

Summary
Objectives
To improve treatment for patients with breast and prostate cancer.
Methods
A number of novel inhibitors of steroidogenic enzymes have been developed. Their biological effects have been evaluated in a variety of preclinical models. Aromatase (estrogen synthetase) inhibitors have now been extensively tested in clinical trials in breast cancer patients. Inhibitors of 17α-hydroxylase/lyase have also been studied in preclinical models and are beginning trials in prostate cancer patients.
Results
The enzyme aromatase (CYP19) has proved to be an important therapeutic target. Inhibitors of aromatase (AIs) as are showing greater benefit than antiestrogens in the treatment of breast cancer. Although effective in other conditions in both women and men, AIs have not been useful in benign prostatic hypertrophy or prostate cancer. However inhibitors of 17αhydroxylase/lyase (CYP17) to block synthesis of androgens may be effective for prostate cancer. Recent clinical trials with abiraterone and preclinical studies with other novel CYP17 inhibitors which also interact with the androgen receptor and cause its down regulation could provide a new approach for treating this disease.
In further studies we optimized treatment with aromatase inhibitors and antiestrogens utilizing an intratumoral aromatase xenograft model. AIs were more effective and sustained growth inhibition longer than antiestrogens. However, inevitably tumors eventually began to grow despite continued treatment. Analysis of breast tumors from mice treated with letrozole revealed upregulation of HER-2 and MAPKinase signaling proteins and downregulation of the estrogen receptor. Our studies showed that tumors adapt to AI treatment by activating alternate signaling pathways, thus enable them to proliferate in absence of estrogen. When mice bearing resistant tumors were treated with trastuzumab the anti-HER-2 antibody (Herceptin), HER-2 was decreased in the tumor but the estrogen receptor and aromatase were restored. Tumor growth was significantly inhibited by treatment with trastuzumab in addition to letrozole.
Conclusions
Aromatase inhibitors are proving to be an effective new class of agents for the treatment and breast cancer. Compounds inhibiting 17αhydroxylase/lyase have potential for the treatment of prostate cancer. Our results suggest that strategies to overcome resistance to these types of agents can restore sensitivity of the tumors to hormone therapy.
Keywords: breast cancer, xenograft models, letrozole, anastrozole, Her2, trastuzumab
Introduction
Aromatase (P-450arom) has a key role in development and reproduction. Androgens (namely, androstenedione and testosterone) are converted to estrogens (estrone and estradiol) in a rate limiting reaction catalyzed by aromatase. In humans, the enzyme is expressed in the granulosa cells of the ovarian follicle, the syncytiotrophoblasts of the placenta during pregnancy [1,2] and in the Leydig cells of the testes. Estrogens produced by aromatase have a number of important physiology and pathologic functions in men and women. One of the most important is the stimulatory role of estrogens breast cancer in women.
While the ovaries are the principal source of circulating estrogens in premenopausal women, the majority of patients with BC are postmenopausal women. About 75% of these patients have estrogen dependent tumors [3]. After menopause, the ovaries cease to produce estrogens but these steroids continue to be synthesized in non-ovarian tissue [4] including the breast tissue [5], albeit in much smaller amounts. Important sites of non-ovarian estrogen synthesis are adipose tissue and muscle [6], where production increases with age and is the primary source of circulating estrogen in postmenopausal women [7]. However, breast tissue has been found to have several-fold higher levels of estrogen than those in serum and are equivalent to levels in premenopausal women [8,9]. A number of reports indicate that aromatase activity as well as aromatase mRNA is present in normal breast tissue and breast tumors [10-16]. Approximately 60% of breast tumors express aromatase [16,17] and have aromatase activity [18]. Although gonadal aromatase is regulated by follicle-stimulating hormone, aromatase in extragonadal sites is regulated by other factors including glucocorticoids, cyclic adenosine monophosphate, and prostaglandin E2 [19]. For example, cycooxygnease-2 expression has been correlated with aromatase expression, suggesting that local production of prostaglandin E2 may stimulate aromatase [17,20]. Thus, in postmenopausal breast cancer patients, estrogen synthesis is independent of feedback regulation between the pituitary and the ovary and involves tissue-specific regulation of P-450arom using alternative promoters [19].
Peripherally synthesized estrogens act locally in an intracrine or paracrine manner without being released into the circulation and act by binding to specific estrogen receptor α (ERα) in the tumor cells thereby mediating transcription of genes regulating proliferation.
Aromatase Inhibitors for Breast Cancer Treatment
Tamoxifen blocks the action of estrogens by binding to the ER. For the past 30 years tamoxifen has proved to be effective first-line or adjuvant endocrine treatment for advanced breast cancer. However, there were concerns initially that it might not be completely effective in inhibiting the tumor as it was a partial agonist or weak estrogen. In addition, its estrogenic actions in some tissues such as the uterus and vasculature could result in increased risk of endometrial cancer [21] and stroke [22]. Moreover, tamoxifen's efficacy proved to be limited to 5 years of treatment and it was considered to be without further benefit beyond that time [22]. Therefore to overcome these problems a different strategy was initiated. Compounds selectively targeting aromatase and lacking estrogenic activity were envisaged as being more effective than the antiestrogens. For the same reason, they were likely to cause fewer side-effects in patients. As one of the first successful targets for therapeutic inhibition, aromatase has several important attributes. Aromatization of androgens to estrogens is the last in the series of reactions in steroid biosynthesis and is rate-limiting for estrogen synthesis. Therefore, there are no steroids produced downstream to be affected by inhibition of aromatase. Also, although aromatase shares common features with other P-450 enzymes, the unique characteristics of the aromatization reaction, involving loss of the C-19 carbon and conversion of the steroidal A ring to an aromatic ring, provide the opportunity to develop inhibitors selective for P450arom. A number of selective aromatase inhibitors of were identified [23] in the early 1970. Among these, 4-OHA (formestane) was developed [24] and became the first selective AI for the treatment of BC [25,26]. Today, three aromatase inhibitors have been approved by the FDA exemestane (Aromasin), a steroidal inhibitor similar to formestane and two non-steroidal inhibitors anastrozole (Arimidex) and letrozole (Femara). All have high potency and specificity for aromatase. The latter two inhibitors were based on inhibition of P-450 enzymes and were derived from drugs used as antifungal agents such as ketoconazole, an inhibitor of fungal P-450 enzymes. Triazole and imidazole compounds possess a heteroatom, such as nitrogen-containing heterocyclic moiety. This interferes with steroidal hydroxylation by binding with the heme iron of cytochrome P-450. These nonsteroidal compounds are reversible inhibitor of aromatase whereas the steroidal inhibitors cause irreversible inactivation of the enzyme. Aromatase inhibitors are proving to be superior to tamoxifen in the advanced setting [27] and now are replacing tamoxifen as first-line therapy.
Aromatase and Steroid Synthesis Inhibitors in Men
In men, aromatase inhibitors may be useful for conditions associated with estrogen excess, such as gynecomastia and oligospermia. It had also been suggested that aromatase inhibitors might be of value in prostatic cancer and benign prostatic hypertrophy (BPH) [28].
While androgens are of primary importance in the growth of normal prostate, benign prostatic hypertrophy (BPH) and prostatic cancer, several lines of evidence suggested that estrogens may also have a role. In BPH, estrogen receptors have been identified and higher than normal concentrations of estradiol have been detected in stroma of the prostate [29]. Based on these findings, we investigated whether aromatase is present in prostatic tissue and whether aromatase inhibitors might have a role in BPH and prostatic cancer treatment. Our results indicated that aromatase is absent or, at the most, very low in the human prostate [30], although detection of aromatase in prostate stroma has recently reported [31]. However, estrogen synthesized in other tissues, such as adipose tissue as well as the testis could influence the prostate. In studies in the cynomolgus monkey [32], 4-OHA and another aromatase inhibitor, 1-methylandrosta-1,4-diene-3,17-dione (atamestane; Schering AG) have been shown to reverse estrogen-induced hyperplastic changes in the prostate. However, atamestane was without effect in a clinical trial of patients with BPH. We, and others, had found that 4-OHA inhibits 5α-reductase in vitro although with less potency than it inhibits aromatase [30]. Because of these two activities, the possibility that 4-OHA might be effective in prostatic cancer was explored in a small group of men with advanced disease. Subjective responses were observed in 80% of these patients, although there was no clear evidence of objective remissions [33]. Estrogen levels were reduced as expected but dihydrotestosterone concentrations were unchanged in the patients. This latter finding in addition to the weak androgenic activity of the compound may have determined the lack of objective responses. The other AI studies yielded negative results [34, 35].
We therefore turned to a different approach. We have designed and synthesized some novel inhibitors of androgen biosynthesis with the aim of providing more effective treatment for patients with prostatic cancer. The majority of patients initially respond to hormone ablative therapy although they eventually relapse, as is typical with all cancer treatments. Nevertheless, more strategic use of androgen ablative approaches could also improve outcome. In an ECOG trial, where patients receiving radical prostatectomies were given immediate or no ablation therapy, survival after 7 years was 17% with treatment versus 30% without [36]. Current treatment by orchidectomy or GnRH agonists result in reduced androgen production by the testis but does not interfere with androgen synthesis by the adrenals. In fact, increased adrenal DHEA and DHEAS (androgen precursors) have been observed in patients treated with GnRH implants [37]. Following 3 months of treatment with a GnRH agonist, testosterone and DHT concentrations in the prostate were found to remain at 25% and 10% respectively, of pretreatment levels [38]. Similarly, about 20% of castrated patients in relapse had significant levels of DHT in their prostatic tissue [39]. These findings suggest that the adrenals may contribute precursor androgens to the prostate. This is supported by clinical studies of patients receiving combined treatment with either GnRH or orchidectomy and an antiandrogens, such as flutamide, to block the actions of androgens via the androgen receptor, including adrenal androgens which are unaffected by GnRH treatment and castration alone. Such patients have increased progression-free survival time compared to patients treated with GnRH agonist or orchidectomy alone [40,41]. While androgen ablation is an effective treatment, patients eventually relapse and their tumors progress despite continued treatment. It has been reported that AR are not lost in “hormone insensitive” prostate cancer. In patients with recurring tumors treated with endocrine therapy, high level androgen receptor (AR) amplification was found in about 30% of cases [42,43]. This suggests that AR amplification may facilitate tumor cell growth in low androgen concentrations. Mutations in the androgen receptor have also been found in a number of human prostatic cancers [44-46].
Further support for the role of AR and androgens in PC is the recent report of increased expression of genes of androgen converting enzymes and persistence of androgen regulated genes in androgen-independent PC [47-49]. These observations suggest that therapies that inhibit production of androgens and target multiple points in the AR signaling cascade could offer a more effective approach for prolonging remission of PC. Thus, total androgen blockade using more potent agents as first line therapy may be more effective than conventional androgen deprivation by achieving maximum suppression of androgen (and estrogen) concentrations and inhibition of androgen action [50]. Furthermore, new agents which act by different mechanisms could produce second responses in a portion of relapsed patients. Although the percentage of patients who respond to second-line hormonal therapy may be relatively low, a substantial number of patients may benefit because of the high incidence of prostatic cancer. Recent studies in breast cancer patients treated with the aromatase inhibitor letrozole (Femara®) raise the possibility that in estrogen-responsive neoplasms, approaches that remove estrogen's presence are a successful subsequent maneuver in patients with progression of disease while receiving estrogen-receptor antagonist treatment [51]. Currently no analogous highly efficient means of accomplishing the same end with expected favorable side effect profile exists to influence the androgenic axis in patients with prostate cancer. The novel androgen synthesis inhibitors we are developing may provide such an opportunity. They may address this truly unmet medical need, and accomplish for patients with prostate cancer what aromatase inhibitors are accomplishing for patients with breast cancer. Androgen synthesis inhibitors may allow clinical investigators to manipulate the “androgen axis” in a way not currently encompassed by any current single agent. The eventual use of our inhibitors might not only occur in patients relapsing form LHRH inhibitors or castration plus androgen receptor antagonists, but it could afford a new dimension for hormonal modulation in newly diagnosed patients.
We have identified a number of novel compounds which inhibit CYP17 and are also potent antiandrogens and inhibit growth of human prostate cancer cells [52,53]. The 17α-hydroxylase/C17,20-lyase (CYP17) is a key enzyme in the biosynthesis of androgens and converts the C21 steroids (pregnenolone and progesterone) to the C19 androgens, dehydroepiandrosteone (DHEA), 5-androstenediol (A-diol), testosterone, and androstenedione in the testis and adrenals. Currently, ketoconazole, an imidazole fungicide, is the only inhibitor used to reduce testosterone biosynthesis in the treatment of patients with advanced prostatic cancer [54,55]. However, ketoconazole is not very potent or specific. Despite its draw backs, careful scheduling of treatment can produce prolonged responses in otherwise hormone-refractory patients [56]. Also, in a study by Small et al. [57], 62.5% of patients with advanced prostate cancer who had progressed following antiandrogens (flutamide) withdrawal, were found to have greater than 50% decrease in PSA values, while 48% had greater than 80% decrease. These findings suggest that more potent and selective inhibitors of this enzyme could provide useful agents for treating this disease, even in advanced stages and in some patients who may appear to be hormone refractory. It is possible that these tumors would be amenable to more potent inhibitors of androgen synthesis or second line therapy with differently acting agents.
We have reported the effects of a number of novel steroidal inhibitors of CYP17. Some have been shown to be strong inhibitors of androgen production and tumor growth in rodent models [52,58-61]. Jarman and colleagues recently described the effects of a similar steroidal CYP17 inhibitor, abiraterone, in patients with PC [62, 63]. Interestingly, our most effective CYP17 inhibitors possess several activities, such as inhibition of 5α-reductase and/or are antiandrogens with potent antitumor efficacy [64,53]. VN/124-1 exhibits potent AR antagonism and downregulates the AR. This compound causes tumor growth suppression in LAPC4 xenografts [Sean, 66] that is significantly greater than due to castration. Development of this compound is in progress for clinical trials in prostate cancer patients.
Effects of Long-Term Treatment with Aromatase Inhibitors
Clinical evidence indicates that AIs are more effective than tamoxifen and provide significant benefit to breast cancer patients. Nevertheless, tumors of some patients are initially refractory while others inevitably become resistance to treatment. Therefore, we have developed a xenograft model suitable for investigating the mechanisms of resistance in tumors that are no longer responsive to treatment with AIs. Our finding parallels our studies in xenograft models of prostate cancer [67] and suggest that tumors adapt and survive treatment by activating alternate signaling pathways such as the HER-2/MAPK pathway [67,68]. Similarly in the LNCap prostate cancer model, loss of androgen dependency was associated with activation of mTOR [67]. We propose that blocking signaling pathways associated with resistance as well as estrogen signaling may restore sensitivity of the tumors to aromatase or CYP17 inhibitors.
In our intratumor aromatase xenograft model letrozole treatment caused marked inhibition of tumor growth for an extended period of time and reduced estrogen levels by 90%. In this model human ER positive breast cancer cells (MCF-7) stably transected with aromatase [69] are inoculated into athymic immune suppressed mice [70]. These tumor cells (MCF-7Ca) served as an autocrine source of estrogen by aromatizing androstenedione. The model simulates the post-menopausal situation where breast and tumor aromatase are the main source of estrogen and not under gonadotropin feedback regulation. Since the tumor cells express ERα as well as aromatase, antiestrogens as well as aromatase inhibitors could be studied in tumors formed from these cells [70,71]. This intratumoral aromatase xenograft model has predicted the outcome of several clinical trials [71]. It has provided data showing that the AI letrozole was more effective than tamoxifen in suppressing breast tumor growth (Figure 1) and for a longer period of time. Also, our findings that combining an AI with tamoxifen was consistent with the ATAC trial where BC patients did not benefit significantly more from the combined treatment of anastrozole and tamoxifen than tamoxifen alone [72]. We have also shown that tumors progressing on tamoxifen remained sensitive to second-line therapy with the AI letrozole compared with those remaining on tamoxifen [72]. In clinical trials where patients received tamoxifen for 2-5 years switched to letrozole showed significant survival benefit (MA-17) [51] or exemestane showed improved disease-free survival [73]. In contrast, switching from letrozole treatment to second-line therapy with the antiestrogens fulvestrant or tamoxifen was less beneficial than remaining on letrozole [74]. Analysis of tumors from the mice in these studies provides a better insight into current trials that are examining the correct sequences of AIs and AEs [75].
Figure 1. The effects of long term treatment with letrozole on the growth of MCF-7Ca xenografts.
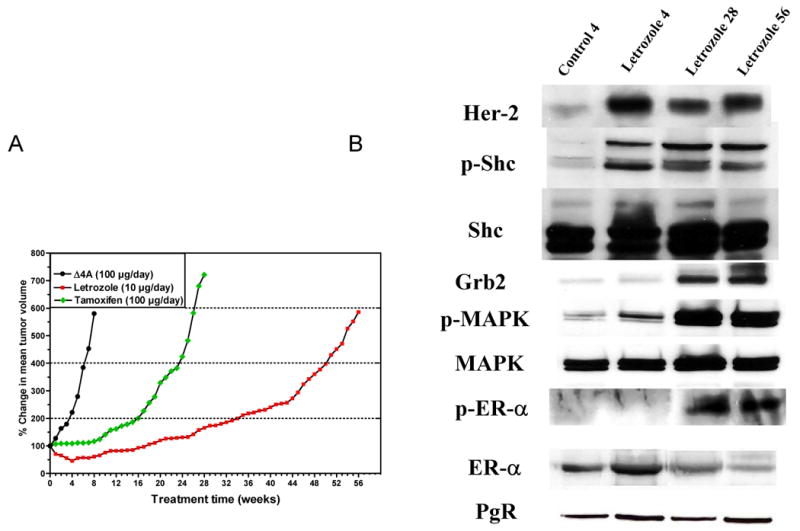
A) Animals were inoculated with MCF-7Ca cells at two sites on each flank and were supplemented with androstenedione (100 mg/day) for the duration of experiment [75]. When the tumors reached a measurable size (∼300 mm3), animals were assigned to 3 groups (n = 20 per group), and injected sc daily with vehicle (control), or tamoxifen (100 mg/day), or letrozole (10 mg/day). Tumor volumes were measured weekly and were expressed as the percent change relative to the initial tumor volume. Two mice per group were sacrificed and tumors were collected for analysis at 4, 28, and 56 weeks as indicated on the graph. B) The effect of letrozole treatment on Her-2, p-Shc, Shc, Grb2, p-MAPK, MAPK, ER-a, p-ER-a and PgR expression in MCF-7Ca tumor xenografts [75]. Letrozole treated tumors collected at 4 weeks, 28 and 56 weeks (above), were analyzed by Western immunoblotting, and were compared to vehicle treated tumors collected at Week 4 (control). Tumors were homogenized in lysis buffer as described previously [75].
Despite marked and sustained suppression of tumor growth, tumors eventually acquired the ability to grow in the presence of letrozole and were unresponsive to subsequent treatment with AEs [74]. Tumors were collected from the mice at specific times (4, 28 and 56 weeks) during the course of treatment and analyzed for changes associated with resistance to treatment (Figure 1A) [75]. Tumors at 4 weeks had regressed in response to treatment. At 28 weeks, tumors had almost doubled in size, whereas those at 56 weeks had increased 6-fold compared to their initial size even though letrozole treatment was continued. Immunoblot analysis revealed that during transition from a responsive to an unresponsive state, the level of the ER was decreased whereas the Her2/Raf/MAPK signaling pathway was activated (Figure 1B) [75]. At 4 weeks of letrozole treatment, when tumors were regressing, expression of Her-2 in the tumor was increased two-fold (Figure 1B). Greater increases in Her-2 expression were seen at 28 and 56 weeks when tumors were growing on letrozole treatment (Figure 1A). The activation of Shc (p-Shc) (Figure 1B) as well as increases in Grb-2 and p-MAPK were also observed (6-fold) (Figure 1B) [75] at these later times.
For further studies, a cell line was isolated from the long term letrozole treated tumors (LTLTCa cells) (Figure 1A) [74]. As in the tumors, the cells showed upregulation of Her-2, Grb-2, p-Shc, p-Raf, p-MEK1/2 and p-MAPK (Figure 2A). In addition, downstream kinesis that are targets for MAPK, such as p90RSK and Elk were also activated by phosphorylation (Figure 2A). It has been shown previously that MAPK can phosphorylate ER directly or indirectly via these downstream targets Elk-1 and p90RSK [76-78]. We found that at 4 weeks when tumors were regressing ERα levels were increased but when they became insensitive to letrozole and were actively growing, ER-α expression was decreased below control levels at 28 and 56 weeks (Figure 2B). Although the total amount of ER-α was reduced, the levels of phosphorylated ER-α were significantly greater than the levels of phosphorylated ER-α seen in letrozole responsive tumors. Furthermore, the level of expression of ER regulated progesterone receptor (PgR) during 56 weeks of treatment was unchanged evenwhen the tumors progressed from a responsive state to a refractory state (Figure 2A). Consistent with findings in the tumors, the LTLTCa cells also have decreased expression of ER-α (Figure 2B) whereas PgR was similar to levels in parental MCF-7Ca cells (Figure 2A). These results suggest that the growth of letrozole resistant tumors becomes independent of estrogen stimulation due to decrease in ER expression via interaction between Her-2/Raf/MAPK and ER that ultimately causes constitutive activation of ER by phosphorylation. These findings are consistent with previous reports that the interaction between growth factor receptor and ER-α results in an increased ER-related transactivation in a ligand-independent manner [79]. In our model, the increase in the phosphorylation of the ER promoted its transactivation even in the absence of the ligand as indicated by the steady expression of the estrogen-regulated PgR (Figures 1B).75 Using LTLTCa cells we have shown that letrozole resistance is similar to resistance to other types of hormonal therapies such as anastrozole, exemestane, tamoxifen and fulvestrant (Figures 3A and 3B).
Figure 2. Profile of protein expression on LTLTCa cells compared to MCF-7Ca cell.
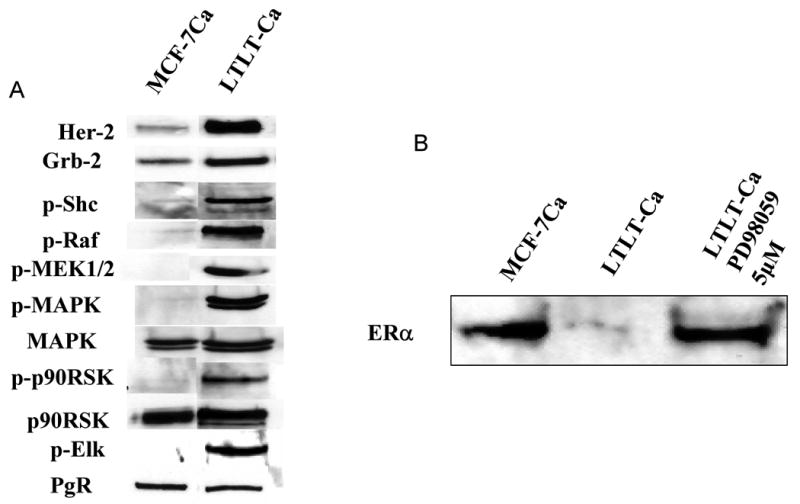
A) Expression of Her-2, p-Shc, Grb2, p-MEK1/2, p-p90RSK, p90RSK, p-Elk, p-MAPK, MAPK, and PgR in MCF-7Ca (letrozole sensitive) and LTLT-Ca (letrozole resistant) cells. Cell lysates were prepared and used for Western Blot as described [75]. B) Effect of 5 mM of PD98059 on increasing ER-a expression in LTLTCa cells compared to MCF-7Ca cells. LTLT-Ca cells were transferred into steroid-free medium for 3 days before plating. The next day cells were washed and treated with steroid-free medium containing androstenedione (25nM) and the indicated concentration of PD 98059. The medium was changed every 3 days, and the cells were lysed 9 days later [75].
Figure 3. Restoration of hormonal sensitivity to LTLTCa cells with trastuzumab (Herceptin).

LTLTCa cells were transferred to IMEM without phenol red before they were plated into 96-well plates (1000 cells/well) and allowed to attach overnight. The next day, they were treated with the indicated agents for a total of 6 days. The cell viability was measured using the MTT assay as described [68]. A) Effect of combination of letrozole and trastuzumab in LTLT-Ca cells. Combination of letrozole plus trastuzumab was significantly better than single drug treatment or control, p < 0.0001 (10-12M-10-9M), p < 0.05 (10-8M-10-5M). B) Effect of combining trastuzumab with AEs tamoxifen, fulvestrant and AIs exemestane, anastrozole in LTLT-Ca cells. The cell viability was found to be significantly lower in groups treated with the combination of trastuzumab plus AI or AE versus control (p<0.0001) or trastuzumab alone (p<0.0001) or the endocrine agent alone (p<0.0001). C) Effect of estradiol (E2) on proliferation of MCF-7Ca and LTLT-Ca cells in presence or absence of trastuzumab (Herceptin) pretreatment. When pre-treated with herceptin (100μg/mL), proliferation of LTLT-Ca cells was significantly stimulated in response to E2 at concentrations of 10-12M - 10-7M when compared to E2 alone (p <0.0001). When MCF-7Ca cells were pretreated with herceptin, E2 stimulated proliferation was increased at concentrations 10-11M - 10-10M (p = 0.02 and 0.03 respectively) [68].
Consistent with the above findings, letrozole resistant LTLTCa cells were also unresponsive to E2 [75] (Figure 3C). Due to the constitutive activation of the ER, the LTLTCa cells became insensitive to proliferative concentrations of E2 and to reduction in E2 levels caused by AIs. The estrogen independence of the LTLTCa was further elucidated when they were transplanted into mice. These cells proliferated and formed tumors in the animals without any hormonal supplements. Thus the tumors grew at the same rate as those receiving androstenodione (Δ4A) or a combination of Δ4A and letrozole.
Overcoming resistance to AI therapy
Tumors that overexpress Her-2 are likely to be ER negative and PgR negative [80]. Our studies suggest that Her-2 causes downregulation of ER expression resulting in resistance to letrozole. We therefore investigated whether inhibition of key proteins in the Her-2 pathway would restore ER expression to normal levels and reverse resistance to AIs. When LTLTCa cells were treated with the clinically used Her-2 antibody, trastuzumab (Herceptin) (Figures 5) or the MAPK inhibitor PD98059 (Figure 2B) there was a marked increase in expression of ER to levels observed in the parental MCF-7Ca cells and much higher than the levels of the untreated LTLTCa cells [75]. In addition, Her-2 and MAPK inhibitors effectively inhibited proliferation of LTLTCa cells indicating their dependency on Her-2/MAPK signaling for stimulating growth. Importantly, treatment of LTLTCa cells with trastuzumab was also very effective in restoring the antiproliferative effect of letrozole treatment in LTLTCa cells (Figure 3A) [68]. Trastuzumab (100μg/mL) showed a synergistic inhibition of proliferation when combined with letrozole. Furthermore, trastuzumab not only reversed the resistance of LTLTCa cells to letrozole but also to tamoxifen, and other AIs exemestane and anastrozole (Figure 3B) [68]. As shown in Figure 3c, LTLTCa cells are unresponsive to estradiol (E2). However, when pre-treated with 100μg/mL of herceptin, they became as sensitive to the effects of E2 as MCF-7Ca cells. Trastuzumab (100μg/mL) also normalized the levels of p-Elk1, p-MAPK and p-p90RSK expression in LTLTCa cells to those of MCF-7Ca cells (Figure 4) [68]. These results indicate that Her-2 is a negative regulator of ER-α and that blockade of the Her-2 signaling pathway restores ER-α expression to normal levels observed in hormone sensitive MCF-7Ca cells. Furthermore, the results indicate that control over tumor growth is achieved by blocking growth factor receptor similarly as well as ER-mediated signaling [68].
Figure 5. The Effect of trastuzumab alone or in combination with letrozole on the growth of MCF-7Ca xenografts.

Tumors of MCF-7Ca cells were grown in mice following procedures described in Fig 1 and treated with vehicle, trastuzumab or letrozole. At week 16, the group receiving letrozole was divided into two groups and received letrozole plus trastuzumab or continued on letrozole. Trastuzumab alone (5mg/kg/week) did not inhibit the growth of MCF-7Ca tumors. The difference in the exponential parameter governing the growth rate of letrozole versus letrozole switched to letrozole plus trastuzumab was 0.21 ± 0.08, p = 0.008. The difference in the exponential parameter governing tumor growth rate of letrozole plus trastuzumab versus letrozole switched to letrozole plus trastuzumab was 0.39 ± 0.09, p <0.0001. The difference in the exponential parameter governing rate of letrozole switched to trastuzumab versus letrozole switched to letrozole plus trastuzumab was 0.2 ± 0.08, p = 0.011, over weeks 15-28. When compared through week 29, the difference in the exponential parameter governing growth rate of letrozole versus letrozole switched to trastuzumab was 0.005 ± 0.08, p value = 0.97 [68].
Figure 4. The effect of trastuzumab treatment on ER-a expression.
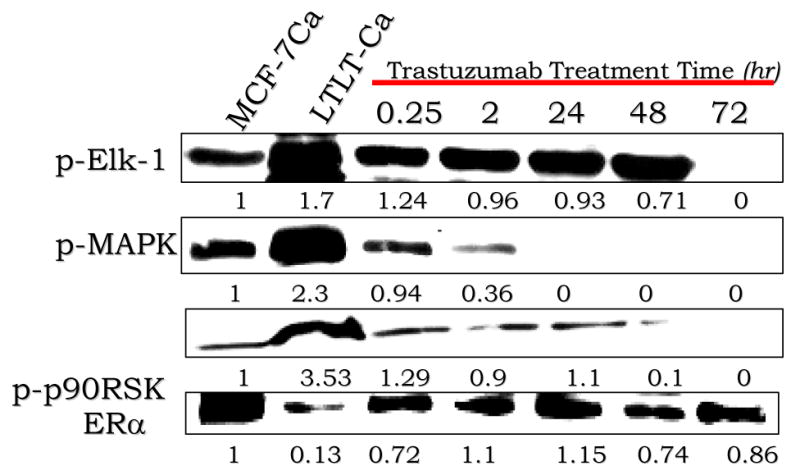
LTLTCa cells were transferred to IMEM without phenol red 24 hours before the beginning of the experiment. The next day, they were treated with the indicated agents for the specified amount of time. The cell lysates were prepared as described [75]. The effect of trastuzumab treatment at various time points on protein expression of p-ELK-1, p-MAPK, p-p90RSK and ER-a in LTLT-Ca cells is shown. Protein expression was examined using western immunoblotting. Blot shows phospho-Elk-,1 at 60 kDa, phospho-p90RSK at 90 kDa, p-MAPK at 42-44 kDa and ER-a at 66 kDa. The blots show a single representative of three independent experiments.
We investigated the hypothesis that combining agents that inhibit estrogen mediated signaling and growth factor receptor signaling will block their interaction and overcome resistance to hormone therapy. When MCF-7Ca tumors reached a measurable size (∼300 mm3) mice in the control group were divided into groups. One group continued as control another was treated with trastuzumab (5 mg/kg/week) and a third group with letrozole (10μg/day). The tumors in the trastuzumab group grew at the same rate as the controls. Tumors in the letrozole group, initially regressed for several weeks but eventually began to grow and had doubled in volume by 18 weeks. The mice were then re-grouped and continued on letrozole, switched to trastuzumab treatment or treated with letrozole plus trastuzumab (5mg/kg/week). Tumors switched to trastuzumab were significantly reduced by the Her-2 blocker for a few weeks but subsequently grew to the same size as control tumors. The addition of trastuzumab to letrozole treatment caused significant regression compared to the tumors that remained on letrozole and all other treatments (Fig 5) [68]. This finding shows that resistance to letrozole can be overcome by adding trastuzumab to the letrozole treatment regime and could be effective in BC patients whose tumors have developed resistance to this AI. A number of clinical trials are in progress with tyrosine kinase inhibitors that will test the hypothesis.
Conclusions
Targeting steroidogenic enzymes such as aromatase (CYP19) has proved an effective strategy for treating breast cancer. Although aromatase inhibitors are having utility in other conditions in both women and men, aromatase inhibitors have not been successful in treating either benign prostatic hypotrophy (BPH) or prostate cancer. New strategies to develop inhibitors of 17α-hydroxylase/lyase (CYP17) have potential on this regard. Clinical trials with abiraterone are currently in progress. Some of these CYP17 inhibitors are of particular interest as they are also androgen receptor antagonists and cause down regulation of this receptor. A lead compound is anticipated to begin clinical trials in the near future. Strategies to meet the challenge of inevitable resistance to these treatments are in progress. Results indicate that tumors adapt by activating alternate signaling pathways. Inhibiting these pathways as well as estrogen/androgen signaling is effective in model systems in restoring and extending the benefits of therapy with these hormonal agents.
Acknowledgments
This work was supported by an NIH grant (CA-62483) to Dr. Angela Brodie.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Inkster SE, Brodie AM. Immunocytochemical studies of aromatase in early and full-term human placental tissues: Comparison with biochemical assays. Biol Reprod. 1989;100:1684–95. doi: 10.1095/biolreprod41.5.889. [DOI] [PubMed] [Google Scholar]
- 2.Inkster SE, Brodie AM. Expression of aromatase cytochrome P-450 in premenopausal and postmenopausal human ovaries: An immunocytochemal study. J Clin Endocrinol Metab. 1991;73:717–26. doi: 10.1210/jcem-73-4-717. [DOI] [PubMed] [Google Scholar]
- 3.McGuire WL. An update on estrogen and progesterone receptors in prognosis for primary and advanced breast cancer. In: Iacobelli S, et al., editors. Hormones and Cancer. Vol. 15. 1980. pp. 337–44. [Google Scholar]
- 4.Grodin JM, Siiteri PK, MacDonald PC. Source of estrogen production in postmenopausal women. Journal of Clin Endo & Metab. 1973;36:207–14. doi: 10.1210/jcem-36-2-207. [DOI] [PubMed] [Google Scholar]
- 5.O'Neill JS, Elton RA, Miller WR. Aromatase activity in adipose tissue from breast quadrants: a link with tumor site. Br Med J Clin Res Ed. 1988;296:741–3. doi: 10.1136/bmj.296.6624.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Longcope C, Pratt JH, Schneider SH, Fineberg SE. Aromatization of androgens by muscle and adipose tissue in vivo. J Clin Endocrinol Metab. 1978;46:146–52. doi: 10.1210/jcem-46-1-146. [DOI] [PubMed] [Google Scholar]
- 7.Hemsell DL, Grodin JM, Brenner PF, Siiteri PK, MacDonald PC. Plasma precursors of estrogen: II. Correlation of the extent of conversion of plasma androstenedione to estrone with age. J Clin Endocrinol Metab. 1974;38:476–9. doi: 10.1210/jcem-38-3-476. [DOI] [PubMed] [Google Scholar]
- 8.Thorsen T, TAngen M, Stoa KF. Concentration of endogenous oestradiol as related to oestradiol receptor sites in breast tumor cytosol. Eur J Cancer Clin Oncol. 1982;18:333–7. doi: 10.1016/0277-5379(82)90002-5. [DOI] [PubMed] [Google Scholar]
- 9.Blankenstein MA, Szymczak J, Daroszewski J, Milewicz A, Thijssen JH. Estrogens in plasma and fatty tissue from breast cancer patients and women undergoing surgery for non-oncological reasons. Gynecol Endocrinol. 1992;6:13–7. doi: 10.3109/09513599209081001. [DOI] [PubMed] [Google Scholar]
- 10.Perel E, Blackstein ME, Killinger DW. Aromatase in human breast carcinoma. Cancer Res. 1982;42:3369s–72s. [PubMed] [Google Scholar]
- 11.James VH, McNeill JM, Lai LC, Newton CJ, Ghilchik MW, Reed MJ. Aromatase activity in normal breast and breast tumor tissues: in vivo and in vitro studies. Steroids. 1987;50:269–79. doi: 10.1016/0039-128x(83)90077-6. [DOI] [PubMed] [Google Scholar]
- 12.Killinger DW, Perel E, Daniilescu D, Kharlip L, Blackstein ME. Aromatase activity in the breast and other peripheral tissues and its therapeutic regulation. Steroids. 1987;50:523–36. doi: 10.1016/0039-128x(87)90036-5. [DOI] [PubMed] [Google Scholar]
- 13.Miller WR, Hawkins RA, Forrest AP. Significance of aromatase activity in human breast cancer. Cancer Res. 1982;42:3365s–8s. [PubMed] [Google Scholar]
- 14.Price T, Aitken J, Head J, Mahendroo M, Means G, Simpson E. Determination of aromatase cytochrome P450 messenger ribonucleic acid in human breast tissue by competitive polymerase chain reaction amplification. J Clin Endocrinol Metab. 1992;74:1247–52. doi: 10.1210/jcem.74.6.1592866. [DOI] [PubMed] [Google Scholar]
- 15.Koos RD, Banks PK, Inkster SE, Yue W, Brodie AM. Detection of aromatase and keratinocyte growth factor expression in breast tumors using reverse transcription-polymerase chain reaction. J Steroid Biochem Mol Biol. 1993;45:217–25. doi: 10.1016/0960-0760(93)90335-t. [DOI] [PubMed] [Google Scholar]
- 16.Lu Q, Nakmura J, Savinov A, et al. Expression of aromatase protein and messenger ribonucleic acid in tumor epithelial cells and evidence of functional significance of locally produced estrogen in human breast cancers. Endocrinology. 1996;137:3061–8. doi: 10.1210/endo.137.7.8770932. [DOI] [PubMed] [Google Scholar]
- 17.Brodie AM, Lu Q, Long BJ, et al. Aromatase and COX-2 expression in human breast cancers. J Steroid Biochem Mol Biol. 2001;79:41–7. doi: 10.1016/s0960-0760(01)00131-5. [DOI] [PubMed] [Google Scholar]
- 18.Lipton A, Santner SJ, Santen RJ, et al. Aromatase activity in primary and metastatic human breast cancer. Cancer Invest. 1987;59:779–82. doi: 10.1002/1097-0142(19870215)59:4<779::aid-cncr2820590419>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 19.Simpson ER, Mahendroo MS, Means GD, Kilgore MW, Corbin CJ, Mendelson CR. Tissue-specific promoters regulate aromatase cytochrome P450 expression. J Steroid Biochem Mol Biol. 1993;44:321–30. doi: 10.1016/0960-0760(93)90235-o. [DOI] [PubMed] [Google Scholar]
- 20.Brueggemeier RW, Quinn AL, Parrett ML, Joarder FS, Harris RE, Robertson FM. Correlation of aromatase and cyclooxygenase gene expression in human breast cancer specimens. Cancer Lett. 1999;140:27–35. doi: 10.1016/s0304-3835(99)00050-6. [DOI] [PubMed] [Google Scholar]
- 21.Fisher B, Costantino JP, Redmond CK, Fisher ER, Wickerham DL, Cronin WM. Endometrial cancer in tamoxifen-treated breast cancer patients: findings from the National Surgical Adjuvant Breast and Bowel Project (NSABP) B-14. J Natl Cancer Inst. 1994;86:527–37. doi: 10.1093/jnci/86.7.527. [DOI] [PubMed] [Google Scholar]
- 22.Jordan VC. Tamoxifen: toxicities and drug resistance during the treatment and prevention of breast cancer. Annu Rev Pharmacol Toxicol. 1995;35:195–211. doi: 10.1146/annurev.pa.35.040195.001211. [DOI] [PubMed] [Google Scholar]
- 23.Schwarzel WC, Kruggel WG, Brodie HJ. Studies on the mechanism of estrogen biosynthesis. 8. The development of inhibitors of the enzyme system in human placenta. Endocrinology. 1973;92:866–80. doi: 10.1210/endo-92-3-866. [DOI] [PubMed] [Google Scholar]
- 24.Brodie AM, Schwarzel WC, Shaikh AA, Brodie HJ. The effect of an aromatase inhibitor, 4-hydroxy-4-androstene-3,17-dione, on estrogen-dependent processes in reproduction and breast cancer. Endocrinology. 1977;100:1684–95. doi: 10.1210/endo-100-6-1684. [DOI] [PubMed] [Google Scholar]
- 25.Coombes RC, Goss P, Dowsett M, Gazet JC, Brodie A. 4-Hydroxyandrostenedione in treatment of postmenopausal patients with advanced breast cancer. Lancet. 1984;2:1237–9. doi: 10.1016/s0140-6736(84)92795-8. [DOI] [PubMed] [Google Scholar]
- 26.Goss PE, Powles TJ, Dowsett M, et al. Treatment of advanced postmenopausal breast cancer with aromatase inhibitor, 4-hydroxyandrostenedione-phase 2 report. Cancer Res. 1986;46:4823–6. [PubMed] [Google Scholar]
- 27.Baum M, Budzar AU, Cuzick J, et al. Anastrozole alone or in combination with tamoxifen versus tamoxifen alone for adjuvant treatment of postmenopausal women with early breast cancer: first results of the ATAC randomized trial. Lancet. 2002;359:2131–9. doi: 10.1016/s0140-6736(02)09088-8. [DOI] [PubMed] [Google Scholar]
- 28.Henderson D. Estrogens and benign prostatic hyperplasia: rationale for therapy with aromatase inhibitors. Ann Med. 1991;23:201–3. doi: 10.3109/07853899109148048. [DOI] [PubMed] [Google Scholar]
- 29.Kozák I, Bartsch W, Krieg M, Voigt KD. Nuclei of stroma: site of highest estrogen concentration in human benign prostatic hyperplasia. Prostate. 1982;3:433–8. doi: 10.1002/pros.2990030503. [DOI] [PubMed] [Google Scholar]
- 30.Brodie AM, Son C, King DA, Meyer KM, Inkster SE. Lack of evidence for aromatase in human prostatic tissues: effects of 4-hydroxyandrostenedione and other inhibitors on androgen metabolism. Cancer Res. 1989;49:6551–5. [PubMed] [Google Scholar]
- 31.Ellem SJ, Risbridger GP. Aromatase and prostate cancer. Minerva Endocrinol. 2006;31:1–12. [PubMed] [Google Scholar]
- 32.Habenicht UF, Schwarz K, Neumann F, el Etreby MF. Induction of estrogen-related hyperplastic changes in the prostate of the cynomolgus monkey (Macaca fascicularis) by androstenedione and its antagonization by the aromatase inhibitor 1-methyl-androsta-1,4-diene-3,17-dione. Prostate. 1987;11:313–26. doi: 10.1002/pros.2990110404. [DOI] [PubMed] [Google Scholar]
- 33.Shearer R, Davis JH. Studies in prostatic cancer with 4-hydroxyandrostenedione. In: Coombes RC, Dowsett M, editors. 4-Hydroxyandrostenedione-A Nw Approach to Hormone-Dependent Cancer. 1991. pp. 41–44. [Google Scholar]
- 34.Smith MR, Kaufman D, George D, et al. Selective aromatase inhibition for patients with androgen-independent prostate carcinoma. Cancer. 2002;95:1864–8. doi: 10.1002/cncr.10844. [DOI] [PubMed] [Google Scholar]
- 35.Santen RJ, Petroni GR, Fisch MJ, Myers CE, Theodorescu D, Cohen RB. Use of the aromatase inhibitor anastrozole in the treatment of patients with advanced prostate carcinoma. Cancer. 2001;92:2095–101. doi: 10.1002/1097-0142(20011015)92:8<2095::aid-cncr1550>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 36.Messing E. The timing of hormone therapy for men with asymptomatic advanced prostate cancer. Urol Oncol. 2003;21:245–54. doi: 10.1016/s1078-1439(03)00016-4. [DOI] [PubMed] [Google Scholar]
- 37.Spitz IM, Chertin B, Fridmans A, et al. A hydrogel implant containing the gonadotropin-releasing hormone superanalog, Histrelin [(ImBzl)D-His6, Pro9-Net] results in medical castration for up to 3 years in patients with prostate cancer (PC) with immediate reversal of testosterone suppression on implant removal. Abs OR11-2, 85th Ann Meet of the Endocrine Soc, Philadelphia, PA. 2003:19–23. [Google Scholar]
- 38.Forti G, Salerno R, Moneti G, et al. Three-month treatment with a long-acting gonadotropin-releasing hormone agonist of patients with benign prostatic hyperplasia: effects on tissue androgen concentration, 5 alpha-reductase activity and androgen receptor content. J Clin Endocrinol Metab. 1989;68:461–8. doi: 10.1210/jcem-68-2-461. [DOI] [PubMed] [Google Scholar]
- 39.Geller J, Albert JD, Nachtsheim DA, Loza D. Comparison of prostatic cancer tissue dihydrotestosterone levels at the time of relapse following orchiectomy or estrogen therapy. J Urol. 1984;132:693–6. doi: 10.1016/s0022-5347(17)49829-6. [DOI] [PubMed] [Google Scholar]
- 40.Crawford ED, Eisenberger MA, McLeod DG, et al. A controlled trial of leuprolide with and without flutamide in prostatic carcinoma. N Engl J Med. 1989;321:419–24. doi: 10.1056/NEJM198908173210702. [DOI] [PubMed] [Google Scholar]
- 41.Labrie F, Belanger A, Simard J, Labrie C, Dupont A. Combination therapy for prostate cancer. Endocrine and biologic basis of its choice as new standard first-line therapy. Cancer Suppl. 1993;71:1059–67. doi: 10.1002/1097-0142(19930201)71:3+<1059::aid-cncr2820711426>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 42.Visakorpi T, Hyytinen E, Koivisto P, et al. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet. 1995;9:401–6. doi: 10.1038/ng0495-401. [DOI] [PubMed] [Google Scholar]
- 43.Culig Z, Hobisch A, Hittmair A, et al. Androgen receptor gene mutations in prostate cancer. Implications for disease progression and therapy. Drugs Aging. 1997;10:50–8. doi: 10.2165/00002512-199710010-00005. [DOI] [PubMed] [Google Scholar]
- 44.Suzuki H, Akakura K, Komiya A, Aida S, Akimoto S, Shimazaki J. Codon 877 mutation in the androgen receptor gene in advanced prostate cancer: relation to antiandrogen withdrawal syndrome. Prostate. 1996;29:153–8. doi: 10.1002/1097-0045(199609)29:3<153::aid-pros2990290303>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 45.Gaddipati JP, McLeod DG, Heidenberg HB, et al. Frequent detection of codon 877 mutation in the androgen receptor gene in advanced prostate cancers. Cancer Res. 1994;54:2861–4. [PubMed] [Google Scholar]
- 46.Taplin ME, Bubley GJ, Shuster TD, et al. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N Engl J Med. 1995;332:1393–8. doi: 10.1056/NEJM199505253322101. [DOI] [PubMed] [Google Scholar]
- 47.Mostaghel EA, Page ST, Lin DW, et al. Intraprostatic androgens and androgen-regulated gene expression persist after testosterone suppression: therapeutic implications for castration-resistant prostate cancer. Cancer Res. 2007;67:5033–41. doi: 10.1158/0008-5472.CAN-06-3332. [DOI] [PubMed] [Google Scholar]
- 48.Suzuki K, Nishiyama T, Hara N, Yamana K, Takahashi K, Labrie F. Importance of the intracrine metabolism of adrenal androgens in androgen-dependent prostate cancer. Prostate Cancer Prostatic Dis. 2007;10:301–6. doi: 10.1038/sj.pcan.4500956. [DOI] [PubMed] [Google Scholar]
- 49.Stanbrough M, Bubley GJ, Ross K, et al. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006;66:2815–25. doi: 10.1158/0008-5472.CAN-05-4000. [DOI] [PubMed] [Google Scholar]
- 50.Kellens RE. Gene amplification in mammalian cells: A comprehensive guide. Marcel Dekker, Inc.; New York: 1993. [Google Scholar]
- 51.Goss PE, Ingle JN, Martino S, et al. A randomized trial of letrozole in postmenopausal women after five years of tamoxifen therapy for early-stage breast cancer. N Engl J Med. 2003;349:1793–802. doi: 10.1056/NEJMoa032312. [DOI] [PubMed] [Google Scholar]
- 52.Grigoryev DN, Long BJ, Nnane IP, Njar VC, Liu Y, Brodie AM. Effects of new 17alpha-hydroxylase/C(17,20)-lyase inhibitors on LNCaP prostate cancer cell growth in vitro and in vivo. Br J Cancer. 1999;81:622–30. doi: 10.1038/sj.bjc.6690739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Long BJ, Grigoryev DN, Nnane IP, Liu Y, Ling YZ, Brodie AM. Antiandrogenic effects of novel androgen synthesis inhibitors on hormone-dependent prostate cancer. Cancer Res. 2000;60:6630–40. [PubMed] [Google Scholar]
- 54.Trachtenberg J. Ketoconazole therapy in advanced prostatic cancer. J Urol. 1984;132:61–3. doi: 10.1016/s0022-5347(17)49464-x. [DOI] [PubMed] [Google Scholar]
- 55.Williams G, Kerle DJ, Ware H, et al. Objective responses to ketoconazole therapy in patients with relapsed progressive prostatic cancer. Br J Urol. 1986;58:45–51. doi: 10.1111/j.1464-410x.1986.tb05426.x. [DOI] [PubMed] [Google Scholar]
- 56.Muscato JJ, Ahmann TA, Johnson KM, Wilding W, Monaghan G, Schlossman DM. Optimal dosing of ketoconazole and hydrocortisone leads to long responses in hormone refractory prostate cancer. Proc Am Assoc Cancer Res. 1994;13:22. [Google Scholar]
- 57.Small EJ, Baron AD, Fippin L, Apodaca D. Ketoconazole retains activity in advanced prostate cancer patients with progression despite flutamide withdrawal. J Urol. 1997;157:1204–7. [PubMed] [Google Scholar]
- 58.Njar VC, Brodie AM. Inhibitors of 17alpha-hydroxylase/17,20-lyase (CYP17): potential agents for the treatment of prostate cancer. Curr Pharm Des. 1999;5:163–80. [PubMed] [Google Scholar]
- 59.Hartmann RW, Ehmer PB, Haidar S, et al. Inhibition of CYP 17, a new strategy for the treatment of prostate cancer. Arch Pharm (Weinheim) 2002;335:119–28. doi: 10.1002/1521-4184(200204)335:4<119::AID-ARDP119>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 60.Hakki T, Bernhardt R. CYP17- and CYP11B-dependent steroid hydroxylases as drug development targets. Pharmacol Ther. 2006;111:27–52. doi: 10.1016/j.pharmthera.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 61.Leroux F. Inhibition of p450 17 as a new strategy for the treatment of prostate cancer. Curr Med Chem. 2005;12:1623–9. doi: 10.2174/0929867054367185. [DOI] [PubMed] [Google Scholar]
- 62.O'Donnell A, Judson I, Dowsett M, et al. Hormonal impact of the 17alpha-hydroxylase/C(17,20)-lyase inhibitor abiraterone acetate (CB7630) in patients with prostate cancer. Br J Cancer. 2004;90:2317–25. doi: 10.1038/sj.bjc.6601879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Attard G, Belldegrun AS, de Bono JS. Selective blockade of androgenic steroid synthesis by novel lyase inhibitors as a therapeutic strategy for treating metastatic prostate cancer. BJU Int. 2005;96:1241–6. doi: 10.1111/j.1464-410X.2005.05821.x. [DOI] [PubMed] [Google Scholar]
- 64.Njar VC, Kato K, Nnane IP, Grigoryev DN, Long BJ, Brodie AM. Novel 17-azolyl steroids, potent inhibitors of human cytochrome 17 alpha-hydroxylase-C17,20-lyase (P450(17) alpha): potential agents for the treatment of prostate cancer. J Med Chem. 1998;41:902–12. doi: 10.1021/jm970568r. [DOI] [PubMed] [Google Scholar]
- 65.Handratta VD, Vasaitis TS, Njar VC, et al. Novel C-17-heteroaryl steroidal CYP17 inhibitors/antiandrogens: synthesis, in vitro biological activity, pharmacokinetics, and antitumor activity in the LAPC4 human prostate cancer xenograft model. J Med Chem. 2005;48:2972–84. doi: 10.1021/jm040202w. [DOI] [PubMed] [Google Scholar]
- 66.Vasaitis TS, Belosay A, Schayowitz A, et al. Androgen recptor inactivation contributes to antitumor efficacy of 17α-hydroxylase/17,20-lyase inhibitor 3β-hydroxy-17-(1H-benzimidazole-1yl)androsta-5,16-diene in prostate cancer. doi: 10.1158/1535-7163.MCT-08-0230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schayowitz A, Sabnis G, Goloubeva O, Njar VC, Brodie AM. Prolonging hormone sensitivity in prostate cancer xenograft through dual inhibition of AR and mTOR. doi: 10.1038/sj.bjc.6605882. in review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sabnis G, Schayowitz A, Goloubeva O, Macedo L, Brodie A. Trastuzumab reverses letrozole resistance and amplifies the sensitivity of breast cancer cells to estrogen. Cancer Res. 2008 doi: 10.1158/0008-5472.CAN-08-0857. in review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhou D, Pompom D, Chen S. Stable expression of human aromatase complementary DNA in mammalian cells: a useful system for aromatase inhibitor screening. Cancer Res. 1990;50:6949–54. [PubMed] [Google Scholar]
- 70.Yue W, Wang J, Savinov A, Brodie A. The effect of aromatase inhibitors on growth of mammary tumors in a nude mouse model. Cancer Res. 1995;55:3073–7. [PubMed] [Google Scholar]
- 71.Brodie A, Jelovac D, Macedo L, Sabnis G, Tilghman S, Goloubeva O. Therapeutic observations in MCF-7 aromatase xenografts. Clin Cancer Res. 2005;11:884s–8s. [PubMed] [Google Scholar]
- 72.Long BJ, Jelovac D, Handratta V, et al. Therapeutic strategies using the aromatase inhibitor letrozole and tamoxifen in breast cancer model. J Natl Cancer Inst. 2004;96:456–65. doi: 10.1093/jnci/djh076. [DOI] [PubMed] [Google Scholar]
- 73.Coombes RC, Hall E, Gibson LJ, et al. A randomized trial of exemestane after two to three years of tamoxifen therapy in postmenopausal women with primary breast cancer. N Eng J Med. 2004;350:1081–92. doi: 10.1056/NEJMoa040331. [DOI] [PubMed] [Google Scholar]
- 74.Long BJ, Jelovac D, Thiantanawat A, Brodie AM. The effect of second-line antiestrogen therapy on breast tumor growth after first-line treatment with the aromatase inhibitor letrozole: long-term studies using the intratumoral aromatase postmenopausal breast cancer model. Clin Cancer Res. 2002;8:2378–88. [PubMed] [Google Scholar]
- 75.Jelovac D, Sabnis G, Long BJ, Macedo L, Goloubeva OG, Brodie AM. Activation of mitogen-activated protein kinase in xenografts and cells during prolonged treatment with aromatase inhibitor letrozole. Cancer Res. 2005;65:5380–9. doi: 10.1158/0008-5472.CAN-04-4502. [DOI] [PubMed] [Google Scholar]
- 76.Kato S, Endoh H, Masuhiro Y, et al. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science. 1995;270:1491–4. doi: 10.1126/science.270.5241.1491. [DOI] [PubMed] [Google Scholar]
- 77.Joel PB, Traish AM, Lannigan DA. Estradiol-induced phosphorylation of serine 118 in the estrogen receptor is independent of p42/p44 mitogen-activated protein kinase. J Biol Chem. 1998;273:13317–23. doi: 10.1074/jbc.273.21.13317. [DOI] [PubMed] [Google Scholar]
- 78.Chen D, Washbrook E, Sarwar N, et al. Phosphorylation of human estrogen receptor alpha at serine 118 by two distinct signal transduction pathways revealed by phosphorylation-specific antisera. Oncogene. 2002;21:4921–31. doi: 10.1038/sj.onc.1205420. [DOI] [PubMed] [Google Scholar]
- 79.Levin ER. Bidirectional signaling between the estrogen receptor and the epidermal growth factor receptor. Mol Endocrinol. 2003;17:309–17. doi: 10.1210/me.2002-0368. [DOI] [PubMed] [Google Scholar]
- 80.Konecny G, Pauletti G, Pegram M, et al. Slamon. Quantitative association between HER-2/neu and steroid hormone receptors in hormone receptor-positive primary breast cancer. J Natl Cancer Inst. 2003;95:142–53. doi: 10.1093/jnci/95.2.142. [DOI] [PubMed] [Google Scholar]