

Abstract
More than 120 mutations in the Myelin Protein Zero gene (MPZ, P0) cause various forms of hereditary neuropathy. Two human mutations encoding either P0S63C or P0S63del have been shown to cause demyelination in mice through different gain of function pathomechanisms. P0S63del, for example, is retained in the endoplasmic reticulum (ER) and elicits a pathogenetic unfolded protein response (UPR). As P0 likely forms oligomers, another gain of abnormal function could include a dominant-negative interaction between P0S63del and normal P0 (P0wt). To test this idea, we generated a transgenic mouse that expressed a form of P0wt with a myc epitope tag at the C terminus (P0ct-myc). We show that P0ct-myc is trafficked and functions like P0wt, thus providing a new tool to study P0 in vivo. In mice that express both P0ct-myc and P0S63del, P0S63del specifically delays the transit of P0ct-myc through the ER and reduces the level of P0wt in the myelin sheath by half—a level previously shown to cause demyelination in mice and humans. Surprisingly, P0ct-myc does not co-immunoprecipitate with P0S63del, suggesting an indirect interaction. Thus, P0S63del causes not only a UPR-related toxic mechanism, but also a dominant-negative effect on P0wt that probably contributes to demyelinating neuropathy.
INTRODUCTION
P0 is an abundant 28 kDa transmembrane glycoprotein synthesized by myelin-forming Schwann cells in peripheral nerves. The P0 extracellular domain (ECD) contains an adhesive immunoglobulin-like fold that promotes myelin compaction (1–4). Accordingly, peripheral myelin lacking P0 is largely uncompacted (5), and mice or patients with half dosage of P0 develop a late-onset demyelinating neuropathy (6). In addition, >120 mutations in MPZ, many in the ECD, are linked to hereditary demyelinating neuropathies (7). However, most patients manifest early-onset Charcot–Marie–Tooth 1B (CMT1B) or other heterogeneous phenotypes—many with dominant inheritance—implicating gain of function (GOF) mechanisms.
Expression of MPZ mutations in transgenic mice confirmed GOF pathogenesis and demonstrated that different mutants act through diverse pathomechanisms (8). Mutations of P0 serine 63 (S63C and S63del) that cause different phenotypes in patients were studied in mice. Interestingly, P0S63C reaches the myelin sheath and is associated with alterations in myelin packing (9), whereas P0S63del is retained in the endoplasmic reticulum (ER) and activates an unfolded protein response (UPR) (8). Ablation of the UPR mediator CHOP [C/EBP (CAAT-enhancer-binding protein) homologous protein] from S63del mice reduced demyelination, indicating that the UPR is pathogenetic (10). However, only half of demyelination was rescued, raising the spectre of other GOF mechanisms.
The crystal structure of the P0ECD (11) and biochemical analysis of Xenopus laevis myelin (12) suggest that P0 forms oligomers—a context for dominant-negative effects by P0 mutants on the P0wt counterpart. However, such effects have not yet been shown, as there are no antibodies that discriminate between P0wt and mutants. In addition, expression of P0 with a myc epitope tag at the mature N terminus (P0myc) in transgenic mice produced a tomacular, CMT1B-like neuropathy (13). Therefore, we generated mice that synthesize P0 with the myc peptide fused to the intracellular C terminus (P0ct-myc). We show that P0ct-myc nerves contain normal myelin, and that P0ct-myc can increase compaction in P0-null myelin. In addition, we analyse transgenic mice expressing P0ct-myc together with different P0 mutants and determine that P0S63del impedes P0wt trafficking, which reduces the amount of P0 in myelin by approximately half.
RESULTS
Generation of P0ct-myc mice
To generate a mouse synthesizing P0 with a myc tag at the C terminus, we exploited an Mpz transgene whose expression parallels that of endogenous Mpz and produces P0, which rescues dysmyelination in Mpz-null mice (14,15) (Fig. 1A). Transgenic offspring from 4 of 12 founders showed low transgenic copy number, normal ambulation without tremor and normal myelin in P28 sciatic nerves. Line Tg140.12 (hereafter P0ct-myc) was maintained for further study.
Figure 1.

P0ct-myc is expressed at low levels and does not cause myelin alterations. (A) Diagram of the P0ct-myc transgene. (B) Semi-quantitative RTPCR analysis shows expression of transgenic (244 bp, DpnII sensitive; arrowhead) compared with endogenous MpzFVB/N (326 bp, DpnII insensitive). Control MpzFVB/N and MpzC57BL/6 alleles are DpnII insensitive and sensitive, respectively. Densitometric quantifications of sciatic nerves from three animals (P0ct-myc I, II and III) were averaged and plotted relative to endogenous MpzFVB/N expression. (C) Western analysis for P0 or myc on sciatic nerve lysates reveals P0ct-myc (arrowhead) at the expected size. Quantification of P0 experiments from three animals is plotted relative to P0wt (Mr = 28). (D) STSs from P0ct-myc and wt sciatic nerves at P28 and 1 year show no significant differences. P0myc = P0ct-myc. Scale bar: 10 μm.
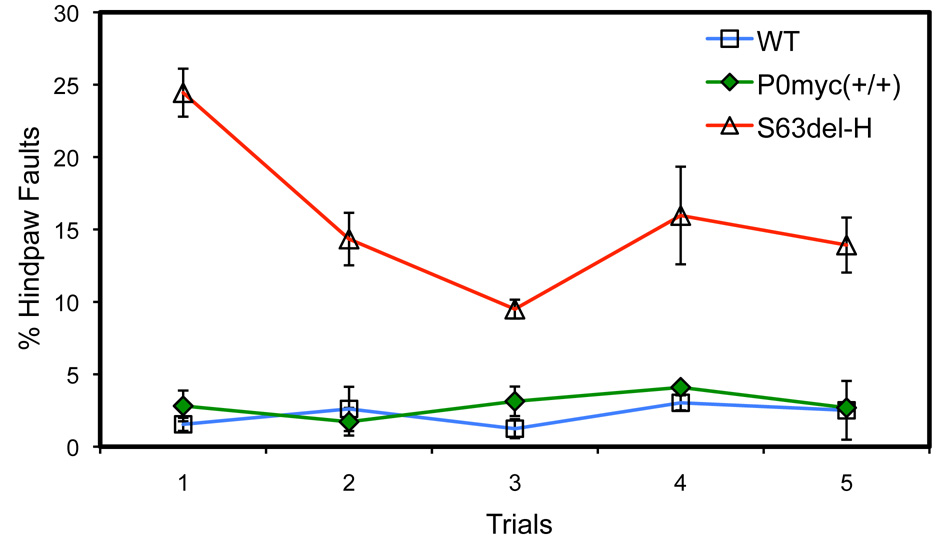
Behaviour of P0ct-myc mice was tested by recording hindpaw placement errors during grid walking; there was no impairment (Supplementary Material, Fig. S1). Transgenic mRNA expression was analysed by reverse transcribed polymerase chain reaction (RTPCR) (Fig. 1B), taking advantage of a polymorphic DpnII site, present only in the transgene. Consistent with the behavioural analysis, transgene expression was 10 ± 0.5% of total endogenous Mpz, well below the previously described pathological threshold of 40% overexpression (15). Western blot analysis of sciatic nerve lysates for P0 showed the presence of a band migrating above endogenous P0, as expected and selectively recognized by antibodies to myc (Fig. 1C). The level of P0ct-myc was 14 ± 0.5% of endogenous P0wt, in accord with the mRNA data.
P0ct-myc does not cause nerve pathology
We analysed semi-thin sections (STSs) of sciatic nerves to investigate whether P0ct-myc causes morphological alterations. No significant differences between P0ct-myc and wild-type (wt) nerves were observed at P28 or at 1 year (Fig. 1D). In P28 P0ct-myc nerves, we observed a small increase in the hypertrophic myelin figures, which are occasionally seen also in wt nerves (not shown). As expected on the basis of the expression data, there was no pathology typical of P0 overexpression [unsorted axons or hypomyelination (15)].
P0ct-myc is correctly trafficked to the myelin sheath
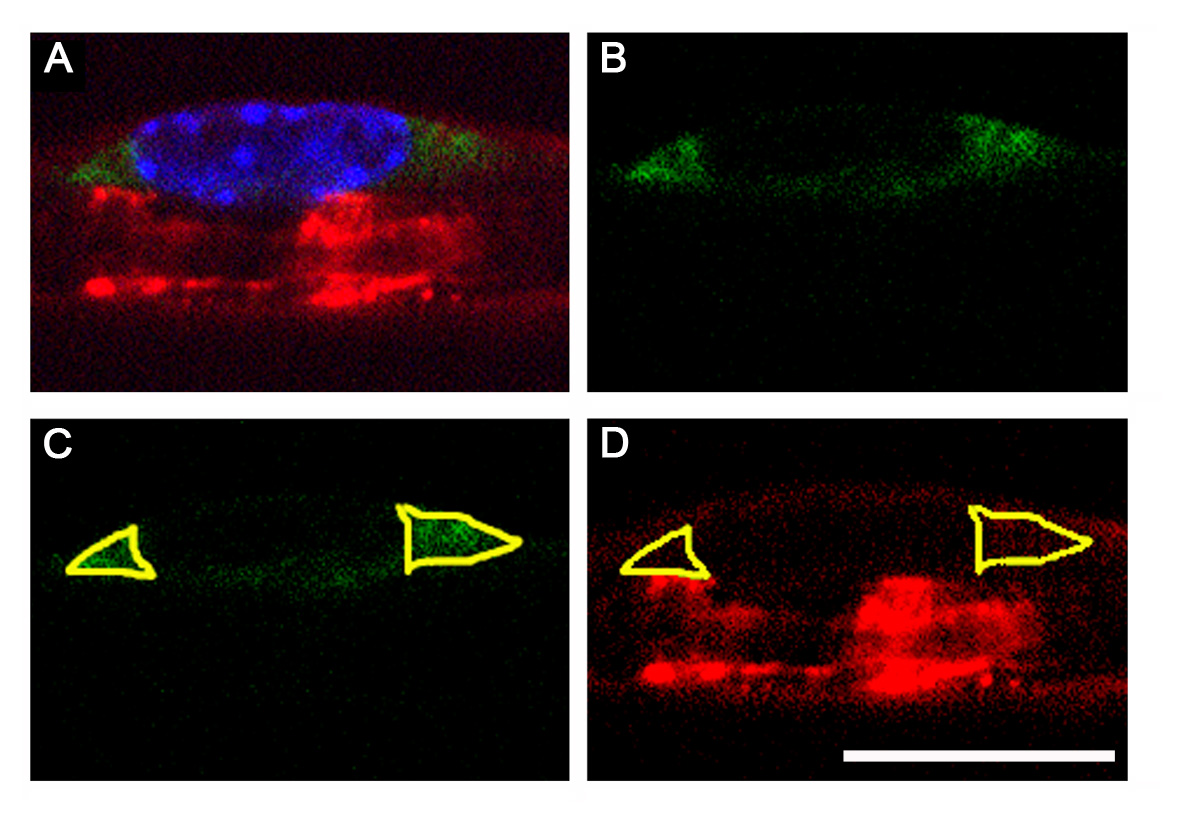
P0ct-myc is targeted to myelin like P0wt, as shown in the confocal images of immunohistochemistry (IHC) on transverse sections or teased fibres of sciatic nerves, using two different antibodies to myc (Fig. 2A and B). Immuno-electron microscopic (IEM) analysis of P0ct-myc sciatic nerves, using rabbit anti-myc antibodies, confirmed the presence of P0ct-myc throughout the thickness of the myelin sheath (Fig. 2C). In IHC and IEM, non-transgenic nerves showed no labelling (data not shown). Further, we investigated whether the presence of P0ct-myc caused an impairment of P0 trafficking. We estimated the amount of P0 staining in the ER of Schwann cells by confocal microscopy of teased fibres (Fig. 2D; for details, see Materials and Methods and Supplementary Material, Fig. S2). As shown in Fig. 2E, there was no significant increase in ER staining for P0 in P0ct-myc when compared with wt nerves, whereas it was increased by >50% in S63del nerves by the same technique [Supplementary Material, Fig. S3, (10)].
Figure 2.

P0ct-myc recapitulates P0wt function. (A and B) IHC for myc (red) in transverse sections (A) and teased fibres (B) from P0ct-myc sciatic nerves shows that P0ct-myc is located in myelin. (C) IEM analysis for myc in P0ct-myc sciatic nerves reveals that P0ct-myc is distributed throughout the myelin sheath. (D and E) Confocal microscopic analysis of teased fibres reveals similar intensity of P0 signal (red) in perinuclear KDEL-positive (green) Schwann cell cytoplasm in P0ct-myc when compared with wt sciatic nerves (note that the P0 antibodies recognize both P0wt and P0ct-myc as in Figure 1C; see Supplementary Material, Figure S2, for sampling area, and Figure S3 for S63del-positive control); ns, not significantly different. (F) Anti-myc antibodies co-immunoprecipitate P0wt (Mr = 28, arrowhead) from P0ct-myc sciatic nerve lysates. wt lysates in IP with myc antibody or with only beads [no myc antibody, wt (−Ab)] serve as negative controls. (G and H) Ultrastructural analysis of myelin sheaths from P0ct-myc/Mpz−/− nerves (no P0wt present) shows regular presence of several compacted myelin wraps (H), which are found much less often in Mpz−/− nerves (G) (Table 1). P0myc, P0ct-myc. Scale bar in H (A and B): 9 μm; (C): 200 nm; (D): 26 μm; (G and H) 50 nm.
P0ct-myc functions like P0wt
P0 is thought to form tetramers in peripheral myelin (11,12). As a first test of P0ct-myc function, we asked whether P0ct-myc interacts with P0wt. We performed co-immunoprecipitation (co-IP) assays on wt and P0ct-myc sciatic nerve lysates, using anti-myc antibodies, followed by western analysis for P0. As expected, no P0 (myc or wt) was precipitated from wt sciatic nerves, whereas both P0ct-myc and P0wt were co-precipitated from the P0ct-myc sciatic nerves (Fig. 2F). Thus, P0ct-myc very likely interacts with P0wt.
Furthermore, we asked whether P0ct-myc contributes to myelin compaction. To this end, we crossed P0ct-myc onto the Mpz-null background (Mpz−/−). By EM analysis, we compared myelin from P0ct-myc/Mpz−/− and Mpz−/− sciatic nerves. Mpz−/− sciatic nerves contain occasional, partially compacted myelin sheaths, but never comprising more than a few completely compacted myelin wraps [Class I units (5)]. P0ct-myc/Mpz−/− nerves showed severe hypomyelination, similar to Mpz−/− nerves. However, P0ct-myc/Mpz−/− nerves contained significantly more fibres with compacted myelin, and the compacted wraps had an increased periodicity in P0ct-myc/Mpz−/− compared with those of Mpz−/−, as would be expected if P0ct-myc had populated and compacted the extracellular apposition in myelin (Fig. 2G and H; Table 1; and reference 16). Thus, P0ct-myc can function in myelin compaction, even if, as expected, its very low dosage is insufficient to fully rescue the P0-null phenotype.
Table 1.
Participation of P0ct-myc in compaction of myelin
| Genotype | Compacta | Non-compact | Period (Å) [mean ± SEM (n)]b |
|---|---|---|---|
| P0ct-myc(−/−) | 170 | 30 | 162 ± 3.0 (11) |
| P0(−/−) | 150 | 59* | 153 ± 2.8 (14)** |
aCompact: fibres with at least some compacted wraps of myelin; non-compact: fibres with no compacted wraps.
bn denotes total myelin sheaths from three nerves (animals) of each genotype.
*P < 0.0001 by χ2 analysis for 1 degree of freedom.
**P < 0.05 by Student's t-test.
P0S63del causes P0ct-myc to be retained in the ER
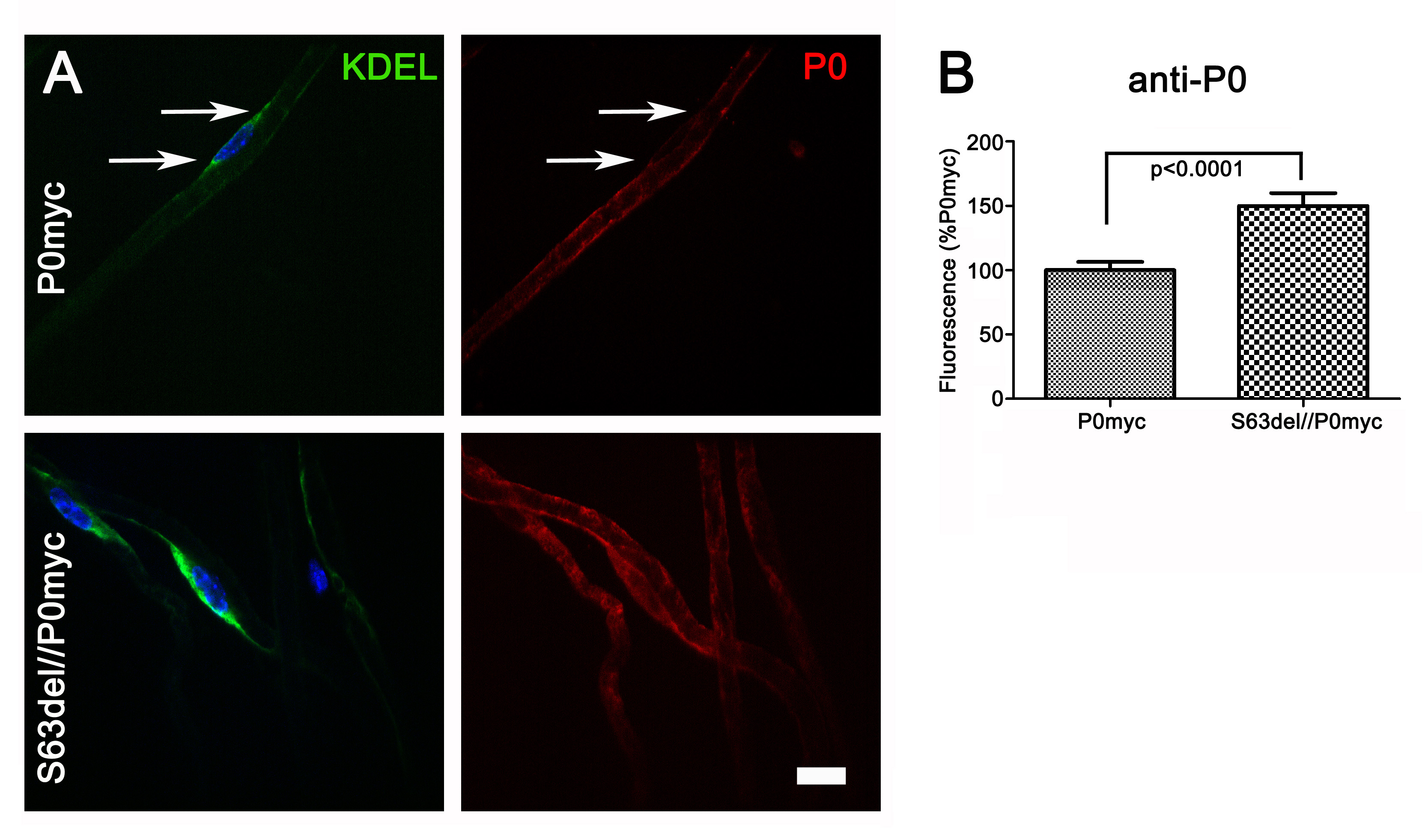
Having determined that P0ct-myc mice are a valid tool to study P0 function in vivo, we investigated P0ct-myc trafficking in the presence of pathogenetic Mpz mutations. For example, even in the presence of P0wt, P0S63del but not P0S63C was previously shown to be retained in the Schwann cell ER in sciatic nerves (8,10). Therefore, we crossed P0ct-myc mice with transgenic lines synthesizing either P0S63del or P0S63C, and we quantified ER retention of P0ct-myc in the presence of either mutant P0 on confocal images of teased fibres from sciatic nerves. Comparable areas of KDEL-positive (ER) perinuclear cytoplasm in Schwann cells were selected and anti-myc fluorescence was quantified (Supplementary Material, Fig. S2). Average intensity was significantly higher (P < 0.0001) in S63del/P0ct-myc mice when compared with either S63C/P0ct-myc or P0ct-myc alone, suggesting that P0S63del induces accumulation of P0ct-myc in the ER (Fig. 3A).
Figure 3.

P0S63del induces ER retention of P0ct-myc, but not MAG. (A and B) Teased fibres from P0ct-myc, S63del//P0ct-myc or S63C//P0ct-myc sciatic nerves were stained for DAPI (blue), KDEL (green) and myc (A; red) or MAG (B; red) and imaged by confocal microscopy. The intensity of myc or MAG staining was measured in comparable perinuclear KDEL-positive areas and expressed as the percent of intensity for P0ct-myc. P0myc, P0ct-myc. Scale bars: 20 μm.
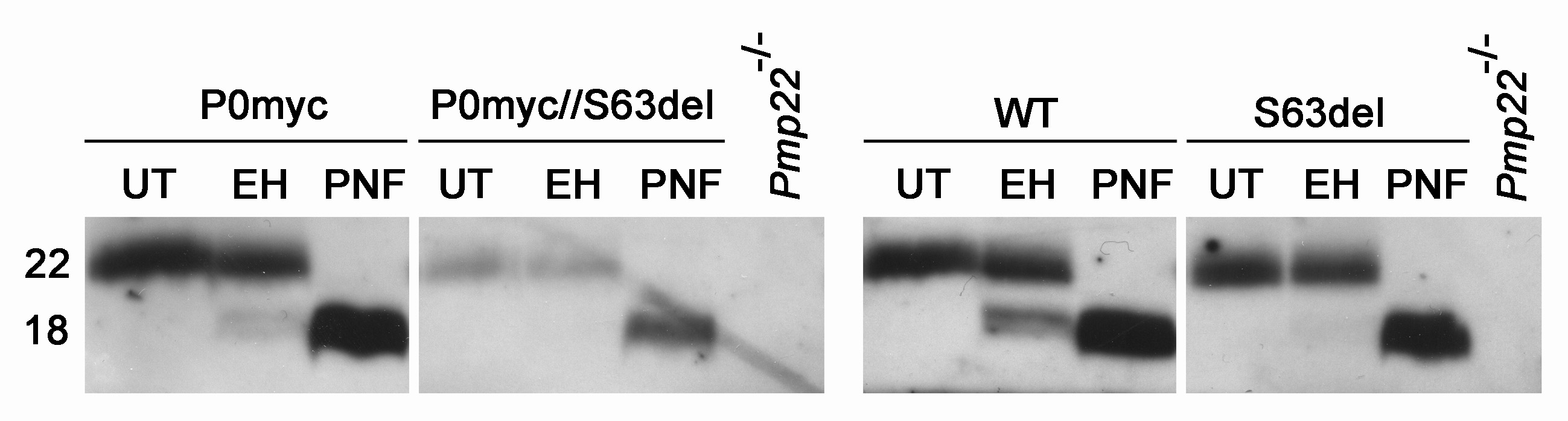
To confirm this, we analysed the glycosylation of P0ct-myc. In myelin, P0wt is partially sensitive to endoglycosidase H (EndoH) treatment, whereas P0S63del is retained in the ER and is therefore entirely sensitive to Endo H treatment (E.T. and L.W., unpublished data). As expected, P0ct-myc (Fig. 4A) recapitulated the Endo H sensitivity of P0wt in P0ct-myc/Mpz−/− nerves (P0ct-myc with no P0wt) (data not shown; 15). In contrast, the EndoH-sensitive form of P0ct-myc was significantly increased (from 55 ± 1.3 to 65 ± 2.5%; P < 0.01) in littermate S63del/P0ct-myc/Mpz−/− nerves (express both P0S63del and P0ct-myc) (Fig. 4A). As a control, PNGase F (deglycosylates all P0 independently of glycosyl maturation) digested 100% of P0ct-myc, producing a band which migrated identically to the EndoH-sensitive band, thus confirming that the EndoH-insensitive band was the mature glycosylated form of the protein (data not shown). These data further support that P0S63del induces P0ct-myc retention in the ER.
Figure 4.

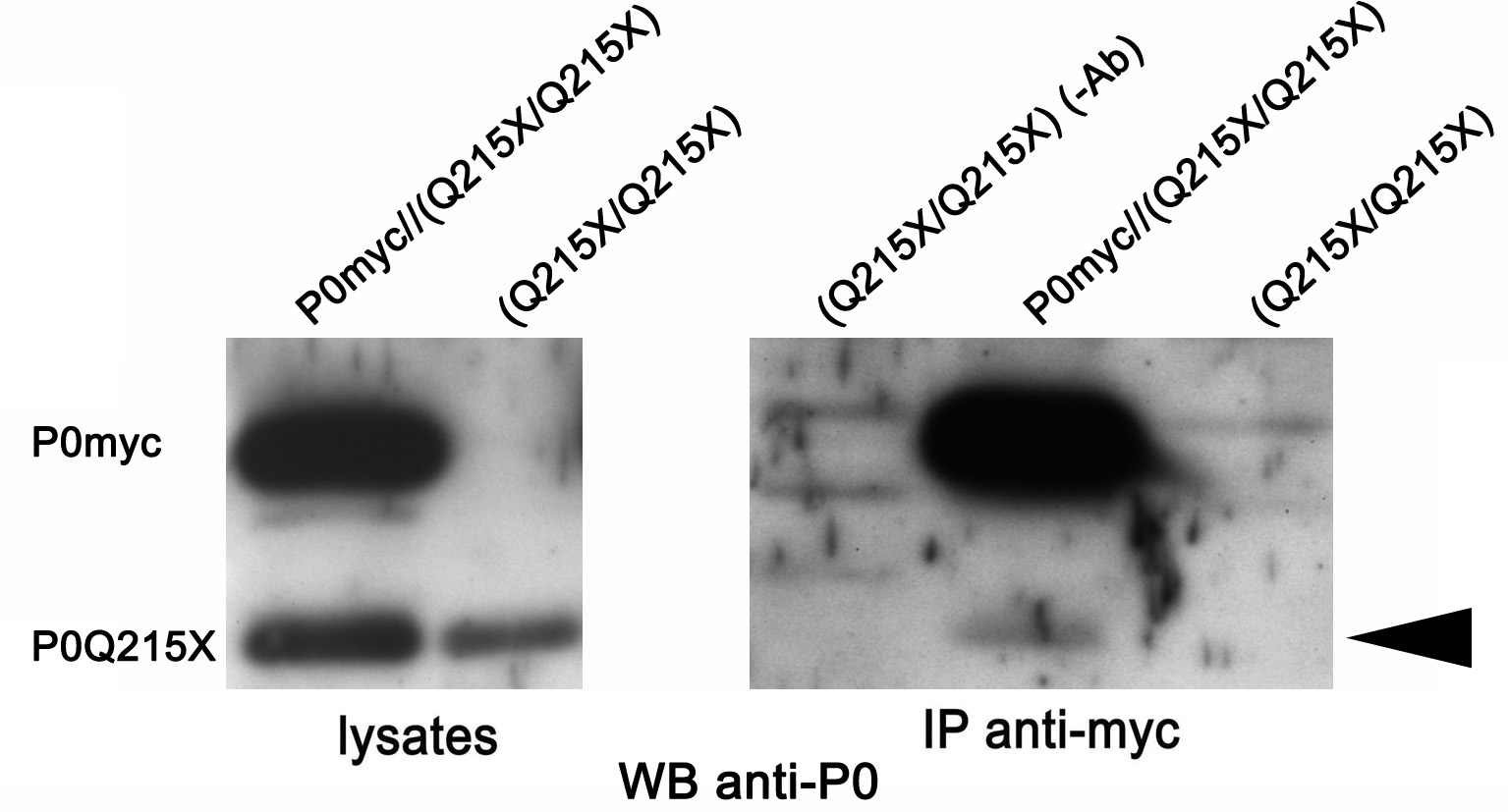
In the presence of P0S63del, P0ct-myc shows more glycosylation typical of the ER, but does not co-immunoprecipitate with P0S63del. (A) Western analysis for myc was performed on EndoH-treated (EH) or -untreated (UT) sciatic nerve lysates of P0ct-myc//Mpz−/− or S63del//P0ct-myc//Mpz−/− mice. EndoH-sensitive and -insensitive bands from four sciatic nerve lysates (four mice) of each genotype were quantified by densitometry and expressed as %total signal. (B) IP of myc followed by western analysis for P0 on SN lysates does not detect P0S63del (arrowhead), whereas direct western analysis for P0 does. P0myc, P0ct-myc.
P0ct-myc retention is not caused by direct interaction with P0S63del
P0 protein is likely to be present in myelin sheaths as dimers and tetramers (11,12), but it is not known where in the secretory pathway these complexes form. If they form in the ER, then retained P0S63del could exert a dominant-negative effect on P0ct-myc and promote its retention in the ER. To test this hypothesis, we performed IP assays on sciatic nerve homogenates from double-transgenic mice in a P0-null background. The P0-null background was necessary so that P0wt would not obscure P0S63del at 28 kDa. Surprisingly, IP of P0ct-myc did not precipitate P0S63del (Fig. 4B), even if IP of P0ct-myc performed with the same conditions could precipitate P0wt (Fig. 2F). To control for a possible effect of dosage, IPs were performed on nerves expressing P0ct-myc and another P0 mutant, MpzQ215X. MpzQ215X is expressed at significantly lower levels than MpzS63del, but P0Q215X is trafficked beyond the ER. In this case, IP of P0ct-myc did precipitate P0Q215X (Supplementary Material, Fig. S4; P.F. and L.W., manuscript in preparation). Together these data suggest that ER retention of P0ct-myc is not due to direct interaction with its P0S63del counterpart.
An alternative explanation is that the UPR associated with P0S63del augments ER retention of multiple proteins (10,17,18). Therefore, we performed immunofluorescence analysis for ER retention of myelin-associated glycoprotein (MAG) and PMP22. Both are myelin proteins that are synthesized in the ER of Schwann cells (19). No significant increase of MAG in the ER was found in the presence of P0S63del (Fig. 3B). Unfortunately, immunofluorescence analysis on wt and S63del teased, myelinated fibres was not sensitive enough to detect PMP22 in the ER above background as defined in Pmp22-null nerves. However, in contrast to P0ct-myc, immature glycosylation of PMP22 was not increased in S63del nerves, suggesting that PMP22 is not retained in the ER (compare Supplementary Material, Fig. S5, with Fig. 4A). The sum of these data suggest that retention is specific to P0ct-myc.
In the presence of S63del, less P0 arrives to myelin
To investigate whether P0S63del could actually reduce the levels of P0 in myelin sufficiently to alter its morphology, we performed quantitative IEM analysis on myelin from S63del/P0ct-myc and control nerves. Since myelin is thinner in S63del nerves, we first analysed P0 density as a function of myelin thickness to rule out any resulting bias. P0 density was found to be constant in myelin of variable thicknesses within each genotype (Supplementary Material, Fig. S6). Second, we analysed P0 heterozygous null (P0ct-myc/Mpz+/−) mice as a positive control for diminished P0 myelin density. P0 density was decreased to 52 ± 3.1% of normal in P0ct-myc/Mpz+/− myelin sheaths (Fig. 5C), as expected on the basis of western blot analysis of Mpz+/− sciatic nerves (15) and the low dosage of P0ct-myc. These data further validate quantitative IEM analysis for P0 in rodent myelin (13,20).
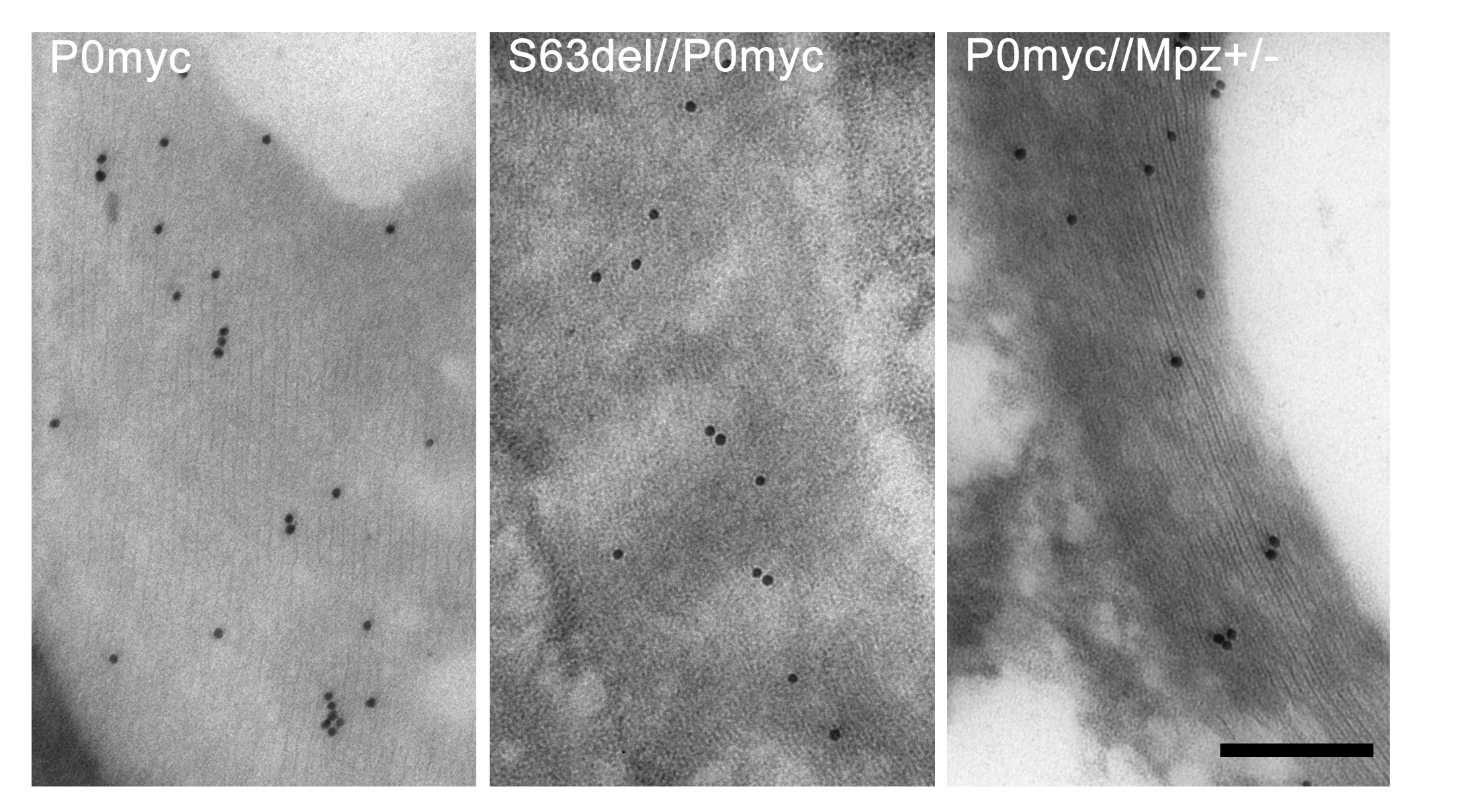
Figure 5.

Total P0 is reduced in S63del myelin. (A–C) IEM analysis of myelin sheaths from P0ct-myc (A), S63del//P0ct-myc (B), P0ct-myc//Mpz+/− (C) sciatic nerves at P28, using anti-P0 antibody. Gold particle density using anti-P0 (D and E) and anti-myc antibodies (F and G) was measured and plotted relative to P0ct-myc = 100% (average of three experiments; anti-myc micrographs are shown in Supplementary Material, Fig. S7). P0−/− or P0wt nerves showed no signal with P0 or myc antibodies, respectively (not shown). ***P < 0.001. P0myc, P0ct-myc. Scale bar (A–C): 200 nm.
Importantly, also in S63del/P0ct-myc mice, P0 density in myelin was found to be significantly reduced to 53 ± 3.7% when compared with controls (P < 0.0001; Fig. 5A and B, and quantified in D and E). In contrast, no comparable reduction was found in S63C/P0ct-myc nerves, where P0S63C is not retained in the ER (data not shown; 8). The reduced P0 density is not simply due to transcriptional alteration, as we have previously shown that Mpzwt mRNA levels are similar in S63del and wt nerves (8). IEM analysis with an anti-myc antibody revealed a similar decrease in myc density in S63del/P0ct-myc when compared with P0ct-myc myelin (Fig. 5F and G; see Supplementary Material, Fig. S7, for micrographs). Interestingly, P0ct-myc/Mpz+/− myelin showed increased myc density, perhaps due to a compensatory increase in expression of the P0ct-myc transgene in P0ct-myc/Mpz+/− nerves (as seen for Mpz+/− in Fig. 5C in reference 8). Thus, P0S63del in the ER reduces P0 density in myelin by half, which is known to cause pathology in both mice and humans.
DISCUSSION
To look for dominant-negative effects of P0S63del on P0wt, we engineered a transgenic mouse that expresses P0wt with a myc tag at the C terminus, and here we exploit it to show that P0S63del also causes selective retention of its P0wt counterpart and reduces P0wt dosage by half in the myelin sheath. Even if this effect is specific to P0 among myelin proteins, co-IP experiments suggest that the dominant-negative effect is not mediated by direct physical interaction between P0wt and P0S63del in the ER.
P0ct-myc mice are a valid tool to study P0wt in vivo
Extensive evaluation supports that P0ct-myc reproduces P0wt expression, trafficking and function. Unlike P0 with myc at the mature N terminus, P0ct-myc does not elicit a steric effect in the myelin sheath with resulting pathological myelin alterations (13). Moreover, here P0ct-myc is expressed at low levels, thus avoiding the congenital hypomyelination phenotype found in mice that overexpress P0wt (15). Furthermore, the similar P0ct-myc:P0wt ratio at both mRNA and protein levels and normal pattern of glycosylation confirm that post-transcriptional regulation and modification of P0ct-myc are normal. No mis-trafficking of P0ct-myc is detectable, and it is correctly targeted to myelin. co-IP analysis shows that P0ct-myc is in complexes with P0wt, as expected for a protein that interacts in polymers. Finally, we demonstrate that P0ct-myc contributes to myelin compaction when it is the only P0 expressed in sciatic nerves.
P0S63del causes selective retention of P0ct-myc
Misfolded P0S63del is retained in the ER. We generated P0S63del/P0ct-myc double-transgenic mice and showed that specifically P0S63del, but not P0S63C (another P0 disease-causing mutation that operates through GOF), causes P0ct-myc to be retained in the ER, although unlike P0S63del, only partially. ER retention was evidenced both by immunofluorescence and by shift of the glycosylation status to a more EndoH-sensitive state, as expected for immature, ER-retained proteins.
Interestingly, P0ct-myc is unable to co-immunoprecipitate P0S63del, suggesting that retention of P0ct-myc is not simply due to binding between the two proteins. We doubt that detergent conditions were excessively stringent, as P0ct-myc was able to co-immunoprecipitate either P0wt or P0Q215X under identical conditions. Also, dosage is an unlikely explanation, as MpzQ215X mRNA is expressed at 20% of one Mpzwt allele (P.F. and L.W., manuscript in preparation), whereas MpzS63del is expressed at 120% of one Mpzwt allele (8), and mRNA levels are reasonable indicators of levels of newly synthesized P0 protein in the ER and cis-Golgi. Since both P0wt and P0Q215X are trafficked beyond the ER, but P0S63del is not, we speculate that P0 dimer/tetramer formation occurs after exit from the ER.
Although the retention mechanism for P0ct-myc is indirect, it appears to be specific and not symptomatic of general ER retention of proteins. In fact, MAG, and probably PMP22—both myelin glycoproteins processed through the ER—are not retained in the presence of P0S63del. Selective, partial retention without binding between P0ct-myc and P0S63del suggests a mechanism involving quality control of P0 synthesis. One possibility is that some unfolded P0wt, which might normally be folded, is lost to degradation because P0S63del competes better for BiP. In this scenario, the high level of P0 synthesis together with misfolded P0S63del represents a special challenge for the ER and limits error correction (17). Alternatively, ER retention of normally folded P0wt may increase, paradoxically because BiP levels are upregulated in response to P0S63del (10,18). In either case, less P0wt would be trafficked to myelin. This fits with a definition of dominant negative that includes effects that do not depend on direct physical interaction between mutant and wt counterparts but may include competition for common partners—e.g. DNA for transcription factors, or the chaperone BiP for P0wt and P0S63del (see reference 21 for examples).
These hypotheses also depend on the idea that different myelin proteins may employ specific ER quality control checkpoints. There is already some evidence for this in Schwann cells (22; E.T. and L.W., manuscript in preparation).
P0S63del significantly limits dosage of P0wt in myelin
Retention of P0S63del in the ER induces corresponding retention of P0ct-myc. But does this retention actually alter dosage of P0wt in myelin? After validating IEM analysis for P0 in myelin (13,20), we found a similar reduction of P0 density in P0S63del/P0ct-myc/Mpz+/+ and P0ct-myc/Mpz+/− myelin. The reduction depends on ER retention of the P0 mutant, as P0S63del, but not P0S63C, produced this effect. Finally, the reduction was confirmed by measuring also the density of P0ct-myc in myelin. So retention of P0S63del in the ER has a significant effect on total P0 density in myelin.
Data from S63del mice predict that the amount of P0wt arriving to patient myelin would be one-fourth of normal in CMT1B due to MPZS63del. This is because P0S63del likely does not arrive to myelin in patients as in mice (thus, one-half of P0 is lost). Also, in contrast to our experimental P0S63del/P0ct-myc/Mpz+/+ mice, patients have only one other MPZwt allele. If half of this P0wt is retained in the ER due to a dominant-negative effect of P0S63del, the total P0wt arriving to myelin would be one-fourth of normal. Although there is no experimental model of one-fourth of P0 dosage, it is likely that the resulting demyelinating/hypomyelinating neuropathy would be significantly worse than the mild neuropathy-associated MPZ haploinsufficiency in humans or mice. These data taken together with partial rescue of demyelination by CHOP ablation in S63del mice suggest that MPZS63del causes neuropathy through two GOF function mechanisms: toxic effect of UPR (10) and a dominant-negative limitation on trafficking of P0wt to myelin. Therapeutic strategies for this neuropathy will need to address both mechanisms.
MATERIALS AND METHODS
Transgenic mice
All experiments with animals were described in experimental protocols approved by local Institutional Animal Care and Use Committees. P0S63del, P0S63C and Mpz-null mice have been described (5,8). Q215X mice contain a targeted mutation in Mpz exon 5 encoding P0Q215X (P.F. and L.W., manuscript in preparation). All transgenic mice were maintained on the FVB/N genetic background (Charles River, Calco, Italy).
Transgene monomers encoding P0 with a myc tag at the C terminus were engineered by altering exon 6 of mP0TOT (14). An FseI site followed by stop codons in each reading frame was introduced by site-directed mutagenesis immediately upstream of the natural Mpz stop codon, and annealed oligonucleotides encoding the myc tag (EQKLISEEDLN) were ligated into mP0TOT digested with FseI. The insertion was confirmed by sequence analysis. After pronuclear injection of FVB/N embryos, 12 independent lines were established. Genotype analysis (Southern blot and PCR) were performed as described previously (15). Animals from several lines developed tremor and hypomyelination typical of P0 overexpression (15). Four lines exhibited normal behaviour and nerve morphology. Of these, Tg140.12 (hereafter called P0ct-myc for P0 C terminus myc) was maintained for further study [genetic designation Tg(Mpz3′Myc)1Wra].
Behavioural analysis was performed as described (23), with the following modifications. Hindpaw placement errors (faults) were determined as a proportion of total hindpaw steps on horizontal grid mesh with openings of 1.2 × 1.2 cm. Errors were defined as passage of the hindpaw through an opening in the grid. Three animals of each genotype were tested in five trials of 5 min each, repeated hourly on a single day.
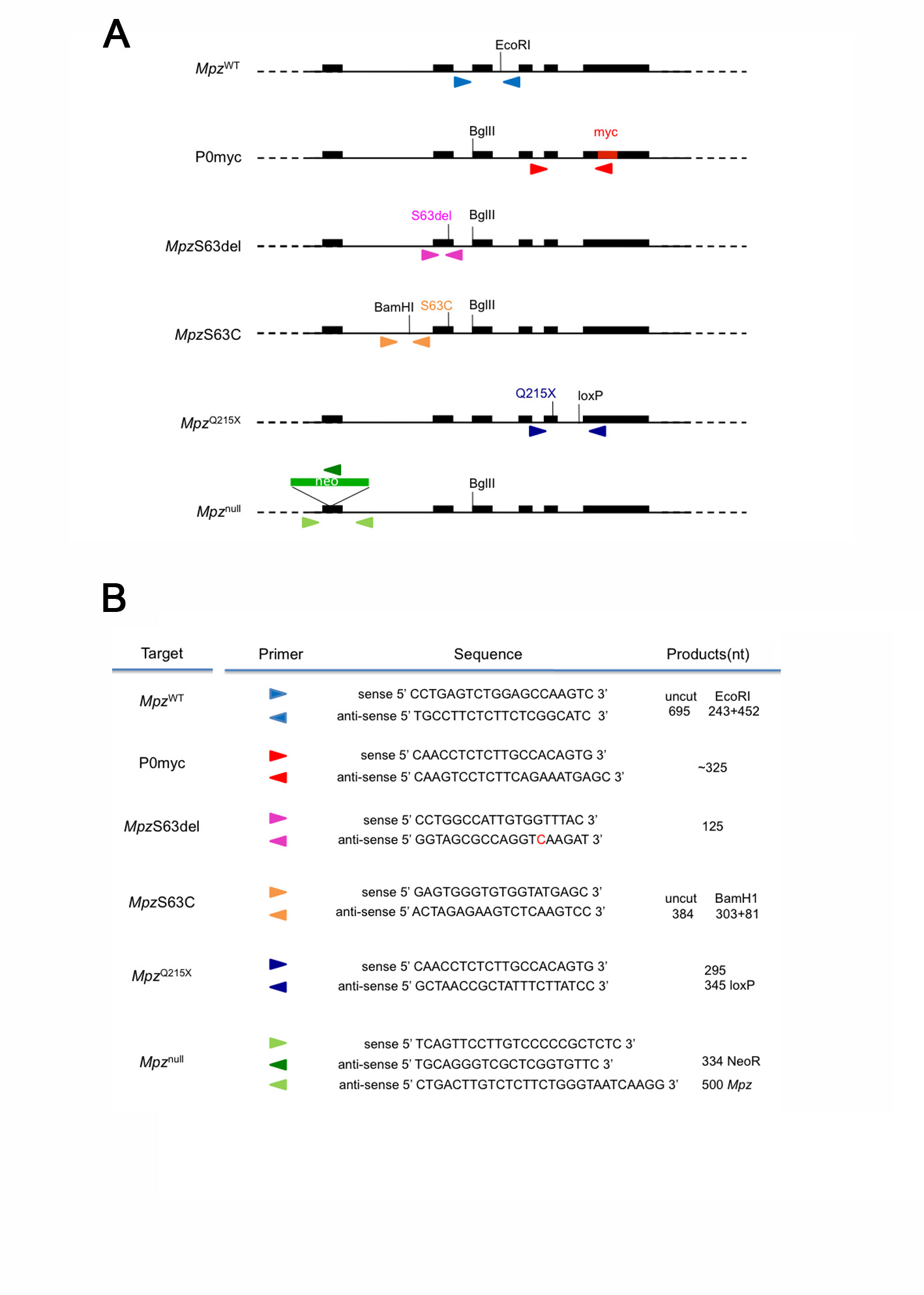
P0ct-myc, S63del, S63C, Q215X and heterozygous Mpz-null animals were crossed to generate breeders for the experiments in the Mpz-null background. PCR analysis of genomic DNA from tail samples distinguished genotypes of offspring by using specific primers for the MpzS63del, MpzS63C, P0ct-myc transgenes, or the MpzQ215X and Mpznull alleles (see Supplementary Material, Fig. S8, for genotyping algorithms and primer sequences). The genotype of Pmp22-null animals was determined by PCR analysis with Pmp22 exon 2F: 5′-ATGCTCCTACTCTTGTTGGG-3′ and Pmp22 IntronR: 5′-AGATTAGCCACAGCCATAGTC-3′ or NeoR: 5′-GCCTTCTATCGCCTTCTTGAC-3′, which amplified 608 or 300 nucleotide fragments, respectively. PCR conditions were: 94°C for 30 s, 55°C for 90 s and 72°C for 60 s (35 cycles), followed by 7 min extension at 72°C, in a standard PCR reaction mix.
RTPCR analysis
RTPCR was performed as described previously (15) on RNA from sciatic nerves. DpnII digestion distinguishes transgenic from wt Mpz (15).
Western blot analysis
Sciatic nerves were analysed as described previously (15). The primary antibodies included chicken anti-P0 (Aves Labs, Inc., New Orleans, LA, USA), rabbit anti-myc (Millipore, Billerica, MA, USA), or mouse monoclonal anti-myc (9E10, gift from R. Sitia, San Raffaele Science Institute, Milan, Italy), and rabbit anti-PMP22 (ET-S2M1 raised against IYTVRHSEWHVNNDY peptide from rat PMP22 and affinity-purified). The intensity of bands was quantified by densitometry. Deglycosylation was performed as described (15).
Immunoprecipitation
Sciatic nerves, usually one per experiment, were lysed in 400 µl of IP buffer [50mm Tris–HCl, pH 7.4; 150 mm NaCl; 1 mm EDTA; 1 mm Na3VO4; 1 mm NaF; Triton X 1%; protease inhibitor cocktail (Sigma-Aldrich) 1:100], incubated on a rotating wheel for 2 h at 4°C with 5 µl of rabbit anti-myc Ab, followed by 2 h at 4°C with 20 µl of Sepharose-Protein A (GE Healthcare). After washes, elution was performed by boiling for 10 min in DTT containing sample buffer. Aliquots of the lysate or the first wash (void) were collected and resolved by PAGE alongside IP samples.
Morphological analysis
STS and EM analyses of sciatic nerves were performed as described previously (24). For myelin compaction analysis, electron micrographs of P0ct-myc//Mpz−/− or Mpz−/− sciatic nerves at ×4000 magnification were obtained using a Leo 912AB Transmission Electron Microscope. Periodicity was measured at ×20 000 magnification as described previously (8).
IHC analysis
Sciatic nerves were dissected from P28 littermates, and teased fibres or cryosections were prepared as described (10) with addition of immersion in cold methanol for 10 min (only for full-thickness myelin; Fig. 2A and B) and two rinses in PBS. Primary antibodies were mouse monoclonal anti-KDEL (Stressgen, Victoria, British Columbia, Canada), chicken polyclonal anti-P0 (1:300, Aves Labs) and mouse monoclonal anti-MAG (1:200, Chemicon International). FITC-, rhodamine- and Cy5-conjugated secondary antibodies were from (Jackson ImmunoResearch, Baltimore, MD, USA). Samples were examined on an UltraVIEW ERS spinning disk confocal microscope (Perkin Elmer), equipped with a Plan Apochromat 63×/1.4 oil-immersion objective. Image analysis was performed with ImageJ (v1.38x) by measuring the average P0, myc or MAG fluorescence in the perinuclear, KDEL-positive triangular area (Supplementary Material, Fig. S2).
Immuno-electron microscopy
IEM was performed as in Previtali et al. (13) but with embedding modified as described (20). Primary antibodies were chicken anti-P0 (Aves) and rabbit anti-myc (Millipore). Gold-conjugated secondary antibodies were 10 nm for P0 and 15 nm for myc (British Biocell International).
Statistical analysis
Results are plotted as mean ± SEM and considered significant if P ≤ 0.05 by Student's t-test.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by grants from Telethon, Italy (GGP08021 to M.L.F., GGP071100 to L.W.); the National Institutes of Health (NS45630 to M.L.F., NS55256 to L.W., Waisman Center HD03352 to A.M.); and the Fondazione Mariani, Italy (to L.W.).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Volkmar Gieselmann and Roberto Sitia for insightful discussions. CFCM Telethon/HSR transgenic mouse service performed pronuclear injections. We thank Roberto Sitia for the 9E10 antibody; Rudolf Martini and Ueli Suter for the Mpz-null and Pmp22-null mice, respectively; Nicolo' Musner for technical suggestions; and Marina Fasolini and Denice Springman for expert technical assistance.
Conflict of Interest statement. None declared.
REFERENCES
- 1.D'Urso D., Brophy P.J., Staugaitis S.M., Gillespie C.S., Frey A.B., Stempak J.G., Colman D.R. Protein zero of peripheral nerve myelin: biosynthesis, membrane insertion, and evidence for homotypic interaction. Neuron. 1990;4:449–460. doi: 10.1016/0896-6273(90)90057-m. doi:10.1016/0896-6273(90)90057-M. [DOI] [PubMed] [Google Scholar]
- 2.Filbin M., Walsh F., Trapp B., Pizzey J., Tennekoon G. Role of myelin Po protein as a homophilic adhesion molecule. Nature. 1990;344:871–872. doi: 10.1038/344871a0. doi:10.1038/344871a0. [DOI] [PubMed] [Google Scholar]
- 3.Lemke G., Axel R. Isolation and sequence of a cDNA encoding the major structural protein of peripheral nerve myelin. Cell. 1985;40:501–508. doi: 10.1016/0092-8674(85)90198-9. doi:10.1016/0092-8674(85)90198-9. [DOI] [PubMed] [Google Scholar]
- 4.Schneider-Schaulies J., Brunn A.V., Schachner M. Recombinant peripheral myelin protein Po confers both adhesion and neurite outgrowth-promoting properties. J. Neurosci. Res. 1990;27:286–297. doi: 10.1002/jnr.490270307. doi:10.1002/jnr.490270307. [DOI] [PubMed] [Google Scholar]
- 5.Giese K.P., Martini R., Lemke G., Soriano P., Schachner M. Mouse P0 gene disruption leads to hypomyelination, abnormal expression of recognition molecules, and degeneration of myelin and axons. Cell. 1992;71:565–576. doi: 10.1016/0092-8674(92)90591-y. doi:10.1016/0092-8674(92)90591-Y. [DOI] [PubMed] [Google Scholar]
- 6.Scherer S.S., Wrabetz L. Molecular mechanisms of inherited demyelinating neuropathies. Glia. 2008;56:1578–1589. doi: 10.1002/glia.20751. doi:10.1002/glia.20751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shy M.E., Jani A., Krajewski K., Grandis M., Lewis R.A., Li J., Shy R.R., Balsamo J., Lilien J., Garbern J.Y., et al. Phenotypic clustering in MPZ mutations. Brain. 2004;127:371–384. doi: 10.1093/brain/awh048. doi:10.1093/brain/awh048. [DOI] [PubMed] [Google Scholar]
- 8.Wrabetz L., D'Antonio M., Pennuto M., Dati G., Tinelli E., Fratta P., Previtali S., Imperiale D., Zielasek J., Toyka K., et al. Different intracellular pathomechanisms produce diverse myelin protein zero neuropathies in transgenic mice. J. Neurosci. 2006;26:2358–2368. doi: 10.1523/JNEUROSCI.3819-05.2006. doi:10.1523/JNEUROSCI.3819-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Avila R.L., D'Antonio M., Bachi A., Inouye H., Feltri M.L., Wrabetz L., Kirschner D.A. P0 (protein zero) mutation S34C underlies instability of internodal myelin in S63C mice. J. Biol. Chem. 2010;285:42001–42012. doi: 10.1074/jbc.M110.166967. doi:10.1074/jbc.M110.166967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pennuto M., Tinelli E., Malaguti M., Del Carro U., D'Antonio M., Ron D., Quattrini A., Feltri M.L., Wrabetz L. Ablation of the UPR-mediator CHOP restores motor function and reduces demyelination in Charcot–Marie–Tooth 1B mice. Neuron. 2008;57:393–405. doi: 10.1016/j.neuron.2007.12.021. doi:10.1016/j.neuron.2007.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shapiro L., Doyle J.P., Hensley P., Colman D., Hendrickson W.A. Crystal structure of the extracellular domain from Po, the major structural protein of peripheral nerve myelin. Neuron. 1996;17:435–449. doi: 10.1016/s0896-6273(00)80176-2. doi:10.1016/S0896-6273(00)80176-2. [DOI] [PubMed] [Google Scholar]
- 12.Thompson A.J., Cronin M.S., Kirschner D.A. Myelin protein zero exists as dimers and tetramers in native membranes of Xenopus laevis peripheral nerve. J. Neurosci. Res. 2002;67:766–771. doi: 10.1002/jnr.10167. doi:10.1002/jnr.10167. [DOI] [PubMed] [Google Scholar]
- 13.Previtali S.C., Quattrini A., Fasolini M., Panzeri M.C., Villa A., Filbin M.T., Li W., Chiu S.Y., Messing A., Wrabetz L., et al. Epitope-tagged P(0) glycoprotein causes Charcot–Marie–Tooth-like neuropathy in transgenic mice. J. Cell Biol. 2000;151:1035–1046. doi: 10.1083/jcb.151.5.1035. doi:10.1083/jcb.151.5.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feltri M.L., D'Antonio M., Quattrini A., Numerato R., Arona M., Previtali S., Chiu S.Y., Messing A., Wrabetz L. A novel P0 glycoprotein transgene activates expression of lacZ in myelin-forming Schwann cells. Eur. J. Neurosci. 1999;11:1577–1586. doi: 10.1046/j.1460-9568.1999.00568.x. doi:10.1046/j.1460-9568.1999.00568.x. [DOI] [PubMed] [Google Scholar]
- 15.Wrabetz L., Feltri M., Quattrini A., Imperiale D., Previtali S., D'Antonio M., Martini R., Yin X., Trapp B., Zhou L., et al. P0 overexpression causes congenital hypomyelination of peripheral nerve. J. Cell Biol. 2000;148:1021–1033. doi: 10.1083/jcb.148.5.1021. doi:10.1083/jcb.148.5.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yin X., Baek R.C., Kirschner D.A., Peterson A., Fujii Y., Nave K.A., Macklin W.B., Trapp B.D. Evolution of a neuroprotective function of central nervous system myelin. J. Cell Biol. 2006;172:469–478. doi: 10.1083/jcb.200509174. doi:10.1083/jcb.200509174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gidalevitz T., Ben-Zvi A., Ho K.H., Brignull H.R., Morimoto R.I. Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science. 2006;311:1471–1474. doi: 10.1126/science.1124514. doi:10.1126/science.1124514. [DOI] [PubMed] [Google Scholar]
- 18.Dorner A.J., Wasley L.C., Kaufman R.J. Overexpression of GRP78 mitigates stress induction of glucose regulated proteins and blocks secretion of selective proteins in Chinese hamster ovary cells. EMBO J. 1992;11:1563–1571. doi: 10.1002/j.1460-2075.1992.tb05201.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pedraza L., Owens G.C., Green L.A., Salzer J.L. The myelin-associated glycoproteins: membrane disposition, evidence of a novel disulfide linkage between immunoglobulin-like domains, and posttranslational palmitylation. J. Cell Biol. 1990;111:2651–2661. doi: 10.1083/jcb.111.6.2651. doi:10.1083/jcb.111.6.2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yin A., Kidd G., Wrabetz L., Feltri M., Messing A., Trapp B. Schwann cell myelination requires timely and precise targeting of P0 protein. J. Cell Biol. 2000;148:1009–1020. doi: 10.1083/jcb.148.5.1009. doi:10.1083/jcb.148.5.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Veitia R.A. Exploring the molecular etiology of dominant-negative mutations. Plant Cell. 2007;19:3843–3851. doi: 10.1105/tpc.107.055053. doi:10.1105/tpc.107.055053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dickson K.M., Bergeron J.J., Shames I., Colby J., Nguyen D.T., Chevet E., Thomas D.Y., Snipes G.J. Association of calnexin with mutant peripheral myelin protein-22 ex vivo: a basis for ‘gain-of-function’ ER diseases. Proc. Natl Acad. Sci. USA. 2002;99:9852–9857. doi: 10.1073/pnas.152621799. doi:10.1073/pnas.152621799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tillerson J.L., Miller G.W. Grid performance test to measure behavioral impairment in the MPTP-treated-mouse model of parkinsonism. J. Neurosci. Methods. 2003;123:189–200. doi: 10.1016/s0165-0270(02)00360-6. doi:10.1016/S0165-0270(02)00360-6. [DOI] [PubMed] [Google Scholar]
- 24.Quattrini A., Previtali S., Feltri M.L., Canal N., Nemni R., Wrabetz L. Beta4 integrin and other Schwann cell markers in axonal neuropathy. Glia. 1996;17:294–306. doi: 10.1002/(SICI)1098-1136(199608)17:4<294::AID-GLIA4>3.0.CO;2-#. doi:10.1002/(SICI)1098-1136(199608)17:4<294::AID-GLIA4>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}