

Abstract
Melanin concentrating hormone (MCH) is a cyclic 19-amino-acid peptide expressed mainly in the hypothalamus. It is involved in the control of feeding behavior, energy homeostasis, and body weight. Administration of MCH-R1 antagonists has been proved to reduce food intake and cause weight loss in animal models. In the present study, a homology model of the human MCH-R1 was constructed using the crystal structure of bovine rhodopsin (PDB: 1u19) as a template. Based on the observation that MCH-R1 can bind ligands of high chemical diversity, the initial model was subjected to an extensive ligand-supported refinement using antagonists of different chemotypes. The refinement process involved stepwise energy minimizations and molecular dynamics simulations. The refined model was inserted into a pre-equilibrated DPPC/TIP3P membrane system and then simulated for 20 ns in complex with structurally diverse antagonists. This protocol was able to explain the SAR of MCH-R1 antagonists with diverse chemical structures. Moreover, it reveals new insights into the critical recognition sites within the receptor. This work represents the first detailed study of molecular dynamics of MCH-R1 inserted into a membrane-aqueous environment.
Keywords: Melanin concentrating hormone, Molecular dynamics, Membrane aqueous environment
INTRODUCTION
Obesity can be defined as a body mass index (BMI) of 30 or more. In 2007, according to the World Health Organization (WHO), there were one billion overweight adults worldwide, 300 million of them were clinically obese).1 It is also reported that the prevalence of obesity (BMI ≥ 30) was doubled between 1986 and 2000.2 Complications include Metabolic syndrome, stroke, osteoarthritis, certain cancers (endometrial, colon, and gall bladder), psychological consequences (low self-esteem and clinical depression). Unfortunately, most of the currently used agent are associated with serious side effects and produce variable clinical results.3
Melanin concentrating hormone (MCH) is a cyclic polypeptide originally isolated from the salmon pituitary as a regulator of skin pigments change.4 Human MCH is a cyclic 19-amino-acid peptide, secreted mainly in the hypothalamus and zona incerta.5 This hormone is involved in the control of energy homeostasis, feeding behavior and body weight. MCH receptor is a G protein-coupled receptor (GPCR) with two subtypes R1 and R2. MCH-R1 is expressed in the hypothalamus of rodents and higher mammals while MCHR2 is found only in the hypothalamus of higher mammals.6, 7 Many studies have demonstrated the role of MCH-R1 in feeding and energy homeostasis. For example, MCH-induced body weight gain and hyperphagia are not observed in MCH-R1 null mice.8, 9 In addition, it was reported that alteration of MCH-R1 signaling through transgenesis or ligands has a significant effect in animal models of anxiety and depression.10 The exact biological role of MCH-R2 is still unclear due to species specific expression. However, it is considered to be involved in physiological processes other than feeding.7, 11
Several studies have proved that oral administration of small molecule MCH-R1 antagonists significantly reduces food intake and causes a dose-dependant weight loss in animal models.10, 12–15. In the past few years, numerous antagonists of the MCH-R1 were reported in the patent literature together with their biological data.16 The reported compounds have various chemical structures. However, the majority of these compounds possess a central amide residue with a hydrophobic aromatic group at one end of the molecule and basic nitrogen at the other end. We noticed that most of the reported MCH-R1 antagonists can be classified into three chemotypes (Fig. 1). These compounds can be linear with terminal basic nitrogen, linear with central basic nitrogen, or branched (Y-shaped) with basic nitrogen on one arm.
Figure 1.

Suggested classification of the reported MCH-R1 antagonists
For any biological target, the availability of an X-ray crystal structure can help in explaining the structure activity relationship (SAR) of the reported ligands as well as screening for new scaffolds. Unfortunately, for the majority of G-protein-coupled receptors (GPCRs), including MCH-R1, crystallization is hampered by the difficulty of generating sufficient protein material and inability to obtain satisfactory crystals.17 A number of MCH-R1 homology models have appeared in the literature over the last few years, depending on the X-ray crystal of bovine rhodopsin (bRho).18–24 Most of these models were generated by pharmaceutical companies to aid in optimizing their leads as part of ongoing MCH-R1 projects.
Therefore, each model considers the binding mode of only a specific series of compounds. Also, to our knowledge, no explanation was given for the SAR of the bulky Y-shaped ligands which possess some of the highest potencies. In addition, there have been no attempts to study the time-dependent behavior of MCH-R1, in complex with structurally diverse ligands, using molecular dynamic (MD) simulations.
Ligand-steered homology modeling (LSHM) is a useful approach for incorporating receptor flexibility during the optimization of GPCR binding sites. Thus, it can lead to a model capable of interpreting the binding behavior of ligands of diverse chemotypes. This technique depends on manual or rigid docking of known ligands into the receptor binding site, followed by iterative minimization and molecular dynamics simulation to obtain a ligand-refined model. Various modifications of this general procedure have been introduced over the last few years.25 Another challenge is the accurate detection of binding sites in a 3D protein structure. Recently, Konc and Janežic introduced the ProBiS algorithm for detection of structurally similar binding sites in proteins. This approach uses the maximum clique technique to detect 3D correspondences between proteins at a sub-residue level.26
In the present study, a homology model of the human MCH-R1 was constructed based on the crystal structure of bovine rhodopsin (PDB: 1u19). This model was refined using iterative energy minimizations and MD simulations with antagonists of different chemotypes (linear and branched). In addition, the final refined model, in complex with two diverse MCH-R1 antagonists, was embedded in a DPPC/TIP3P membrane environment and subjected to a 20 ns MD simulation. This protocol led to a model capable of explaining the SAR of all the aforementioned structural classes of antagonists. Moreover, it revealed new interaction sites within the receptor that are potentially critical for ligand recognition. To the best of our knowledge, this is the first MCH-R1 model relaxed in an explicit membrane-aqueous environment.
MATERIALS AND METHODS
i. Multiple Sequence Alignment
The amino acid sequence of the MCH-R1 receptor (MCH-R1_HUMAN) was extracted from the SWISS-PROT and TrEMBL databases of the ExPASy molecular biology server27 (Primary accession number: Q99705). The structure template used for homology modeling was the crystal structure of the bovine rhodopsin (PDB ID: 1U19)28 resolved to 2.2 Å. The sequence alignment with bovine rhodopsin was generated, initially, using CLUSTALW2 web server with all parameters set as default.29 Afterwards, the resultant sequence alignment was adjusted manually using the software BIOEDIT. During this step, Ballesteros’ conserved residues were exploited to verify and adjust the initial alignment leading to unambiguous sequence alignment, despite the low sequence identity (19%). Whenever appropriate, gaps were introduced into the sequence to optimize the alignment taking into consideration the absence of gaps in the structurally conserved regions (SCRs). The length of the TM helices of the MCH-R1 was decided depending on the corresponding bovine rhodopsin helices and considering the prediction by TMPRED web interface.30
ii. Homology Modeling
The final multiple sequence alignment was submitted to MODELLER 9v3 for generating the homology model of the MCH-R1. MODELLER software uses sequence alignment to extract a large number of spatial restraints for homology modeling of the target protein. These spatial restraints are then expressed as PDFs, which are combined into an objective function that is optimized by a combination of conjugate gradients and MD-simulated annealing. Seven different 3D models were built. Following the selection of the best generated model, the helical regions of the receptor were extracted to be used in the next steps. All hydrogen atoms were added to the model using the “ADD HYDROGEN” command within Sybyl v7.2.31 Next, the N-termini of the 7 TM helices were capped with ACE while the C-termini were capped with NMA groups. Side chains of ASN and GLN residues were reoriented to optimize the H-bonding network. Initial side chains orientation was determined using information from the template. Visual examination of the side chains of residues lining the binding pocket was carried out to insure proper orientation. In the upper part, a number of residues were found to have several possible side chain conformations. Whenever appropriate, these side chains were assigned different conformations using the Lovell30 rotamer library in order to open up the binding pocket. This led to the first crude Model I.
iii. Initial Model Refinement
The whole model was subjected to a stepwise energy refinement in Macromodel using the OPLS-2005 force field and PRCG method with distance-dependent dielectric constant and extended cutoff. First, all heavy atoms were kept fixed, and the hydrogen atoms were allowed to optimize their positions using 500 steps of steepest descent algorithm to a gradient of 0.01 kcal/mol/Å followed by 1000 steps of conjugate gradient algorithm to a gradient of 0.001 kcal/mol/Å with a distance dependent dielectric function and a non-bond cutoff distance of 10 Å. Second, the entire backbone of the protein was kept fixed and the side chains were minimized using the same parameters. Finally, the whole molecule including the backbone was tethered with a gradually reducing positional restraint of 100, 50, 25 10, and 5 kcal/mol/Å2. We decided to refrain from reducing the positional restraints below 5 kcal/mol/Å2 to maintain the conserved pattern of the 7 TM helices. This procedure yielded the refined Model II.
iv. Ligand-Supported Model Refinement
Conformational Analysis
Compound T-226296, compound 4, and 5 (Fig. 1) were sketched with their amide bonds in trans conformation. Atomic charges were calculated by the Gasteiger-Huckel method33 and the structure was minimized using the Tripos force field and the Powell conjugate gradient algorithm34 with a convergence of 0.001 kcal/mol/Å. The basic nitrogen was assigned a formal positive charge as would be expected at the normal physiological PH of 7.2. Then, the grid conformational search feature in SYBYL was used to located energy minima for the torsional potential around all the rotatable bonds in each molecule. Selected bonds were rotated in increments of 10° through the range of 360° followed by 300 Powell minimization steps. The resulting conformations were analyzed to identify the global energy minimum. The conformer with the lowest energy was used in the docking step.
Refinement with T-226296
Energy minimized T-226296 was manually docked in Model II using the known SAR information.35 Rigid docking of this ligand was performed using graphical manipulation with continuous energy monitoring (DOCK module within Sybyl v7.2). The starting point for docking was the presumed salt bridge between the positively charged nitrogen of the ligand and Asp192 in the third transmembrane helix. The ligand was then translated and rotated trying to embed the biphenyl group as deeply as possible into the putative binding pocket of Model I while avoiding severe steric overlap with the amino acid residues within the pocket. The ligand-MCH-R1 complex was then minimized using OPLS-2005 force field to relieve any steric clashes. The minimization protocol involved 500 and 1000 steps of steepest descent and conjugate gradient algorithm, up to the gradient of 0.01 and 0.001 kcal/mol/Å, respectively, using a distance dependent dielectric function and a non-bond cutoff distance of 10 Å. During MD simulation, harmonic restraints of 25 kcal/mol/Å2 were applied on the Cα atoms of the protein backbone. Moreover, a distance constraints of 10 kcal/mol/Å2 were applied between helices ends to inhibit artificial opening of helices. In addition, the charge assisted hydrogen bonding interaction between the carboxylic group of Asp192 and the basic amine of the ligand was used as a distance restraint with a force constant of 25 kcal/mol/Å2 to guide the MD simulation. Finally, the side chain torsions of Asp192 were restrained to the values of (x1 = −177°, x2 = 0.00°) using a force constant of 25 kcal/mol/Å2. An NVT (constant-volume/constant-temperature) MD simulation of 7 ns at 300 K was carried out; with a time step of 1.5 fs and a non-bonded update every 25 fs. Shake algorithm was employed to keep all bonds involving hydrogen atoms rigid. The average structures from the last 1 ns trajectory of MD were minimized as mentioned earlier. This procedure results in a significant opening of the binding pocket of MCH-R1 receptor model, leading to the refined model III.
Refinement with compound 5
Compound 5 was docked into Model III using GOLD software. Distance and Hydrogen bond constraints were applied with Asp192 and Gln196, respectively, to orient this compound in a binding pose similar to that of T-226296. The resulting complex was minimized initially using the protocol described earlier and then submitted to MD simulation. The simulation protocol used an NVT canonical ensemble and a time step of 1.5 fs (applying Shake algorithm) for a period of 7 ns at 300 k. The entire protein was kept free during these simulations, except for a mild positional restraint of 5 kcal/mol/Å2 on Cα atoms of the backbone in order to preserve the 3D fold of the TM helices. The complex obtained after MD was minimized, as described earlier, to get the final refined MCH-R1 receptor model (Model IV).
v. Automated Docking Simulations
A total of 18 reported MCH-R1 antagonists were built and energy-minimized using MMFF charges and the MMFF forcefield36 as implemented in Sybyl 7.2 with 2000 steps of conjugate gradient method to a gradient of 0.001 kcal/mol/Å. Formal charges were assigned to ionizable groups of ligands as would be expected at physiological pH. GOLD 3.2 software37 was used for docking of the minimized compounds into the putative binding pocket of the refined Model IV. This software uses a genetic algorithm (GA) to explore possible ligand binding modes by changing dihedrals of ligand rotatable bonds, ligand ring geometries, and dihedrals of protein OH and NH2 groups. The method used for placing the ligand in the binding site depends on fitting points. The program generates hydrogen bonding and hydrophobic fitting points which are used to map ligand hydrogen bonding fitting points and CH groups, respectively. The binding site was defined to include all amino acid residues within 12 Å of the side chain nitrogen of Gln196. Docking was carried out using standard mode settings and the GoldScore function which is a molecular mechanics-like function depending on protein-ligand hydrogen bonding, protein-ligand van der Waals score, ligand intramolecular hydrogen bonding and intramolecular strain of the ligand. A total of 10 genetic algorithm runs were performed for each ligand with early termination criteria set to 1.5 Å root-mean-square deviations (RMSD) value. Distance restraints between Asp192 and ligands basic nitrogens were used to guide the docking simulations. All the obtained docking poses were thoroughly analyzed to insure the validity of the docking poses and critical interactions.
vi. MD simulation of receptor-membrane complexes
The complexes of Model IV with compounds 4 and 5 were used for further study. They were individually inserted in a pre-equilibrated DPPC/TIP3P membrane system using the system builder tool within Desmond software38 as implemented in Maestro graphical interface (Schrodinger Inc., Portland, OR, USA). The longest axis of the Model IV as well as the membrane normal was aligned with Z-axis.
During the system building, lipids that were in close contact with the protein atoms were automatically removed, leaving a gap between the protein and the DPPC interface. Subsequently, chlorine atoms were added to the complex until entire system attained a net neutral charge. The membrane system composed of 74 DPPC molecules, 6 chloride ions, and 5312 water molecules. The number of atoms in the system, including protein, lipids, water, and ions was 28775.
MD simulations were carried out using Desmond application within Maestro. The all-atom optimized potential for liquid simulations (OPLS-AA) force field currently implemented in Desmond software was used for all molecules of the system. Prior to MD simulations, three steps of minimization were carried out. In the first step, protein, ligand, and lipids were kept fixed (force constant: 500 kcal/mol/Å2), and only water and ions are allowed to minimize and relax their positions. In the second step, protein and ligand were kept fixed (force constant: 500 kcal/mol/Å2), and positions of water, ions, and lipids were minimized. In the third step, protein backbone atoms were restrained using a force constant of 50 kcal/mol/Å2, and the rest of the system were kept free. The three minimization steps consisted of 6000 steps each, in which first 1000 steps were steepest descents, and last 5000 were L-BFGS. MD simulations were performed using the minimized structures. The Particle-mesh Ewald (PME) method was used to handle long-range electrostatic interactions. Time step of simulation was 2.0 fs and a 10 Å cut off was used for nonbonded interactions. Shake algorithm was employed to keep all bonds involving hydrogen atoms rigid. Constant-Volume (NVT) MD simulation was performed for the first 100 ps during which temperature of the system was raised from 0 to 300 K. The system temperature was maintained at 300 K for the remainder of the simulation using Langevin dynamics. Subsequently, the system was equilibrated in an NPT ensemble using the protocol depicted in Table 1. During the equilibration, the harmonic constraints were gradually reduced to achieve a smooth relaxation. The production run was then carried out for 20 ns using Langevin dynamics to maintain the temperature at 300 K and a pressure of 1.0132 bar (see Table 1). The evaluation of the trajectory was carried out using Maestro trajectory player and VMD 1.8.7.39 The final structure, after the 20 ns production run, was minimized using the same protocol mentioned above.
Table 1.
Parameters used for the MD equilibration and production runs
| Steps | Timestep | Harmonic Constraints (kcal/mol/Å2) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Numstep | Temperature | Backbone | Ligand | Side Chains | Lipids | Water and Ions | Temperature Control | ||
| Heating | |||||||||
| 1 | 2.0 fs | 100 ps | 300 K | 50 | 50 | 50 | 50 | 50 | NVT |
| Equilibration Steps | |||||||||
| 2 | 2.0 fs | 100 ps | 300 K | 50 | 50 | 50 | 50 | 50 | NPT |
| 3 | 2.0 fs | 100 ps | 300 K | 50 | 50 | 50 | 50 | 10 | NPT |
| 4 | 2.0 fs | 100 ps | 300 K | 50 | 50 | 10 | 10 | 5 | NPT |
| 5 | 2.0 fs | 100 ps | 300 K | 50 | 10 | 5 | 5 | 1 | NPT |
| 6 | 2.0 fs | 100 ps | 300 K | 50 | 5 | 1 | 1 | - | NPT |
| 7 | 2.0 fs | 100 ps | 300 K | 25 | - | - | - | - | NPT |
| 8 | 2.0 fs | 100 ps | 300 K | 10 | - | - | - | - | NPT |
| 9 | 2.0 fs | 100 ps | 300 K | 5 | - | - | - | - | NPT |
| 10 | 2.0 fs | 250 ps | 300 K | 1 | - | - | - | - | NPT |
| Production Run | |||||||||
| 11 | 2.0 fs | 20 ns | 300 K | - | - | - | - | - | NPT P=1.0132 bar T = 300 K |
RESULTS AND DISCUSSION
i. Multiple Sequence Alignment
An accurate sequence alignment is necessary for building a valid 3D homology model. This is even more important in case of MCH-R1 where sequence identity with bRho is as low as 19%. This value reaches 21.1% within the TM helical regions. Multiple sequences of class A GPCRs were used in the alignment process. Sequence of human MCH-R1, human MCH2, human dopamine receptor 1 (DRD1), and bovine rhodopsin were aligned using the web interface ClustalW. The resulting alignment was refined by manual editing to fill any gaps within the TM regions as defined by Palczewski et al.40 This refinement was guided by the conserved motifs in every TM helix specific for class A GPCR which are Asn46 (N1.50) in TMI, Asp74 (D2.50) in TMII, Arg126 (R3.50) in TMIII, Trp153 (W4.50) in TMIV, Pro207 (P5.50) in TMV, Pro255 (P6.50) in TMVI, and Pro299 (P7.50) in TMVII. Additionally, the final multiple sequence alignment were verified using the alignment of Several functional microdomains, such as the LAxxD motif in TMII, the D/ERY motif in TMIII, and the NPxxY motif in TMVII which are known to be conserved across the members of the class A GPCR superfamily.
ii. Homology Modeling
Till the date of our study, there were only three available templates for the building of GPCR homology models; bovine rhodopsin, Turkey β-1, and human β-2 adrenergic receptors. We decided to use bovine rhodopsin as template because, compared to β-2 receptor, it has a higher sequence identity with our target MCH-R1 (ClustalW score of 16% compared to 13% in case of β-2 adrenergic receptor). Moreover, bRho has been used successfully for the construction of several useful GPCR models.41 The multiple sequence alignment obtained in the previous step was used as an input for the building of the homology model of the MCH-R1. Seven different models were generated by MODELLER 9v3 software.42 These models were carefully analyzed for energy value, PDF violations, and violations of geometrical features. The third model displaying lowest violations and lowest MODELLER energy was selected for further studies (see supporting information). The TM helices of this model were extracted. The length of the helical regions was determined according to the helices of bRho as determined by Palczewski et al.40 After all the hydrogen atoms were added to the initial model, visual examination revealed several issues that could affect ligand binding. Two residues, Gln196 and Gln345, were found to form 2 hydrogen bonds closing the entry of the binding pocket. Moreover, side chain of Tyr342 was directed into the hydrophobic pocket, limiting its size. In addition, four residues, namely Asp192, Gln196, Tyr342, and Gln281 possess three, four, two, and four rotamer states, respectively, that are free of steric clashes with other protein atoms. It is worth noting that no information could be drawn from the template structure because the corresponding residues are different. Therefore, to address these problems, Gln196 and Gln281 were moved away of each other breaking the two hydrogen bonds and opening the binding site. Similarly, the above mentioned residues were assigned all combinatorial side chain conformations using the Lovell32 rotamer library and the combination that open the binding pocket as much as possible was used for the next step. The crude model (Model I) generated in this procedure was subjected to energy minimization. Overview of the protocol used for homology modeling, refinement, and validation is illustrated in figure 2.
Figure 2.

Protocol used for the modeling, refinement, and validation of MCH-R1.
iii. Initial Model Refinement and Validation
Model I was minimized in a stepwise manner using OPLS-2005 force field41 in Macromodel v8.0 (Schrodinger Inc., Portland, OR, USA). The refinement process involved rounds of consecutive minimizations, in order to relieve any steric clashes or distorted geometries in the crude model. Positional restraints were applied on the backbone atoms to preserve the pattern of the 7 TM bundle. These restraints were not removed completely because the absence of restraints could lead to unrealistic movement of the TM helices. This protocol allows the model to evolve according to specific structural characteristics, while at the same time to retain the original structure information contained in the template. This initially refined model (Model II) was validated using Procheck software to ensure the absence of any steric clashes and/or distorted geometries. Moreover, the Ramachandran plot generated by Procheck was carefully inspected and more than 97.1% of residues were found in the most favored regions and 2.9% of residues appeared in the additionally allowed regions. In addition, the backbone RMSD between Model II and the bovine rhodopsin crystal structure was found to be 1.76 Å, calculated using Compare Structures command within the Biopolymer module in SYBYL v7.2 (Tripos Inc., St. Louis, MO, USA).
iv. Ligand-Supported MCH-R1 Model refinement
Visual examination of the Model II reveals that the binding site is too small for most of the reported MCH-R1 antagonists, which is common among homology models derived from the bovine rhodopsin crystal structure. This is attributed to (1) improper orientation of some residues side chains within the binding pocket (we have dealt partly with this problem before energy minimization, see model generation step), and (2) flat nature of 11-cis retinal in the binding pocket of bovine rhodopsin crystal structure.44 To help expanding the binding site, the MCH-R1 model (Model II) was subjected to an extensive ligand-supported homology model refinement protocol.
v. Refinement with T-226296
This compound was the first reported MCH-R1 antagonist.12 It was selected in this study due to its relative rigidity compared to other reported MCH-R1 antagonists and lack of unnecessary functional groups. Compound T-226296 was manually docked in the approximate binding site of Model II using the available SAR information. It was reported that benzanilides exist in trans conformation in crystal as well as in solution.45 Moreover, the trans-amide structure was reported to be essential for the biological activity of retinobenzoic acids.46 Therefore, T-226296 was initially sketched with its amide bond in the trans-conformation. The final conformation used as input structure was established from a grid conformational search to locate energy minima for the torsional potential of the rotatable bonds as described in the experimental section. The manual docking of this ligand was based on the assumption that its basic amine should form a charge assisted hydrogen bond with the carboxyl of Asp192. According to the Lovell rotamer library, two favored rotameric states were identified for this residue, the first (x1 = −70°, x2 = −15°) in 51% and the second (x1 = −177°, x2 = 0.00°) in 21% of the structures. Interestingly, the corresponding residue Asp3.32 in β1- and β2-adrenergic receptors shows very close side chain conformations of (x1 = −178°, x2 = −18.8°) and (x1 = −164°, x2 = −30.8°) respectively.47 Thus, we decided to assign Asp192 the second rotameric state of (x1 = −177°, x2 = 0.00°) due to its similarity to the experimental values for Asp3.32 in β1- and β2-adrenergic receptors. The amide group of the ligand was placed in the proximity of Gln196 and the biphenyl part was embedded into the hydrophobic pocket. The bound pose of T-226296 had some steric clashes with certain residues within the binding site, which is expected due to the tight and closed nature of the binding pocket of Model II. The ligand-MCH-R1 complex was subjected to energy minimization using OPLS-2005 force field leading to the resolution of most of the steric clashes. The minimized complex showed the expected interaction between the ligand basic amine and Asp192. However, no other H-bonding was noticed between the ligand and protein and some bad contacts were still noticed in the tight hydrophobic pocket.
It was reported that MD simulations can lead to the evolution of the initially closely related models into alternative structures. Thus, abstaining from MD simulations would result in insufficient coverage of the conformational space and a misleading conformity of structures.48 Therefore, the resultant MCH-R1-T226296 complex was subjected to a restrained MD simulation of 7 ns at a temperature of 300 K. In case of in-vacuo MD simulation, some form of positional restraints should be applied to the model to replace the natural stabilizing effect of the lipid bilayer and to avoid spurious movements of the TM helices.49 The detailed procedure followed for MD simulation is described in the experimental section. The average structures from the last 1 ns trajectory of MD simulation were minimized using OPLS-2005 forcefield. Again, a force constraint of 10 kcal/mol/Å2 was maintained on the backbone of the protein to maintain the critical geometry of α-helices. This protocol resulted in a significant increase in the binding site volume giving the refined model III. This model showed significant correlation with reported SAR information. However, it is still not able to explain the binding of larger ligands.
vi. Refinement with compound 5
Model III was further refined by docking of the Y-shaped compound 5 followed by minimization and MD simulation. The presence of common pharmacophoric features allowed us to use the bound conformation of T-226296 in Model III as a reference for the docking of this branched compound. Docking was performed using the Gold software by applying a distance constraint between the ligand basic nitrogen and Asp192 as well as a hydrogen bond constrain between the ligand urea carbonyl and Gln196 (as noticed with T-226296). As expected, the biphenyl moiety in this compound displayed steric clashes with the side chains of some binding site amino acid residues. This initial complex was minimized and then subjected to a 7 ns MD simulation using NVT ensemble at 300 K with mild restraints of 10.0 kcal/mol/Å2 on the backbone atoms to preserve the proper 3D fold. We believe that the 7 ns MD simulation was sufficient for the refinement of the model as the potential energy of the complex decreased steadily during the first 5 ns of the simulation, and then remained constant for the last 2 ns (Fig. 3). The resulting complex was minimized to give the final refined model (Model IV). This model displayed desirable ligand–receptor interactions (vide infra). The binding pose of the branched compound 5 in the Model IV revealed the presence of critical charge-assisted H-bonding interaction of Asp192 side chain carboxylate with the ligand basic nitrogen. The RMSD values for the backbone atoms as well as for all the heavy atoms, for Model II, Model III, and Model IV were calculated and listed in Table 2. It was noticed that the transition from Model II (initial refined model) to Model III (T-226296 refined) caused more pronounced changes than the transition from Model III to Model IV (compound 5 refined). Changes in Model IV were observed mainly with the side chains of the binding site residues which were tilted outwards to accommodate the bulky compound 5.
Figure 3.

Potential energy fluctuation of Compound 5 - Model III complex during the 7ns MD simulation.
Table 2.
RMSD comparison between BRho (1u19) and MCH-R1 models.
| Models | Backbone (Å) | Heavy Atoms (Å) |
|---|---|---|
| 1u19 – Model II | 1.76 | 2.35 |
| 1u19 – Model IV | 2.23 | 2.87 |
| Model II – Model III | 1.29 | 2.04 |
| Model III – Model IV | 1.13 | 1.44 |
| Model II – Model IV | 1.52 | 2.25 |
vii. Protein Structure Validation
Proper evaluation of the model quality is of utmost importance to gauge its reliability for use in SAR study and/or structure based drug design. The first factor we considered is the overall fold of the final model. Care should have been taken to preserve the conserved α-helicl geometry of the template crystal structure. It was found that the backbone RMSD between the final refined Model IV and bovine rhodopsin was 2.23 Å, which is a reasonable value indicating proper maintenance of the template structural information. Another essential requirement for any 3D model is to have a good stereochemistry. For this purpose, Procheck software was used to assess the overall quality of the protein and highlight the regions that may need further investigation. Careful check of the Procheck output revealed that no steric clashes or distorted geometry were observed. Main chain and side chain parameters were acceptable. Only one rotamer outlier was detected and it was found to be far from the putative binding site. Table 3 summarizes the results of Ramachandran plot analysis and PROCHECK structure validation for Model IV.
Table 3.
Results of Protein Structure Check by PROCHECK.
| Parameter | 1U19 | Model IV | ||
|---|---|---|---|---|
| Residues in most favored regions | 163 | 95.9% | 63 | 93.7% |
| Residues in additional allowed regions | 6 | 3.5% | 11 | 6.3% |
| Residues in generously allowed regions | 1 | 0.6% | 0.0 | 0% |
| Residues in disallowed regions | 0 | 0.0% | 0.00% | |
| PROCHECK score* | 0.35 | 0.08 | ||
| Bad contacts | 3 | 0 | ||
| Chi1-chi2 plots outliers | 1 | 1 | ||
| Main chain bond lengths within limits | 99.90% | 100% | ||
| Main chain bond angles within limits | 99.70% | 95.70% | ||
Ideally, scores should be above −0.5. Values below −1.0 may need investigation.
viii. Docking Simulation
Docking of 18 MCH-R1 antagonists was carried out to assess the validity of our MCH-R1 model and examine its ability to guide the structure-based drug design. GOLD software which depends on genetic algorithm approach was used to perform the automated docking. 18 compounds, of diverse chemical structures and binding affinities ranging from 0.98 nM to 3000 nM, were selected from the literature. Docking was performed in the final Model IV using the salt bridge between Asp192 and ligand basic nitrogen as an anchor point. Top scoring poses for all the ligands showed the expected relevant binding interactions, including the formation of the mandatory salt bridge with Asp192 and H-bond with Gln196. It was also noticed that ligands aromatic groups are impeded in the tight hydrophobic pocket formed by Tyr342, Phe286, Phe290, and Leu203. The plot of GOLD score versus the binding affinities (Fig. 4) shows a reasonable correlation (r2 = 0.70) taking into consideration that a homology-derived GPCR model was used for the docking simulations and only GOLDscore was used as the scoring function. Therefore, the model can be considered successful in rationalizing the variation of the ligands binding affinity and giving insights into the binding modes of ligands of different chemotypes.
Figure 4.

Correlation of binding affinity and GOLD scores. Binding affinity is represented as pki [-log ki].
ix. Molecular dynamics simulation of receptor-membrane complexes
As MCH-R1 is a membrane protein, thus a more accurate computational representation of MCH-R1 model would be one where it is embedded in a membrane aqueous environment. Subsequently, molecular dynamics simulation of this membrane-model complex can refine the system in a biologically relevant manner. Therefore, we decided to perform an explicit membrane MD simulation of the refined model IV in complex with diverse antagonists. This strategy could allow better understanding of the SAR of different structural classes of antagonists and reveal new insights into the potential interaction sites within the receptor. In addition, it will provide a relevant MCH-R1 model relaxed in a membrane environment that can aid in structure-based drug design of novel MCH-R1 antagonists.
In this explicit membrane MD simulation study, we considered only compounds 4 and 5, as representatives of chemotypes II and III, respectively. We decided to refrain from further simulation of T-226296 as this compound is fairly rigid and the binding mode of this type of compounds (chemotype I) was extensively studied in the literature.18, 21, 23, 50 Therefore, we believe that the initial 7 ns MD simulation was sufficient to explore the behavior of T-226296 in complex with MCH-R1. The GOLD-derived complexes of compounds 4 and 5 with Model-IV were embedded in a rectangular box composed of DPPC bilayer solvated with water at intracellular and extracellular ends forming system I and system II, respectively. These two systems were equilibrated and then simulated for 20 ns each in an NPT ensemble. After the equilibration run, it was noticed that the gaps, created due to the deletion of the overlapping lipids during system preparation, were removed yielding a more favorable tightly packed system for the production run (Figure 5). A Side view of the full MCH-R1-membrane system after the 20 ns MD simulation is depicted in figure 6.
Figure 5.

MCH-R1-Membrane system before (A) and after (B) equilibration MD simulation
Figure 6.
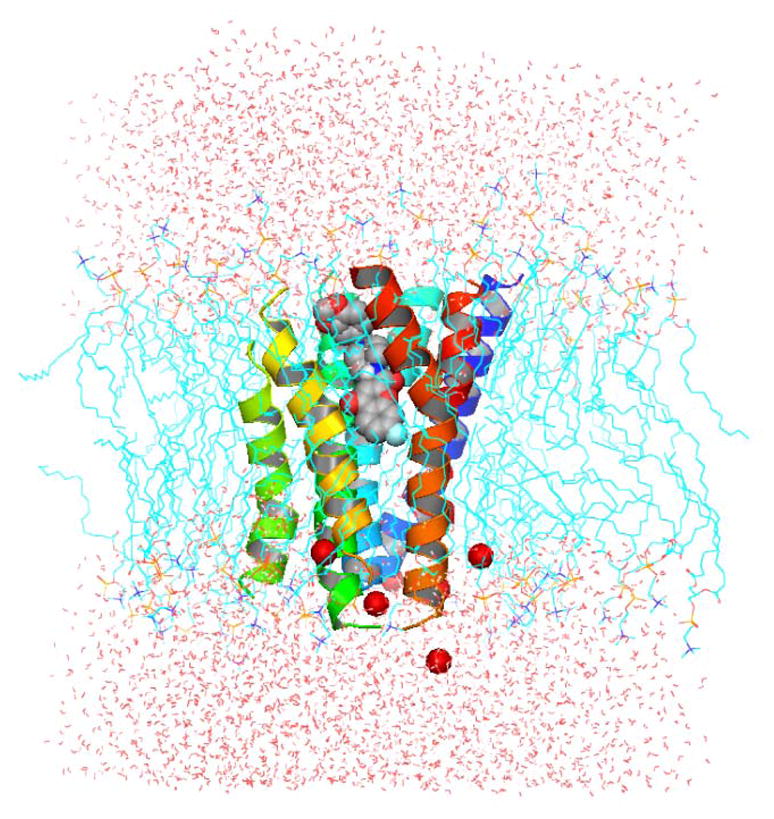
Side view of the full MCH-R1-membrane-aqueous system (I) after the 20 ns MD simulation. Helices are represented as cartoon. Lipids are displayed as cyan lines. Chloride ions are displayed as red spheres. Compound 4 is displayed as spheres colored by atom types.
There are several parameters which need to be monitored to confirm physical stability of MD simulation, including temperature, volume, and total energy. The temperature and the volume of the two systems were stable during the production run, keeping around values of T = 300.035 ± 1.082 K and V = 284010.59 ± 1919.525 A3 for Model IV-compound 4 complex (system I), and T = 299.967 ± 1.092 K and V = 287979.72 ± 1571.406 A3 for Model IV-compound 5 complex (system II). The energy profile of the two systems during the course of MD simulation is shown in figure 7. The total energy of system I decreased initially for the first 8 ns, and then remained stable over the last 12 ns of simulation. Similarly, the total energy of system II decreased steadily until the 8th ns. Then, it stabilized between 8 and 14 ns before slightly decreasing and stabilizing until the end of simulation.
Figure 7.

Total energy vs time plot during the production run (20 ns) of system I (a), and II (b).
The RMSD of the backbone atoms can give information about the structural stability of the system under simulation. Figure 8 shows the RMSD values of the backbone of system I and system II, computed against the starting structures. In both systems, the RMSD of the backbone showed a significant stability over the course of MD simulation. It increased gradually until the 10th ns, and then stabilized for the rest of the simulation around 2.6 Å. For both systems, the final complex was minimized and then analyzed, in order to ensure the presence of key interactions with the binding site residues. The complex was also analyzed using Procheck and Ramachandran plot analysis, to ensure appropriate geometrical features. Together, these results indicate that both systems were stable during the course of the MD simulation.
Figure 8.

Backbone RMSD vs time plot during the production run (20 ns) of system I (a), and II (b).
x. Conserved Microdomains in GPCRs: Bovine rhodopsin Vs MCH-R1 receptor
The overall homology of different class A GPCRs to bovine rhodopsin is relatively low. However, the presence of 7 TM domains containing highly conserved residues suggests an evolutionary relationship among these receptors. In addition to these residues, there are several conserved inter-helical interactions across the super-family of GRCR.51 Our final model, after the 20 ns MD simulation, was examined for the presence of these common structural features.
In the crystal structure of bovine rhodopsin, the side-chain of Asn55 (N1.50) forms an H-bond with the backbone carbonyl groups of Ala299 (A7.46) and Asp83 (D2.50).40 In MCH-R1 model, TMVII had moved away from TMI during refinement. Therefore, the side chain of Asn127 (N1.50) showed H-bonding with the side-chain carboxylate of Asp158 (D2.50), but it had no direct interaction with Ser372 (7.46). Instead, this interaction was mediated by the carboxylate of Asp158 (D2.50). TMII is characterized by the presence of the LAxxD motif which is conserved in several class A GPCRs. The corresponding residues in MCH-R1 model, namely Leu154 (L2.46) and Asp158 (D2.50), were found to adopt the same orientation observed in bRho structure with their side-chains directed towards the core of the receptor. Asn78 (N2.45) in bovine rhodopsin forms H-bonds with Ser127 (S3.42), Thr160 (T4.49), and Trp161 (W4.50).40 Similarly, in the MCH-R1 receptor model, Asn153 (N2.45) appeared to form a H-bond with the side-chain of the conserved Trp153 (W4.50), while, showing interactions with hydrophobic Ile202 (I3.42) and Leu236 (L4.49), instead of the corresponding polar residues in bovine rhodopsin. Another highly conserved microdomain observed in several class A GPCRs is the “(D/E)R(Y/W)” motif in TMIII.41 In bovine rhodopsin, Arg135 (R3.50) may form H-bonds with either one, or both, of the two residues, Glu247 (E6.30) or Thr251 (T6.34), both located in TMVI.38 These interactions between Arg135 of TMIII and residues of TMVI play a role in preserving the relative positions of these TM helices and maintain the receptor in its inactive state.41 Analogous to bovine rhodopsin, Arg210 (R3.50), in the MCH-R1 model, showed H-bonding interactions with the side-chains of the corresponding amino acid residues, Thr319 (T6.30), and Thr323 (T6.34). To our delight, the stability of these interactions throughout the 20 ns simulation indicates that the system, in presence of antagonists, was stabilized in its inactive state. NPxxY is also a conserved microdomain found in TMVII of class A GPCRs. In bovine rhodopsin, the hydroxyl group of Tyr306 (Y7.53) forms a H-bond the side-chain carbonyl of Asn73 (N2.40). The NPxxY motif was found to be conserved in the MCH-R1 receptor model with Tyr379 (Y7.53) showing H-bonding interaction with the corresponding amino acid residue in MCH-R1, Asp148 (D2.40). In addition to the interhelical H-bonding interactions, a network of highly conserved hydrophobic residues further stabilizes the 7Tm bundle of the class A GRCRs. These residues are conserved in the MCH-R1 receptor model and include Trp337 (W6.48) displaying hydrophobic interactions with Phe333 (F6.44) on one side, and Tyr340 (Y6.51) on the other side. This conservation of the characteristic Microdomains and interhelical interactions gives an additional validity to our model.
xi. Non-Peptide Antagonist Binding Site
The putative antagonist binding site of the MCH-R1 receptor was found to be formed by TMIII, TMV, TMVI and TMVII. The binding cavity was expanded and refined using ligand-supported model refinement with the non-peptide MCH-R1 antagonists, T-226296 and the Y-shaped compound 5. These ligands were noticed to be embedded within the top half of the helical domain with axis parallel to TMIII. A close inspection of the binding site reveals several critical ligand-receptor interactions. Generally, the ligand binding site can be divided into three regions, ionic interaction region, H-bonding region, and hydrophobic pocket. The first region involves interaction with Asp192 (3.32) in the third TM domain of the receptor. This residue was reported to be conserved in all biogenic amine GPCR and essential for ligand recognition as suggested by site-directed mutagenesis.35, 52 In our proposed binding pose, the ligand protonated amine forms an ionic and hydrogen bond with Asp192. During the model generation, Asp192 was assigned the second rotamer state, in the Lovell rotamer library, for the reasons mentioned earlier. The ligand basic amine occupies a relatively wide hydrophobic pocket (P1) formed by Thr189, Cys185 and Ile188 on TM3, Met165 and Ile169 on TMII, and Tyr362 and Ile366 on TMVII.
In addition to the essential salt bridge formed between MCH-R1 antagonists and Asp192, most of the reported docking studies suggest the formation of an H-bond between carbonyl oxygen of the ligand and one of the binding site residues. In our model, an H-bond was noticed between the ligand carbonyl group and the side-chain of Gln196 on TMIII. Four different amino acid residues were suggested by different research groups to participate in this H-bonding, namely Gln196 in TMIII, Gln281 in TMV, Tyr342 in TMVI and Gln345 in TMVI.19–21, 24 Comparison of the relative orientation of these residues reveals that the involvement of Gln281 or Gln345 in H-bonding could only explain the binding of flexible ligands like compound 2 (Fig. 1). These ligands possess a flexible linker between the carbonyl group and the terminal hydrophobic moiety. While in case of more rigid molecules, like T-226296, binding with Gln281 or Gln345 would cause the hydrophobic western part of the molecule to protrude from the side of the receptor between TMV and TMVI. This problem is less pronounced in case of binding with Tyr342 and absent with Gln196. Formation of H-bond with Gln196 allows the ligand to probe deeper into the receptor without significant steric clashes.
Therefore, the assumption that MCH-R1 antagonists form H-bond with Gln196 is able to interpret the binding of compounds of almost all chemotypes. In addition to the aforementioned potential H-bonds, our docking studies showed that some ligands are able to form H-bond with the side chain NH of Trp248 (discussed later). We also noticed the presence of a relatively wide hydrophobic pocket (P2) away from the axis of TMIII, surrounding the side chain NHs of Gln281 and Trp248. This pocket is formed by Phe282 and Thr278 on TMV and Val247 and Trp248 on TMIV. It can provide an additional hydrophobic interaction site for certain type of ligands (see later).
Lower in the binding site is the hydrophobic pocket (P3) that extends downwards toward the center of the TM domain. The residues that constitute this pocket are Thr200, Leu203, and Pro289 on TMIII, Phe282, Phe286, and Phe290 on TMV, and Phe334, Trp338, and Tyr342 on TMVI. It is worth noting that the size of this hydrophobic pocket is tight compared to the cavity available around Asp192. The hydroxy group of Thr200 on the inner side of this hydrophobic pocket offers a site for additional H-bonding interaction. The most important residues for the hydrophobic interaction in this pocket are Tyr342, Phe286, Phe290, and Leu203.
xii. Binding Mode of Different Chemotypes
Chemotype I (Linear with Terminal Basic Nitrogen)
As mentioned earlier, compound T-226296 binds with axis parallel to TMIII and basic nitrogen bound to the carboxylic group of Asp192 (Fig. 9a). The biphenyl moiety occupies a relatively tight binding pocket primarily between TMIII from one side and TMV and TMVI from the other side. Aromatic-stacking interaction can be noticed between Phe286 and the biphenyl moiety. The compound is slightly bent so that the central tetralin moiety can make hydrophobic contact with P2 pocket especially Phe282. Chemotype II (Linear with Central Basic Nitrogen): In this type of compounds, the basic nitrogen and carbonyl oxygen form ionic interaction and H-bond with Asp192 and Gln196, respectively (Fig. 9b, 9c). The hydrophobic moiety on the basic nitrogen has the choice to fill either the hydrophobic pocket P1 or P2. In the latter case, it can form H-bond with Gln281 or Trp248 if it carries a suitable H-bond acceptor. Throughout the MD simulation, the H-bond between the basic nitrogen of compound 4 and Asp192 remained stable around 1.8 Å as well as the H-bond between the carbonyl oxygen and Gln196 which fluctuated between 2.5 Å and 3.3 Å (Figure 10). However, the distance between the piperonyl oxygen and Trp248 side chain NH was too long for a stable H-bond. We believe that the hydrogen bonding interaction here can be mediated by a water molecule. Also, derivatives with slightly different chemical structures may reach the proper distance required to form a stable H-bond with Trp248. On the other side, the chromone moiety fills the hydrophobic pocket P3. The presence of OMe or F substituent on this moiety can provide a site for an extra H-bond with Thr200 and that interprets the improved activity with H-bond accepting groups on this hydrophobic terminal of the molecule.
Figure 9.

a) Proposed binding mode of compound 1 (T-226296) in the final model (Model IV). b) and c) Possible binding poses of compound 4 making contacts with hydrophobic pockets P2 or P3. d) Proposed binding interactions of compound 5. Cartoon representation was used for the backbone atoms in purple. Only residues interacting with the ligands were shown as cyan sticks. Ligands are shown as capped sticks and colored by atom types with carbon atoms in yellow. The upper part of helix 6 was truncated and only polar hydrogens are displayed for clarity.
Figure 10.
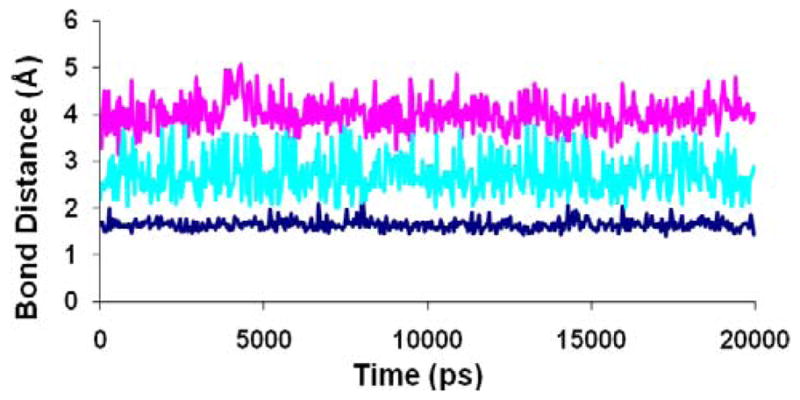
Hydrogen bond distances between compound 4 and critical polar residues in the binding site of Model IV during the 20ns MD simulation. Blue: (NH--O) basic nitrogen and Asp192. Cyan: (C=O--HN) amide carbonyl and Gln196. Pink: (O--HN) piperonyl oxygen and Trp248.
Chemotype III (Branched or Y-Shaped)
Despite its high potency, no docking studies were reported for this type of compounds till date. These compounds have a Y-shaped structure with basic nitrogen at one arm and two hydrophobic groups at the two other arms. In addition to the expected ionic and H-bonding interactions, the two hydrophobic arms fill the hydrophobic pockets P2 and P3. Figure 9d shows the binding mode of compound 5 in the minimized complex after MD simulation. The phenyl ring, directly attached to the urea group of the compound, occupies the P3 pocket, while the biphenyl moiety fills the P2 pocket. Hydrogen accepting groups on these two hydrophobic arms improve the inhibitory activity by forming H-bonds with Glan281 or Trp248 in pocket P2, and Thr200 in pocket P3. Hydrogen bonds formed between compound 5 and Asp192, Gln196, and Trp248 were monitored during the course of the 20ns MD simulation and found to be stable (Fig. 11). This is the first assumption of the involvement of Trp248, Thr200, and the hydrophobic pocket P2 in the binding of MCH-R1 with certain ligands.
Figure 11.
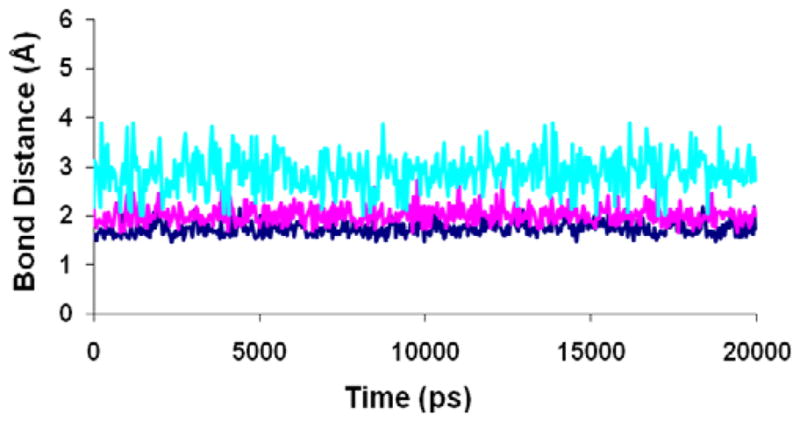
Hydrogen bond distances between compound 5 and critical polar residues in the binding site of Model IV during the 20ns MD simulation. Blue: (NH--O) basic nitrogen and Asp192. Cyan: (C=O--HN) urea carbonyl and Gln196. Pink: (CN--HN) cyano nitrogen and Trp248.
As a rule of thumb, we can conclude that the ideal binding to MCH-R1 requires ionic interaction with Asp192, at least one H-bonding, and hydrophobic interactions with 2 pockets. One of the hydrophobic pockets has to be P3 that exists in the center of the TM domain among TMIII, TMV, and TMVI, and the other one can be P1 or S2 depending on the compound chemotype.
CONCLUSION
In this study, homology modeling was used to develop a 3D model of MCH-R1, utilizing the highest resolution X-ray crystal structure of bovine rhodopsin (PDB ID: 1u19). Previously reported MCH-R1 models were not able to explain the binding mode of compounds of diverse chemical structures. To address this problem, the initial MCH-R1 model was subjected to a strategic ligand supported refinement protocol using compounds of different chemotypes. This model was validated using Procheck software and Ramachandran plot. The final model, in complex with two structurally diverse compounds, was embedded in a membrane aqueous environment and then simulated for 20 ns in an NPT ensemble. This protocol allowed the explanation of several reported SAR data. We illustrated that the antagonistic activity on MCH-R1 requires ionic interaction, H-bonding, and 2 hydrophobic interactions. We also noticed for the first time the presence of an extra hydrophobic pocket P2 and two residues forming additional H-bonds with certain ligands (Thr200 and Trp248). The knowledge gained from the developed 3D model of MCH-R1 can be useful in the identification and structure based design of novel antagonists that can represent promising anti-obesity agents.
Supplementary Material
Acknowledgments
This investigation was conducted in a facility constructed with support from research facilities improvement program grant number C06 Rr-14503-01 from the National Center for Research Resources, National Institutes of Health.
Footnotes
Multiple sequence alignment and structures and docking scores of the compounds used for docking validation are given in the supporting information. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.World Health Organization. [(accessed December 28, 2010)]; http://www.who.int/nutrition/topics/obesity/en/
- 2.Dannenberg AL, Burton DC, Jackson RJ. Economic and environmental costs of obesity: the impact on airlines. Am J Prev Med. 2004;27:264–271. doi: 10.1016/j.amepre.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 3.Haslam DW, James WP. Obesity. Lancet. 2005;366:1197–1209. doi: 10.1016/S0140-6736(05)67483-1. [DOI] [PubMed] [Google Scholar]
- 4.Kawauchi H, Kawazoe I, Tsubokawa M, Kishida M, Baker BI. Characterization of melanin-concentrating hormone in chum salmon pituitaries. Nature. 1983;305:321–323. doi: 10.1038/305321a0. [DOI] [PubMed] [Google Scholar]
- 5.Bittencourt JC, Presse F, Arias C, Peto C, Vaughan J, Nahon JL, Vale W, Sawchenko PE. The melanin-concentrating hormone system of the rat brain: an immuno- and hybridization histochemical characterization. J Comp Neurol. 1992;319:218–245. doi: 10.1002/cne.903190204. [DOI] [PubMed] [Google Scholar]
- 6.Chambers J, Ames RS, Bergsma D, Muir A, Fitzgerald LR, Hervieu G, Dytko GM, Foley JJ, Martin J, Liu WS, Park J, Ellis C, Ganguly S, Konchar S, Cluderay J, Leslie R, Wilson S, Sarau HM. Melanin-concentrating hormone is the cognate ligand for the orphan G-protein-coupled receptor SLC-1. Nature. 1999;400:261–265. doi: 10.1038/22313. [DOI] [PubMed] [Google Scholar]
- 7.Hill J, Duckworth M, Murdock P, Rennie G, Sabido-David C, Ames RS, Szekeres P, Wilson S, Bergsma DJ, Gloger IS, Levy DS, Chambers JK, Muir AI. Molecular cloning and functional characterization of MCH2, a novel human MCH receptor. J Biol Chem. 2001;276:20125–20129. doi: 10.1074/jbc.M102068200. [DOI] [PubMed] [Google Scholar]
- 8.Kokkotou EG, TN, Mastaitis JW, Slieker L, Maratos-Flier Melanin-Concentrating Hormone Receptor Is a Target of Leptin Action in the Mouse Brain. Endocrinology. 2002;142:680–688. doi: 10.1210/endo.142.2.7981. [DOI] [PubMed] [Google Scholar]
- 9.Chen Y, Hu C, Hsu CK, Zhang Q, Bi C, Asnicar M, Hsiung HM, Fox N, Slieker LJ, Yang DD, Heiman ML, Shi Y. Targeted disruption of the melanin-concentrating hormone receptor-1 results in hyperphagia and resistance to diet-induced obesity. Endocrinology. 2002;143:2469–2477. doi: 10.1210/endo.143.7.8903. [DOI] [PubMed] [Google Scholar]
- 10.Borowsky B, Durkin MM, Ogozalek K, Marzabadi MR, DeLeon J, Lagu B, Heurich R, Lichtblau H, Shaposhnik Z, Daniewska I, Blackburn TP, Branchek TA, Gerald C, Vaysse PJ, Forray C. Antidepressant, anxiolytic and anorectic effects of a melanin-concentrating hormone-1 receptor antagonist. Nat Med. 2002;8:825–830. doi: 10.1038/nm741. [DOI] [PubMed] [Google Scholar]
- 11.Tan CP, Sano H, Iwaasa H, Pan J, Sailer AW, Hreniuk DL, Feighner SD, Palyha OC, Pong SS, Figueroa DJ, Austin CP, Jiang MM, Yu H, Ito J, Ito M, Guan XM, MacNeil DJ, Kanatani A, Van der Ploeg LH, Howard AD. Melanin-concentrating hormone receptor subtypes 1 and 2: species-specific gene expression. Genomics. 2002;79:785–792. doi: 10.1006/geno.2002.6771. [DOI] [PubMed] [Google Scholar]
- 12.Takekawa S, Asami A, Ishihara Y, Terauchi J, Kato K, Shimomura Y, Mori M, Murakoshi H, Suzuki N, Nishimura O, Fujino M. T-226296: a novel, orally active and selective melanin-concentrating hormone receptor antagonist. Eur J Pharmacol. 2002;438:129–135. doi: 10.1016/s0014-2999(02)01314-6. [DOI] [PubMed] [Google Scholar]
- 13.Tavares FX, Al-Barazanji KA, Bishop MJ, Britt CS, Carlton DL, Cooper JP, Feldman PL, Garrido DM, Goetz AS, Grizzle MK, Hertzog DL, Ignar DM, Lang DG, McIntyre MS, Ott RJ, Peat AJ, Zhou HQ. 6-(4-chlorophenyl)-3-substituted-thieno[3,2-d]pyrimidin-4(3H)-one-based melanin-concentrating hormone receptor 1 antagonist. J Med Chem. 2006;49:7108–7118. doi: 10.1021/jm060814b. [DOI] [PubMed] [Google Scholar]
- 14.Ito M, Ishihara A, Gomori A, Egashira S, Matsushita H, Mashiko S, Ito J, Nakase K, Haga Y, Iwaasa H, Suzuki T, Ohtake N, Moriya M, Sato N, MacNeil DJ, Takenaga N, Tokita S, Kanatani A. Melanin-concentrating hormone 1-receptor antagonist suppresses body weight gain correlated with high receptor occupancy levels in diet-induced obesity mice. Eur J Pharmacol. 2009;624:77–83. doi: 10.1016/j.ejphar.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 15.Ito M, Ishihara A, Gomori A, Matsushita H, Metzger JM, Marsh DJ, Haga Y, Iwaasa H, Tokita S, Takenaga N, Sato N, MacNeil DJ, Moriya M, Kanatani A. Mechanism of the anti-obesity effects induced by a novel melanin-concentrating hormone 1-receptor antagonist in mice. Br J Pharmacol. 2010;159:374–383. doi: 10.1111/j.1476-5381.2009.00536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Handlon AL, Zhou H. Melanin-concentrating hormone-1 receptor antagonists for the treatment of obesity. J Med Chem. 2006;49:4017–4022. doi: 10.1021/jm058239j. [DOI] [PubMed] [Google Scholar]
- 17.Becker OM, Shacham S, Marantz Y, Noiman S. Modeling the 3D structure of GPCRs: advances and application to drug discovery. Curr Opin Drug Discov Devel. 2003;6:353–361. [PubMed] [Google Scholar]
- 18.Vitale RM, Pedone C, De Benedetti PG, Fanelli F. Structural features of the inactive and active states of the melanin-concentrating hormone receptors: insights from molecular simulations. Proteins. 2004;56:430–448. doi: 10.1002/prot.20125. [DOI] [PubMed] [Google Scholar]
- 19.Clark DE, Higgs C, Wren SP, Dyke HJ, Wong M, Norman D, Lockey PM, Roach AG. A virtual screening approach to finding novel and potent antagonists at the melanin-concentrating hormone 1 receptor. J Med Chem. 2004;47:3962–3971. doi: 10.1021/jm040762v. [DOI] [PubMed] [Google Scholar]
- 20.Ulven T, Frimurer TM, Receveur JM, Little PB, Rist O, Norregaard PK, Hogberg T. 6-Acylamino-2-aminoquinolines as potent melanin-concentrating hormone 1 receptor antagonists. Identification, structure-activity relationship, and investigation of binding mode. J Med Chem. 2005;48:5684–5697. doi: 10.1021/jm050103y. [DOI] [PubMed] [Google Scholar]
- 21.Tavares FX, Al-Barazanji KA, Bigham EC, Bishop MJ, Britt CS, Carlton DL, Feldman PL, Goetz AS, Grizzle MK, Guo YC, Handlon AL, Hertzog DL, Ignar DM, Lang DG, Ott RJ, Peat AJ, Zhou HQ. Potent, selective, and orally efficacious antagonists of melanin-concentrating hormone receptor 1. J Med Chem. 2006;49:7095–7107. doi: 10.1021/jm060572f. [DOI] [PubMed] [Google Scholar]
- 22.Witty DR, Bateson JH, Hervieu GJ, Jeffrey P, Johnson CN, Muir AI, O’Hanlon PJ, Stemp G, Stevens AJ, Thewlis KM, Wilson S, Winborn KY. SAR of biphenyl carboxamide ligands of the human melanin-concentrating hormone receptor 1 (MCH R1): discovery of antagonist SB-568849. Bioorg Med Chem Lett. 2006;16:4865–4871. doi: 10.1016/j.bmcl.2006.06.056. [DOI] [PubMed] [Google Scholar]
- 23.Cavasotto CN, Orry AJ, Murgolo NJ, Czarniecki MF, Kocsi SA, Hawes BE, O’Neill KA, Hine H, Burton MS, Voigt JH, Abagyan RA, Bayne ML, Monsma FJ., Jr Discovery of novel chemotypes to a G-protein-coupled receptor through ligand-steered homology modeling and structure-based virtual screening. J Med Chem. 2008;51:581–588. doi: 10.1021/jm070759m. [DOI] [PubMed] [Google Scholar]
- 24.Giordanetto F, Karlsson O, Lindberg J, Larsson LO, Linusson A, Evertsson E, Morgan DG, Inghardt T. Discovery of cyclopentane- and cyclohexane-trans-1,3-diamines as potent melanin-concentrating hormone receptor 1 antagonists. Bioorg Med Chem Lett. 2007;17:4232–4241. doi: 10.1016/j.bmcl.2007.05.034. [DOI] [PubMed] [Google Scholar]
- 25.Cavasotto CN, Phatak SS. Homology modeling in drug discovery: current trends and applications. Drug Discov Today. 2009;14(13–14):676–683. doi: 10.1016/j.drudis.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 26.Konc J, Janezic D. ProBiS algorithm for detection of structurally similar protein binding sites by local structural alignment. Bioinformatics. 2010;26(9):1160–11688. doi: 10.1093/bioinformatics/btq100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bairoch A, Apweiler R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000;28(1):45–48. doi: 10.1093/nar/28.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Okada T, Sugihara M, Bondar AN, Elstner M, Entel P, Buss V. The retinal conformation and its environment in rhodopsin in light of a new 2.2 A crystal structure. J Mol Biol. 2004;342(2):571–583. doi: 10.1016/j.jmb.2004.07.044. [DOI] [PubMed] [Google Scholar]
- 29.Clustalw2. [accessed December 28, 2010]; http://www.ebi.ac.uk/Tools/clustalw2/index.html.
- 30.TMPRED. [accessed December 28, 2010]; http://ch.embnet.org./software/TMPRED_form.html.
- 31.Matthew Clark RDCI, Van Opdenbosch Nicole. Validation of the general purpose tripos 5.2 force field. J Comp Chem. 1989;10:982–991. [Google Scholar]
- 32.Lovell SC, JW, Richardson JS, Richardson DC. The Penultimate Rotamer Library. Proteins: Structure Function and Genetics. 2000;40:389–408. [PubMed] [Google Scholar]
- 33.Johann Gasteiger MM. Iterative partial equalization of orbital electronegativity—a rapid access to atomic charges. Tetrahedron. 1980;36(22):3219–3228. [Google Scholar]
- 34.Fletcher R, CMR Function Minimization by Conjugate Gradients. Comp J. 1965;7:149–154. [Google Scholar]
- 35.Macdonald D, Murgolo N, Zhang R, Durkin JP, Yao X, Strader CD, Graziano MP. Molecular characterization of the melanin-concentrating hormone/receptor complex: identification of critical residues involved in binding and activation. Mol Pharmacol. 2000;58(1):217–225. doi: 10.1124/mol.58.1.217. [DOI] [PubMed] [Google Scholar]
- 36.Halgren T. Merck molecular force field. I Basis, form, scope, parametization, and performance of MMFF94. J Comp Chem. 1996;17:490–498. [Google Scholar]
- 37.Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol. 1997;267(3):727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 38.Bowers KJCE, Xu H, Dror RO, Eastwood MP, Gregersen BAK, JL, Kolossvary I, Moraes MA, Sacerdoti FD, Salmon JKSY, Shaw DE. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. Proceedings of the 2006 ACM/IEEE Conference on Supercomputing (SC06); Tampa, FL. 2006; New York: ACM Press; 2006. [Google Scholar]
- 39.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14(1):33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 40.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289(5480):739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 41.Filipek S, Teller DC, Palczewski K, Stenkamp R. The crystallographic model of rhodopsin and its use in studies of other G protein-coupled receptors. Annu Rev Biophys Biomol Struct. 2003;32:375–397. doi: 10.1146/annurev.biophys.32.110601.142520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234(3):779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 43.Tirado-Rives WLJaJ. The OPLS Force Field for Proteins. Energy Minimizations for Crystals of Cyclic Peptides and Crambin. J Am Chem Soc. 1988;110:1657–1666. doi: 10.1021/ja00214a001. [DOI] [PubMed] [Google Scholar]
- 44.Kimura SR, Tebben AJ, Langley DR. Expanding GPCR homology model binding sites via a balloon potential: A molecular dynamics refinement approach. Proteins. 2008;71(4):1919–1929. doi: 10.1002/prot.21906. [DOI] [PubMed] [Google Scholar]
- 45.Akiko Itai YT. Nobuo Tomioka, Hiroyuki Kagechika, Isao Azumaya, Koichi Shudo, Stereochemistry of N-methylbenzanilide and benzanilide. Tet Lett. 1989;30:6177–6180. [Google Scholar]
- 46.Kagechika H, Himi T, Kawachi E, Shudo K. Retinobenzoic acids. 4. Conformation of aromatic amides with retinoidal activity. Importance of trans-amide structure for the activity. J Med Chem. 1989;32(10):2292–2296. doi: 10.1021/jm00130a011. [DOI] [PubMed] [Google Scholar]
- 47.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318(5854):1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Strahs D, Weinstein H. Comparative modeling and molecular dynamics studies of the delta, kappa and mu opioid receptors. Protein Eng. 1997;10(9):1019–1038. doi: 10.1093/protein/10.9.1019. [DOI] [PubMed] [Google Scholar]
- 49.Boeckler F, Lanig H, Gmeiner P. Modeling the similarity and divergence of dopamine D2-like receptors and identification of validated ligand-receptor complexes. J Med Chem. 2005;48(3):694–709. doi: 10.1021/jm049612a. [DOI] [PubMed] [Google Scholar]
- 50.Meyers KM, Mendez-Andino J, Colson AO, Hu XE, Wos JA, Mitchell MC, Hodge K, Howard J, Paris JL, Dowty ME, Obringer CM, Reizes O. Novel pyrazolopiperazinone- and pyrrolopiperazinone-based MCH-R1 antagonists. Bioorg Med Chem Lett. 2007;17(3):657–661. doi: 10.1016/j.bmcl.2006.10.096. [DOI] [PubMed] [Google Scholar]
- 51.Mirzadegan T, Benko G, Filipek S, Palczewski K. Sequence analyses of G-protein-coupled receptors: similarities to rhodopsin. Biochemistry. 2003;42(10):2759–2767. doi: 10.1021/bi027224+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kristiansen K, Kroeze WK, Willins DL, Gelber EI, Savage JE, Glennon RA, Roth BL. A highly conserved aspartic acid (Asp-155) anchors the terminal amine moiety of tryptamines and is involved in membrane targeting of the 5-HT(2A) serotonin receptor but does not participate in activation via a “salt-bridge disruption” mechanism. J Pharmacol Exp Ther. 2000;293(3):735–746. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.