of T. brucei FPP synthase complexed with Mg2+3-BPH-721 [25] reveals conservation of the trinuclear magnesium cluster for substrate binding and catalysis (Fig. 3d). The first carboxylate of the D103DXXD107 motif on helix D coordinates to Mg2+A and Mg2+C with syn,syn-bidentate geometry, and one oxygen atom of D107 bridges Mg2+A and Mg2+C with syn,anti-coordination stereochemistry. The first aspartate in the D255DXXD motif on helix H is the only residue that directly coordinates to Mg2+B; however, D256 and D259 indirectly interact with Mg2+B via bridging water molecules. Oxygen atoms of the two phosphonate groups of the inhibitor BPH-721 also accept hydrogen bonds from the side chains of R112, K212, and K269 [26].
Geranylgeranyl Diphosphate Synthase, Nonspecific Prenyl Synthase
Geranylgeranyl disphosphate synthase (GGPP synthase) catalyses the condensation of IPP and FPP to form GGPP (Fig. 1). Recently, GGPP synthase has emerged as a pharmaceutical target for the treatment of cancer since geranylgeranylation is involved in Rac, Rap and Rho signaling pathways [27]. The crystal structures of GGPP synthases from Thermus thermophilus [28], Sinapis alba [29], and Saccharomyces cerevisiae [30] have been determined in addition to that of human GGPP synthase [31]. However, a crystal structure containing a complete trinuclear magnesium cluster has only been observed in the active site of monomer B of the S. cerevisiae GGPP synthase-Mg2+3-BPH-252 complex (Fig. 3e) [32]. The first aspartate of the D80DIED84 motif coordinates to Mg2+A and Mg2+C with syn,syn-bidentate geometry, and one oxygen atom of D84 bridges Mg2+A and Mg2+C with syn,anti-coordination stereochemistry; the first aspartate of the second D214DYLN motif coordinates to Mg2+B. Each Mg2+ ion is coordinated with octahedral geometry, with non-protein coordination sites occupied by oxygen atoms of the inhibitor phosphonate groups and water molecules. Oxygen atoms of the two phosphonate groups of the inhibitor BPH-252 also accept hydrogen bonds from R89, K174 and K238.
Recently, the crystal structure of a nonspecific prenyl synthase from Cryptosporidium parvum has been determined [33]. C. parvum causes livestock infections and is classified as a bioterrorism threat by the Centers for Disease Control and Prevention [34]. The enzyme has a unique ability to catalyze chain elongation reactions with isoprenoid substrates of various lengths to generate C20-C45 linear isoprenoids products. The crystal structure of the enzyme reveals conservation of the classic α-helical terpenoid synthase fold, and its complex with the inhibitor risedronate reveals that a complete trinuclear magnesium cluster is coordinated by DDXXD and NDXXD motifs (Fig. 3f) [33]. The first carboxylate of the D115DXXD119 motif on helix D is oriented for coordination to Mg2+A and Mg2+C with syn,syn-bidentate geometry; however, the distance between D115 and Mg2+C is 3.14 Å, thus too long to be considered an inner-sphere metal coordination interaction. One oxygen atom of the third aspartate, D119, bridges Mg2+A and Mg2+C with syn,anti-coordination stereochemistry. N254 of the N254DXXD motif on helix H coordinates to Mg2+B, and D255 and D258 indirectly interact with Mg2+B via bridging waters. The diphosphate moiety additionally accepts hydrogen bonds from K210 and G251, and the closed active site conformation is stabilized by hydrogen bonds between D116 of the DDXXD motif and R124, and D255 of the NDXXD motif and Q251.
ISOPRENOID CYCLIZATION ENZYMES
Fungal cyclases
The sesquiterpene cyclase trichodiene synthase from Fusarium sporotrichioides catalyzes the first committed step in the biosynthesis of nearly 100 different trichothecene mycotoxins. Trichodiene synthase is one of the most thoroughly studied terpenoid cyclases, and enzymological and crystallographic studies have illuminated important features in the cyclization mechanism (reviewed in [15]). Recent computational studies have also provided new insight on the catalytic mechanism [35]. The structures of unliganded trichodiene synthase and the trichodiene synthase-Mg2+3-PPi complex were the first to reveal the binding of a trinuclear magnesium cluster in the active site of a terpenoid synthase (Fig. 4a) [20]. The first aspartate of the D100DXXD motif on helix D coordinates to Mg2+A and Mg2+C with syn,syn-bidentate geometry. The second metal binding motif N225DLMS229FYKE333 is located on helix H and coordinates to Mg2+B. All three metal ions are additionally coordinated by PPi and solvent molecules to complete octahedral coordination polyhedra.
Fig. 4.

Conservation of Mg2+3-PPi and -diphosphate binding motifs among terpenoid cyclases. Metal coordination (black) and hydrogen bond (red) interactions with phosphate(s) are indicated. (a) F. sporotrichioides trichodiene synthase-Mg2+3-PPi complex (PDB code 1JFG); (b) A. terreus aristolochene synthase-Mg2+3-PPi complex (PDB 2OA6); (c) S. coelicolor epi-isozizaene synthase-Mg2+3-PPi complex (PDB code 3KB9); (d) N. tabacum 5-epi-aristolochene synthase-Mg2+3-farnesyl hydroxyphosphonate complex (PDB code 5EAT; note that many of the metal-phosphate interactions indicated are too long to be considered inner-sphere metal coordination interactions); (e) S. officinalis (+)-bornyl diphosphate synthase-Mg2+3-PPi complex (PDB code 1N22; metal ions are labeled according to the convention first established for trichodiene synthase); (f) M. spicata limonene synthase-Mn2+3-FLPP complex (PDB code 2ONG; conserved hydrogen bonding is indicated between D353 and R315 despite poor geometry).
Superposition of unliganded and Mg2+3-PPi complexed trichodiene synthase structures reveals conformational changes that cap the active site upon ligand binding. Overall, the r.m.s. deviation between the native and liganded structures is 1.4 Å for 349 Cα atoms. Interestingly, upon ligand binding, D101 in the aspartate-rich motif forms a salt bridge with R304, which donates a hydrogen bond to PPi. In addition to Mg2+ coordination interactions, the PPi anion also accepts hydrogen bonds from R182, K232, and Y305. The D101-R304-PPi hydrogen bond network appears to link substrate binding with the transition between the open and closed active site conformations. Assuming that the diphosphate group of FPP triggers the same structural changes as observed for PPi, the substrate is sequestered from bulk solvent and the complete trinuclear magnesium cluster triggers departure of the diphosphate leaving group to generate the carbocation that initiates the cyclization cascade. The seemingly conservative D100E mutation results in a 22-fold loss in catalytic activity (measured by kcat/KM) and structural studies indicate that the additional methylene group of E100 perturbs the Mg2+3-PPi complex such that Mg2+A binding is weakened, E233 breaks its coordination interaction with Mg2+B, and Mg2+C is dissociated; additionally, hydrogen bond interactions between PPi and R182 and R304 are broken [36,37].
The role of the D101-R304 salt bridge in closing the trichodiene synthase active site has been explored in mutagenesis studies. The D101E mutation results in a moderate 5-fold decrease in catalytic activity; however, there are no crystal structures of this mutant available for study [38]. In contrast, the R304K mutant results in a 5000 fold decrease in catalytic activity, and the crystal structure of R304K trichodiene synthase complexed with Mg2+3-PPi-(R)-azabisabolene reveals the loss of the expected hydrogen bond between K304 and D101 [39]. Although the PPi binding motif remains intact in the R304K mutant, it is evident that the R304-D101 hydrogen bond is critical for properly activating the substrate diphosphate group. In contrast, while Y305 donates a hydrogen bond to PPi in the wild-type enzyme complex with Mg2+3-PPi, catalytic activity and PPi binding are not significantly affected in the Y305F mutant [37,40].
Another fungal cyclase that has been the subject of extensive structural and functional study is aristolochene synthase, which is a sesquiterpene cyclase that catalyzes the cyclization of FPP to form (+)-aristolochene. Structures of aristolochene synthases from Penicillium roqueforti [41] and Aspergillus terreus [42] have been solved; these enzymes are related by 61 % amino acid sequence identity. Although there is no crystal structure of P. roqueforti aristolochene synthase complexed with Mg2+3-PPi, the structure of the A. terreus aristolochene synthase Mg2+3-PPi complex [42] (Fig. 4b) indicates conservation of the Mg2+3-PPi binding motif first observed in trichodiene synthase [20].
The aspartate-rich motif of aristolochene synthase on helix D appears as D90DXXE. The carboxylate side chain of D90 coordinates to Mg2+A and Mg2+B with syn,syn-bidentate geometry, and is the only residue in the aspartate motif that coordinates to the Mg2+ ions. The carboxylate group of D91 makes a salt bridge with R304, and the final carboxylate in the motif is E119, which accepts a hydrogen bond from a water molecule coordinated to Mg2+A. The second metal binding motif N219DIYS223YEKE227 is located on helix H and chelates Mg2+B [42], consistent with the structures of terpenoid cyclases from plants, bacteria, and fungi [20,21,43-45]. As found in the active sites of trichodiene synthase [20] and epi-isozizaene synthase [43], PPi binding in aristolochene synthase is similarly accommodated by hydrogen bonds donated from two arginines (R175 and F314), one lysine (K226), and one tyrosine (Y315) (Fig. 4b).
Bacterial cyclases
In recent years, prokaryotes have emerged as sources of diverse isoprenoids. Specifically, a large number of novel isoprenoids have been isolated from organisms belonging to the taxonomical order Actinomycetales [46]. The crystal structures of two bacterial sesquiterpene cyclases have been solved, and both derive from Actinomycetales: pentalenene synthase from Streptomyces UC5319 [47] and epi-isozizaene synthase from Streptomyces coelicolor [43,48]. Pentalenene synthase catalyzes the cyclization of FPP to form the tricyclic sesquiterpene pentalenene in the first committed step in the biosynthesis of the pentalenolactone family of antibiotics [49]. Although the structure of pentalenene synthase was the first to be reported of a terpenoid cyclase and demonstrated that the terpenoid cyclase shared the FPP synthase fold first observed for avian FPP synthase [17], no structure of this bacterial terpenoid cyclase complexed with metal ions is available.
However, the crystal structure of epi-isozizaene synthase complexed with Mg2+3-PPi-BTAC (BTAC is the benzyltriethylammonium cation, a crystallization additive) reveals that Mg2+3-PPi binding motifs are conserved between fungal and bacterial terpenoid cyclases (Fig. 4c) [43]. The first aspartate of the aspartate-rich motif D99DRHD coordinates to Mg2+A and Mg2+C with syn,syn-bidentate geometry, and Mg2+B is chelated by N240DLCS244LPKE248. Each Mg2+ ion is coordinated with octahedral geometry and nonprotein coordination sites are occupied by oxygen atoms of PPi and water molecules. The PPi anion also accepts hydrogen bonds from the side chains of R194, K247, R338, and Y339 [43] which correspond to R182, K232, R304 and Y305 of trichodiene synthase [20] and R175, K226, R314 and Y315 of aristolochene synthase [42]. Pentalenene synthase and epi-isozizaene synthase share 24 % amino acid sequence identity, and residues that interact with Mg2+ ions or PPi in epi-isozizaene synthase are conserved in pentalenene synthase. Superposition of the liganded closed structure of epi-isozizaene synthase with unliganded pentalenene synthase reveals a very similar alignment of the metal binding motifs, with pentalenene synthase helices D and H 1.5 Å further apart than in epi-isozizaene. Accordingly, upon substrate or PPi binding the active site of pentalenene synthase presumably undergoes a conformational change to a closed conformation comparable to that observed for the epi-isozizaene synthase Mg2+3-PPi-BTAC complex.
Although the second aspartate of the epi-isozizaene synthase aspartate-rich motif, D100, does not directly interact with the Mg2+ ions or PPi, it does accept a hydrogen bond from R338, which also donates a hydrogen bond to PPi [43]. Site-directed mutagenesis reveals that the D100N mutation causes a >95 % loss of activity with respect to the native enzyme [50], suggesting that the D100N mutation disrupts the D100-R338-PPi hydrogen bond network presumed to be important for substrate recognition. As previously discussed, the second aspartate in the aspartate-rich motifs of trichodiene synthase and aristolochene synthase similarly stabilizes a hydrogen bond network with R304 and PPi [20,42], so it appears that the bacterial and fungal cyclases share the same molecular strategy for linking the molecular recognition of the substrate diphosphate group with the active site closure mechanism.
The third aspartate in the aspartate rich motif of epi-isozizaene synthase, D103, points away from the active site and makes no hydrogen bond interactions that are involved in substrate binding, as also observed in Mg2+3-PPi complexes of trichodiene synthase. The absence of a structural or catalytic role for the terminal aspartate in the aspartate-rich motif of bacterial terpenoid cyclases is supported by mutagenesis of the corresponding residue in pentalenene synthase: the D84E mutation results in a mere 3-fold loss of catalytic activity (as measured by kcat/KM), whereas the D80E and D81E mutations yield 3500- and 400-fold reductions in activity, respectively [51].
Plant cyclases
5-Epi-aristolochene synthase from Nicotiana tabacum catalyzes the cyclization of FPP to form 5-epi-aristolochene in the first committed step in the biosynthesis of the antifungal phytoalexin capsidiol [44]. The first crystal structure determined of a plant terpenoid cyclase and the second terpenoid cyclase structure to be reported, the structure of 5-epi-aristolochene synthase reveals the presence of 2 domains [44]: a catalytically active C-terminal domain that adopts the α-helical class I terpenoid synthase fold, and an N-terminal domain of unknown function that nevertheless exhibits an α-helical fold similar to that of a class II terpenoid synthase [9]. Two metal-binding motifs are identified: an aspartate-rich motif D301DXXD305, and a D444DTAT448YEVE452 motif. While the binding of a trinuclear magnesium cluster was identified in the 5-epi-aristolochene synthase farnesyl hydroxyphosphonate complex (Fig. 4d), analysis of the structure reveals that many of the coordination interactions with Mg2+ ions range 2.2 Å – 3.7 Å, longer than expected for ideal Mg2+ coordination [52]. This could suggest that the structure is that of a partially closed conformation. Nonetheless, D301 and D305 of the aspartate-rich motif coordinate to Mg2+A and Mg2+C, while the “DTE” motif chelates Mg2+B.
Interestingly, metal binding motifs are shared between sesquiterpene cyclases and monoterpene cyclases from plants. The monoterpene cyclase (+)-bornyl diphosphate synthase catalyzes the cyclization of geranyl diphosphate (GPP) to form (+)-bornyl diphosphate. This cyclization is unusual in that the substrate diphosphate group is reincorporated into the product. The structure of (+)-bornyl diphosphate synthase from Salvia officinalis was the first of a monoterpene cyclase [21], and remains the only monoterpene cyclase for which structures have been solved in unliganded and liganded states. The crystal structure of (+)-bornyl diphosphate synthase reveals the two-domain α-helical architecture first observed for the plant sesquiterpene synthase 5-epi-aristolochene synthase: a catalytically active C-terminal domain adopting the class I terpenoid synthase fold, and an N-terminal domain adopting the class II terpenoid synthase fold (however, the N-terminal polypeptide caps the active site of the C-terminal domain in ligand complexes) [21]. The (+)-bornyl diphosphate synthase-Mg2+3-PPi complex reveals that conserved metal-binding motifs and the PPi anion (or the diphosphate group of the product itself, (+)-bornyl diphosphate) coordinate to 3 Mg2+ ions (Fig. 4e). The first carboxylate of the D351DXXD355 motif coordinates to Mg2+A and Mg2+C with syn,syn-bidentate geometry, and D355 bridges Mg2+A and Mg2+C with syn,anti-coordination stereochemistry. Interestingly, unlike metal binding in the active sites of bacterial and fungal cyclases, both the first and third aspartates in the DDXXD motif of plant terpenoid cyclases coordinate to the catalytic metal ions. The second metal binding motif, D496DKGT500SYFE504, chelates Mg2+B [21]. In addition to metal ion coordination interactions, the PPi anion accepts hydrogen bonds from R314, R493, and K512. Comparison of the structures of unliganded and Mg2+3-PPi complexed (+)-bornyl diphosphate synthase reveals several Mg2+3-PPi induced conformational changes; however, the r.m.s. deviation of 306 Cα atoms in the catalytic C-terminal domain is only 0.6 Å [21], significantly lower than observed for ligand-induced conformational changes in trichodiene synthase (1.4 Å) [20] and aristolochene synthase (1.8 Å) [42].
The recent structure determination of another plant monoterpene cyclase, limonene synthase from Mentha spicata [45], similarly reveals conservation of a trinuclear metal cluster in a cyclization reaction that generates 94% (−)-(4S)-limonene, and ~2% myrcene, (−)-α-pinene, and (−)-β-pinene [53]. Limonene synthase shares the 2-domain α-helical fold common to plant terpenoid cyclases. Limonene synthase displays similar activity with Mg2+ or Mn2+ (a common feature of some terpenoid cyclases), and the structure of the enzyme has been determined in complex with 3 Mn2+ ions and the intermediate analogue 2-fluorolinalyl diphosphate (FLPP) (Fig. 4f) [45]. Metal coordination interactions are similar to those observed in (+)-bornyl diphosphate synthase [21]. In limonene synthase, the first carboxylate of the D352DXXD356 motif coordinates to Mn2+A and Mn2+C with syn,syn-bidentate geometry, and one oxygen atom of D356 bridges Mn2+A and Mn2+C with syn,anti-coordination stereochemistry. Two out of three residues in the second metal binding motif, D496DLGT500SVEE504, chelate Mg2+B; the position of the side chain of E504 is not indicated and is presumably disordered. Additionally, the γ-hydroxyl of T500 is 3.2 Å away from Mg2+B, which is too long to be considered an inner sphere coordination interaction. The diphosphate group of the bound intermediate analogue FLPP accepts hydrogen bonds from R315, R493, and K512 [45].
Finally, the sesquiterpene cyclase (+)-δ-cadinene synthase from Gossypium arboreum (tree cotton) catalyzes the first committed step in the biosynthesis of the triterpene phytoalexin gossypol, a major defense metabolite synthesized by cotton plants [54]. The recently determined structure of the unliganded enzyme and its complex with 2-fluorofarnesyl diphosphate (2F-FPP) reveals that minimal structural deviations result from ligand binding (the r.m.s. deviations are 0.28 Å and 0.50 Å between unliganded and liganded monomers A (514 Cα atoms) and B (494 Cα atoms), respectively) [55]. In contrast with the plant terpenoid cyclases previously discussed [21,44,45], (+)-δ-cadinene synthase contains a second aspartate-rich motif in place of the DTE motif on helix H. As previously discussed, this motif on helix H is common to chain elongation enzymes such as farnesyl diphosphate synthase, and (+)-δ-cadinene synthase is unique among known class I terpenoid cyclases in that it contains two aspartate-rich motifs for metal coordination. The structure of the liganded enzyme reveals a putative Mg2+3 cluster (weak electron density characterizes the three Mg2+ ions); Mg2+A and Mg2+C are coordinated by D307 and D311 of the first D307DXXD311 motif, and Mg2+B is coordinated by D451 and E455 of the second aspartate-rich motif, D451DVAE455 (Fig. 5). However, many of the carboxylate-Mg2+ distances observed are too long for inner sphere metal coordination interactions; therefore, the structure may reflect an incomplete transition between the “open” and “closed” active site conformations. The diphosphate moiety of 2F-FPP accepts one hydrogen bond from a nearby basic residue, R448.
Fig. 5.
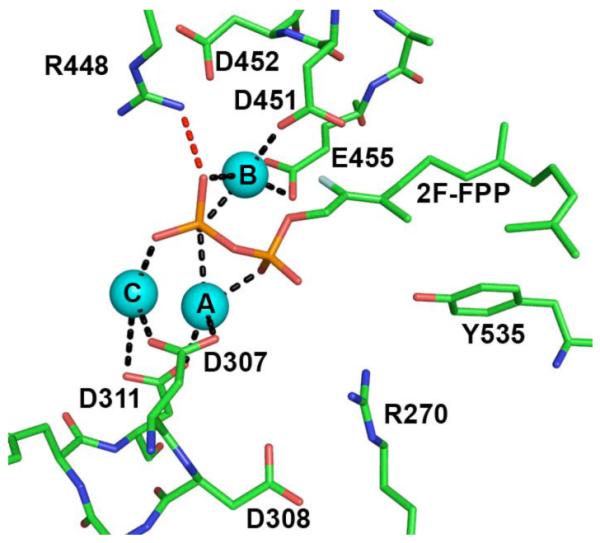
The diphosphate binding site of (+)-δ-cadinene synthase from G. arboreum (PDB code 3G4F) with a putative Mg2+3 cluster and 2F-FPP bound. Metal ions are labeled according to convention established for trichodiene synthase. Some metal-phosphate interactions are too long to be considered inner-sphere coordination interactions, which could be a consequence of the nonproductive binding mode observed for 2F-FPP.
DISCUSSION
Although the metal-dependence of catalysis by class I terpenoid synthases has been known for decades [56], it was not until 2001 that the crystal structure of a terpenoid cyclase-Mg2+3-PPi complex (trichodiene synthase) revealed that a trinuclear metal cluster accommodates PPi binding; this trinuclear metal cluster is similarly implicated in binding and activating substrate farnesyl diphosphate for catalysis [20]. Since then, many X-ray crystal structures of isoprenoid coupling enzymes and terpenoid cyclases have been determined containing Mg2+3 (or Mn2+3) clusters. Comparisons of these structures reveal significant conservation in the constellation of metal ions and the residues that coordinate to these metal ions (Figs. 3 and 4) despite generally insignificant amino acid sequence identity among these enzymes.
Trinuclear metal cluster coordination in FPP synthases is conserved among humans, bacteria and protozoans. Two aspartate-rich DDXXD binding motifs coordinate to 3 Mg2+ ions, which are also coordinated by the substrate diphosphate group. The first and last aspartate in the first DDXXD motif coordinate to Mg2+A and Mg2+C, and the first aspartate of the second DDXXD motif coordinates to Mg2+B. Additionally conserved are one arginine and two lysine residues that donate hydrogen bonds to diphosphate oxygens; the conserved arginine residue also donates hydrogen bond(s) to the second aspartate in the first DDXXD motif (Fig. 3a-d). The crystal structures of other isoprenoid coupling enzymes, GGPP synthase and nonspecific prenyl synthase, similarly reveal conservation of Mg2+3 binding motifs. Hydrogen bond interactions with PPi are also conserved (Fig. 3e-f).
It is notable that the constellation of three metal ions and hydrogen bond donors is also conserved, with minor variations, in terpenoid cyclases from plants, bacteria, and fungi (Fig. 3a-f). First, Mg2+A and Mg2+C are coordinated by the first DDXXD motif: bacterial and fungal cyclases utilize only the first aspartate of this motif, whereas plant cyclases utilize the first and third aspartates of this motif (analogous to isoprenoid coupling enzymes). Second, the second aspartate-rich motif is usually replaced by an NDXXSXXXE motif in bacterial and fungal terpenoid cyclases and a DXXXTXXXE motif in plant terpenoid cyclases, in which boldface residues chelate Mg2+B (although there can be some variations in this sequence, e.g., see [57]). One exception, however, is (+)-δ-cadinene synthase, in which two aspartate-rich motifs coordinate to the trinuclear metal cluster. Third, residues that donate hydrogen bonds to PPi are conserved in terpenoid cyclases across different domains of life. Specifically, two arginines donate hydrogen bonds to diphosphate oxygens: one appears to replace a conserved lysine serving this function in the isoprenoid coupling enzymes, and the other also donates a hydrogen bond to the second aspartate of the first DDXXD motif (as observed in the isoprenoid coupling enzymes). In bacterial and fungal terpenoid cyclases, conserved lysine and tyrosine residues additionally donate hydrogen bonds to PPi.
In all cases in which a complete Mg2+3-PPi cluster is bound, two 6-membered ring chelates are formed with Mg2+A and Mg2+B (Fig. 6). The conformations of these 6-membered rings can vary, e.g., sofa, half-chair, etc. Such 6-membered ring chelates are occasionally observed in metal-diphosphate binding interactions, e.g., in the binding of the substrate analogue imidodiphosphate to inorganic pyrophosphatase [58]. Although the class I terpenoid synthase fold and metal-pyrophosphate binding motif are conserved among isoprenoid coupling enzymes and terpenoid cyclases, it is interesting that the substrate orientation with regard to the trinuclear metal cluster appears to be opposite in these two groups of enzymes that have yielded structures to date: that is, the terminal phosphate group of an isoprenoid diphosphate group does not coordinate to Mg2+C in an isoprenoid coupling enzyme (Fig. 3e), but it does in a terpenoid cyclase (Fig. 4f).
Fig. 6.

Stereoview of the Mg2+3-PPi cluster from epi-isozizaene synthase [43]. Dashed lines (black) represent metal-coordination interactions. The PPi anion forms 6-membered ring chelates with Mg2+A and Mg2+B, both of which adopt distorted sofa conformations.
In summary, conservation of a trinuclear metal cluster is critical for catalysis by class I terpenoid synthases. This cluster not only serves to bind and orient the flexible isoprenoid substrate in the precatalytic Michaelis complex, but it also triggers leaving group departure and initial carbocation formation. Conserved hydrogen bond donors in the terpenoid synthase active site assist the metal cluster in this function. That the trinuclear metal cluster is conserved for catalysis by terpenoid synthases from many domains of life suggests a common ancestry for this family of enzymes in the evolution of terpenoid biosynthesis.
ACKNOWLEDGEMENTS
This work was supported by an NIH Chemical Biology Interface training grant (to J.A.A.) and NIH Grant GM56838 (to D.W.C.).
REFERENCES
- 1.Christianson DW. Science. 2007;316:60–61. doi: 10.1126/science.1141630. [DOI] [PubMed] [Google Scholar]
- 2.Aharoni A, Jongsma MA, Bouwmeester HJ. Trends Plant Sci. 2005;10:594–602. doi: 10.1016/j.tplants.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 3.Pichersky E, Noel JP, Dudareva N. Science. 2006;311:808–811. doi: 10.1126/science.1118510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rondeau J-M, Bitsch F, Bourgier E, Geiser M, Hemmig R, Kroemer M, Lehmann S, Ramage P, Rieffel S, Strauss A, Green JR, Jahnke W. ChemMedChem. 2006;1:267–273. doi: 10.1002/cmdc.200500059. [DOI] [PubMed] [Google Scholar]
- 5.Agrawal AG, Somani RR. Mini Rev. Med. Chem. 2009;9:638–652. doi: 10.2174/138955709788452702. [DOI] [PubMed] [Google Scholar]
- 6.Licata AA. Ann. Pharmacother. 2005;39:668–677. doi: 10.1345/aph.1E357. [DOI] [PubMed] [Google Scholar]
- 7.Ebetino FH, Roze CN, McKenna CE, Barnett BL, Dunford JE, Russell RGG, Mieling GE, Rogers MJ. J. Organomet. Chem. 2005;690:2679–2687. [Google Scholar]
- 8.Thulasiram HV, Erickson HK, Poulter CD. Science. 2007;316:73–76. doi: 10.1126/science.1137786. [DOI] [PubMed] [Google Scholar]
- 9.Wendt KU, Schulz GE. Structure. 1998;6:127–133. doi: 10.1016/s0969-2126(98)00015-x. [DOI] [PubMed] [Google Scholar]
- 10.Lesburg CA, Caruthers JM, Paschall CM, Christianson DW. Curr. Opin. Struct. Biol. 1998;8:695–703. doi: 10.1016/s0959-440x(98)80088-2. [DOI] [PubMed] [Google Scholar]
- 11.Croteau R, Cane DE. Methods Enzymol. 1985;110:383–405. [Google Scholar]
- 12.Cane DE. Chem. Rev. 1990;90:1089–1103. [Google Scholar]
- 13.Yoshikuni Y, Ferrin TE, Keasling JD. Nature. 2006;440:1078–1082. doi: 10.1038/nature04607. [DOI] [PubMed] [Google Scholar]
- 14.Austin MB, O’Maille PE, Noel JP. Nat. Chem. Biol. 2008;4:217–222. doi: 10.1038/nchembio0408-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Christianson DW. Chem. Rev. 2006;106:3412–3442. doi: 10.1021/cr050286w. [DOI] [PubMed] [Google Scholar]
- 16.Christianson DW. Curr. Opin. Chem. Biol. 2008;12:141–150. doi: 10.1016/j.cbpa.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tarshis LC, Yan M, Poulter CD, Sacchettini JC. Biochemistry. 1994;33:10871–10877. doi: 10.1021/bi00202a004. [DOI] [PubMed] [Google Scholar]
- 18.Ashby MN, Edwards PA. J. Biol. Chem. 1990;265:13157–13164. [PubMed] [Google Scholar]
- 19.Hosfield DJ, Zhang Y, Dougan DR, Broun A, Tari LW, Swanson RV, Finn J. J. Biol. Chem. 2004;279:8526–8529. doi: 10.1074/jbc.C300511200. [DOI] [PubMed] [Google Scholar]
- 20.Rynkiewicz MJ, Cane DE, Christianson DW. Proc. Natl. Acad. Sci. U. S. A. 2001;98:13543–13548. doi: 10.1073/pnas.231313098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Whittington DA, Wise ML, Urbansky M, Coates RM, Croteau RB, Christianson DW. Proc. Natl. Acad. Sci. U. S. A. 2002;99:15375–15380. doi: 10.1073/pnas.232591099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tanowitz HB, Machado FS, Jelicks LA, Shirani J, de Carvalho AC, Spray DC, Factor SM, Kirchhoff LV, Weiss LM. Prog. Cardiovasc. Dis. 2009;51:524–539. doi: 10.1016/j.pcad.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garzoni LR, Caldera A, Meirelles M. d. N., de Castro SL, Docampo R, Meints GA, Oldfield E, Urbina JA. Int. J. Antimicrob. Agents. 2004;23:273–285. doi: 10.1016/j.ijantimicag.2003.07.020. [DOI] [PubMed] [Google Scholar]
- 24.Gabelli SB, McLellan JS, Montalvetti A, Oldfield E, Docampo R, Amzel LM. Proteins: Struct. Funct. Bioinf. 2006;62:80–88. doi: 10.1002/prot.20754. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Y, Cao R, Yin F, Hudock MP, Guo R-T, Krysiak K, Mukherjee S, Gao Y-G, Robinson H, Song Y, No JH, Bergan K, Leon A, Cass L, Goddard A, Chang T-K, Lin F-Y, Van Beek E, Papapoulos S, Wang AH, Kubo T, Ochi M, Mukkamala D, Oldfield E. J. Am. Chem. Soc. 2009;131:5153–5162. doi: 10.1021/ja808285e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mao J, Mukherjee S, Zhang Y, Cao R, Sanders JM, Song Y, Zhang Y, Meints GA, Gao YG, Mukkamala D, Hudock MP, Oldfield E. J. Am. Chem. Soc. 2006;128:14485–14497. doi: 10.1021/ja061737c. [DOI] [PubMed] [Google Scholar]
- 27.Russell RGG. Ann. N.Y. Acad. Sci. 2006;1068:367–401. doi: 10.1196/annals.1346.041. [DOI] [PubMed] [Google Scholar]
- 28.Nishio K, Nodake Y, Hamada K, Suto K, Nakagawa N, Kuramitsu S, Miura K. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004;60:178–180. doi: 10.1107/s0907444903025496. [DOI] [PubMed] [Google Scholar]
- 29.Kloer DP, Welsch R, Beyer P, Schulz GE. Biochemistry. 2006;45:15197–15204. doi: 10.1021/bi061572k. [DOI] [PubMed] [Google Scholar]
- 30.Chang T-H, Guo R-T, Ko T-P, Wang AH-J, Liang P-H. J. Biol. Chem. 2006;281:14991–15000. doi: 10.1074/jbc.M512886200. [DOI] [PubMed] [Google Scholar]
- 31.Kavanagh KL, Dunford JE, Bunkoczi G, Russell RGG, Oppermann U. J. Biol. Chem. 2006;281:22004–22012. doi: 10.1074/jbc.M602603200. [DOI] [PubMed] [Google Scholar]
- 32.Chen CK-M, Hudock MP, Zhang Y, Guo R-T, Cao R, No JH, Liang P-H, Ko T-P, Chang T-H, Chang S-C, Song Y, Axelson J, Kumar A, Wang AH, Oldfield E. J. Med. Chem. 2008;51 doi: 10.1021/jm800325y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Artz JD, Dunford JE, Arrowood MJ, Dong A, Chruszcz M, Kavanagh KL, Minor W, Russell RGG, Ebetino FH, Oppermann U, Hui R. Chem. Biol. 2008;15:1296–1306. doi: 10.1016/j.chembiol.2008.10.017. [DOI] [PubMed] [Google Scholar]
- 34.Hashsham SA, Wick LM, Rouillard J-M, Gulari E, Tiedje JM. Biosens. Bioelectron. 2004;20:668–683. doi: 10.1016/j.bios.2004.06.032. [DOI] [PubMed] [Google Scholar]
- 35.Hong YJ, Tantillo DJ. J. Am. Chem. Soc. 2009;131:7999–8015. doi: 10.1021/ja9005332. [DOI] [PubMed] [Google Scholar]
- 36.Rynkiewicz MJ, Cane DE, Christianson DW. Biochemistry. 2002;41:1732–1741. doi: 10.1021/bi011960g. [DOI] [PubMed] [Google Scholar]
- 37.Vedula LS, Rynkiewicz MJ, Pyun HJ, Coates RM, Cane DE, Christianson DW. Biochemistry. 2005;44:6153–6163. doi: 10.1021/bi050059o. [DOI] [PubMed] [Google Scholar]
- 38.Cane DE, Xue Q, Fitzsimons BC. Biochemistry. 1996;35:12369–12376. doi: 10.1021/bi961344y. [DOI] [PubMed] [Google Scholar]
- 39.Vedula LS, Cane DE, Christianson DW. Biochemistry. 2005;44:12719–12727. doi: 10.1021/bi0510476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cane DE, Shim JH, Xue Q, Fitzsimons BC, Hohn TM. Biochemistry. 1995;34:2480–2488. doi: 10.1021/bi00008a011. [DOI] [PubMed] [Google Scholar]
- 41.Caruthers JM, Kang I, Rynkiewicz MJ, Cane DE, Christianson DW. J. Biol. Chem. 2000;275:25533–25539. doi: 10.1074/jbc.M000433200. [DOI] [PubMed] [Google Scholar]
- 42.Shishova EY, Di Costanzo L, Cane DE, Christianson DW. Biochemistry. 2007;46:1941–1951. doi: 10.1021/bi0622524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aaron JA, Lin X, Cane DE, Christianson DW. Submitted for publication. [Google Scholar]
- 44.Starks CM, Back K, Chappell J, Noel JP. Science. 1997;277:1815–1820. doi: 10.1126/science.277.5333.1815. [DOI] [PubMed] [Google Scholar]
- 45.Hyatt DC, Youn B, Zhao Y, Santhamma B, Coates RM, Croteau RB, Kang C. Proc. Natl. Acad. Sci. U. S. A. 2007;104:5360–5365. doi: 10.1073/pnas.0700915104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Daum M, Herrmann S, Wilkinson B, Bechthold A. Curr. Opin. Chem. Biol. 2009;13:180–188. doi: 10.1016/j.cbpa.2009.02.029. [DOI] [PubMed] [Google Scholar]
- 47.Lesburg CA, Zhai G, Cane DE, Christianson DW. Science. 1997;277:1820–1824. doi: 10.1126/science.277.5333.1820. [DOI] [PubMed] [Google Scholar]
- 48.Lin X, Hopson R, Cane DE. J. Am. Chem. Soc. 2006;128:6022–6023. doi: 10.1021/ja061292s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Seto H, Yonehara H. J. Antibiot. (Tokyo) 1980;33:92–93. doi: 10.7164/antibiotics.33.92. [DOI] [PubMed] [Google Scholar]
- 50.Lin X, Cane DE. J. Am. Chem. Soc. 2009;131:6332–6333. doi: 10.1021/ja901313v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Seemann M, Zhai G, de Kraker J-W, Paschall CM, Christianson DW, Cane DE. J. Am. Chem. Soc. 2002;124:7681–7689. doi: 10.1021/ja026058q. [DOI] [PubMed] [Google Scholar]
- 52.Zheng H, Chruszcz M, Lasota P, Lebioda L, Minor W. J. Inorg. Biochem. 2008;102:1765–1776. doi: 10.1016/j.jinorgbio.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Williams DC, McGarvey DJ, Katahira EJ, Croteau R. Biochemistry. 1998;37:12213–12220. doi: 10.1021/bi980854k. [DOI] [PubMed] [Google Scholar]
- 54.Chen X-Y, Chen Y, Heinstein P, Davisson VJ. Arch. Biochem. Biophys. 1995;324:255–266. doi: 10.1006/abbi.1995.0038. [DOI] [PubMed] [Google Scholar]
- 55.Gennadios HA, Gonzalez V, Di Costanzo L, Li A, Yu F, Miller DJ, Allemann RK, Christianson DW. Biochemistry. 2009;48:6175–6183. doi: 10.1021/bi900483b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Robinson DR, West CA. Biochemistry. 1970;9:70–79. doi: 10.1021/bi00803a010. [DOI] [PubMed] [Google Scholar]
- 57.Zhou K, Peters RJ. Phytochemistry. 2009;70:366–369. doi: 10.1016/j.phytochem.2008.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fabrichniy IP, Lehtio L, Tammenkoski M, Zyryanov AB, Oksanen E, Baykov AA, Lahti R, Goldman A. J. Biol. Chem. 2007;282:1422–1431. doi: 10.1074/jbc.M513161200. [DOI] [PubMed] [Google Scholar]