

Abstract
Epigenetic mechanisms play essential roles in mammalian neurodevelopment and genetic mutations or chromosomal deletions or duplications of epigenetically regulated loci or pathways result in several important human neurodevelopmental disorders. Postnatal mammalian neurons have among the most structured and dynamic nuclear organization of any cell type. Human chromosome 15q11-13 is an imprinted locus required for normal neurodevelopment and is regulated by a plethora of epigenetic mechanisms in neurons, including multiple noncoding RNAs, parentally imprinted transcription and histone modifications, large-scale chromatin decondensation, and homologous pairing in mature neurons of the mammalian brain. Here, we describe the multiple epigenetic layers regulating 15q11-13 gene expression and chromatin dynamics in neurons and propose a model of how noncoding RNAs may influence the unusual neuronal chromatin structure and dynamics at this locus. We also discuss the need for improved neuronal cell culture systems that model human 15q11-13 and other neurodevelopmental disorders with epigenetic bases in order to test the mechanisms of chromatin dynamics and nuclear organization in neurons. Induced pluripotent stem cells and other stem cell technologies hold promise for improved understanding of and therapeutic interventions for multiple human neurodevelopmental disorders.
Keywords: epigenetic, imprinting, nucleolar, noncoding RNA, snoRNA, neuronal
Postnatal neurodevelopmental events leading to mammalian cognitive learning and memory are among the most complex biological processes in nature, requiring multiple layers of tightly regulated gene transcription that are genetically determined yet responsive to environmental inputs. Epigenetic mechanisms appear to play a particularly important role in neurodevelopment, as evident by the growing number of human neurodevelomental disorders with genetic mutations in genes encoding chromatin factors and in epigenetically regulated loci [Lasalle and Yasui, 2009; Schanen, 2006]. In addition, the subset of mammalian genes that are parentally imprinted display a predominant expression in brain tissue [Gregg et al.; Royo and Cavaille, 2008; Wilkinson et al., 2007]. Furthermore, mature postnatal neurons exhibit a unique and highly organized nuclear architecture [Manuelidis, 1985; Martou and De Boni, 2000].
Epigenetics is defined as inherited and reversible modifications to DNA and/or chromatin that alter gene expression without associated changes in DNA sequence. Epigenetic gene regulation is a highly ordered process involving multiple layers of regulation, including DNA methylation, histone modifications, nucleosome assembly affecting DNA compaction, and chromatin loop structures [Cook, 2003; Horn and Peterson, 2002; Kosak and Groudine, 2004; Wang et al., 2004]. Higher order epigenetic states affect the spatial positioning of chromosome territories [Cremer et al., 2006] and long noncoding RNAs such as XIST can impact the epigenetic state of an entire chromosome [Brown et al., 1992]. There is also evidence to suggest that environmental triggers and signals can lead to epigenetic modifications [Jirtle and Skinner, 2007]. The landscape of epigenetic modifications controls the tissue-specific and temporal gene expression events necessary for normal mammalian development.
Parental imprinting is a form of epigenetic regulation by which chromosomes retain an epigenetic memory of parental origin, resulting in differential expression of maternal and paternal alleles of the same genetic locus. To date there are over eighty known imprinted transcripts, most of which are involved in either early development or neurodevelopment [Morison et al., 2005; Wilkinson et al., 2007]. However, a recent genomic sequencing analysis of parentally biased gene expression in mouse embryos and brain has suggested the presence of a larger number of transcripts with parent-of-origin bias compared to previous reports [Gregg et al., 2010]. Parent-of-origin specific transcription of imprinted genes is most commonly regulated by differential DNA methylation either at a gene promoter or at an imprinting center (IC) that can regulate gene expression across chromosomal segments of 1 Mb or more [Lewis and Reik, 2006].
Neurodevelomental disorders associated with imprinting and DNA methylation
Abnormalities of the imprinted 15q11-13 locus have been implicated in Angelman and Prader Willi syndromes [Williams et al., 1990], as well as autism [Cook et al., 1997], schizophrenia [Stefansson et al., 2008] and epilepsy [Ben-Shachar et al., 2009; Helbig et al., 2009]. Rett Syndrome, caused by mutations in the methyl binding protein 2 gene, MECP2 [Amir et al., 1999] is also a striking example of a neurodevelopmental disorder caused by a loss of normal epigenetic regulation.
Prader Willi syndrome (PWS) presents as failure to thrive in infants, followed by mental retardation, respiratory abnormalities, short stature, respiratory distress, and child-onset insatiable hunger, and represents the second most common genetic cause of obesity [Butler et al., 2002; Webb et al., 1995]. PWS is caused by 15q11-13 paternal deficiency through deletion or maternal uniparental disomy (UPD). Analysis of PWS patients with rare deletions of paternal 15q11-13 have determined that paternal deficiency of the locus surrounding a cluster of small nucleolar RNAs (snoRNAs, HBII-85/SNORD116), is sufficient to cause PWS [de Smith et al., 2009; Sahoo et al., 2008].
Angelman Syndrome (AS) is caused by maternal deficiency of the same 15q11-13 region as PWS. AS produces a drastically different phenotype, however, with severe mental retardation, developmental delay, almost complete lack of speech, ataxia, a happy disposition, and often severe seizures [Clayton-Smith and Laan, 2003]. The AS disease gene within 15q11-q13 is ubiquitin protein ligase E3A (UBE3A). UBE3A is imprinted only in neurons with both parental alleles being expressed with a maternal bias in all other tissues [Gustin et al., 2010; Yamasaki et al., 2003]. That only neurons exhibit exclusive maternal UBE3A expression highlights the potential role of imprinting in brain development.
Autism is a neurodevelopmental disorder characterized by deficits in language and social engagement as well as repetitive stereotyped interests and behaviors. Genetics is believed to play a central role in autism as the disorder has a high heritability and a high concordance in monozygotic twins [Veenstra-Vanderweele et al., 2004]. Yet, the genetic basis of autism remains complex with multiple chromosomal regions implicated and a high frequency of de novo copy number variations (CNV) observed [Anney et al., 2010; Pinto et al., 2010]. However, 1–3% of autism cases contain maternal 15q11-13 duplications, the most common cytogenetic and CNV abnormality observed in autism [Schanen, 2006]. Intersitial duplications most likely arise from reciprocal recombination events involving segments of low copy repetitive DNA sequences associated with the common 15q11-q13 deletion breakpoints. More commonly, duplications occur as U-type meiotic cross-over events and form isodicentric marker chromosomes, termed isodicentric chromosome 15 or idic(15) [Hogart et al., 2010]. Association and linkage studies have also indicated a role for 15q11-13 polymorphisms and rearrangements in larger autistic cohorts, schizophrenia, biopolar disorder and epilepsy [Flomen et al., 2006; Helbig et al., 2009; Stefansson et al., 2008].
Rett syndrome (RTT) is characterized by normal infant development, followed by severe regression at 6 to 18 months of age leading to loss of speech and purposeful hand movements, stereotyped behaviors, ataxia, breathing abnormalities, severe mental retardation and gastro-intestinal difficulties, as well as autistic features. Mutations or deletions of MECP2 encoding for methylated CpG binding protein 2 (MeCP2) are the cause of most cases of RTT. Phenotypic overlaps with AS and autism include stereotypic behaviors, language impairments, seizures, and mental retardation. While historically defined as a transcriptional repressor of methylated genes [Bird and Wolffe, 1999], recent studies point to MeCP2 having a role in chromatin organization, binding throughout the genome to intergenic DNA as well as both active and inactive gene promoters [Chahrour et al., 2008; Skene et al., 2010; Yasui et al., 2007]. This has led to the model of MeCP2 as an epigenetic modulator, having roles in both gene activation and silencing.
15q11-13 epigenetic regulation and chromatin organization
The imprinted 15q11-q13 region displays an unusually complex gene structure, regulation, and chromatin organization. Figure 1 shows the gene content and chromosome banding of the ~13 Mb locus which resides on the proximal long arm of the acrocentric chromosome 15, a position close to the ribosomal genes on the p arm. Human chromosome 15 is enriched in segmental duplications, with at least five blocks mapping to 15q11-13 (Figure 1). These repetitive blocks form the common breakpoints (BPs) associated with deletions and duplications at the locus [Hogart et al.,2010]. BP1-BP3 deletions are the most frequent rearrangements observed in PWS and AS deletion cases, idic(15) cases are more variable, but often extend from BP1 to BP5, and BP4-BP5 microdeletions and duplications are associated with mental retardation, autism, epilepsy, and schizophrenia [Ben-Shachar et al., 2009; Cook et al., 1997; Cook and Scherer, 2008; Helbig et al., 2009; Stefansson et al., 2008]. In addition to their established role in disease associated chromosomal rearrangements, these primate-specific segmental duplications surrounding 15q11-13 may have a previously unexplored role in the complex chromatin organization and dynamics of this locus.
Figure 1. Location and epigenetic features of the human 15q11-13 locus.
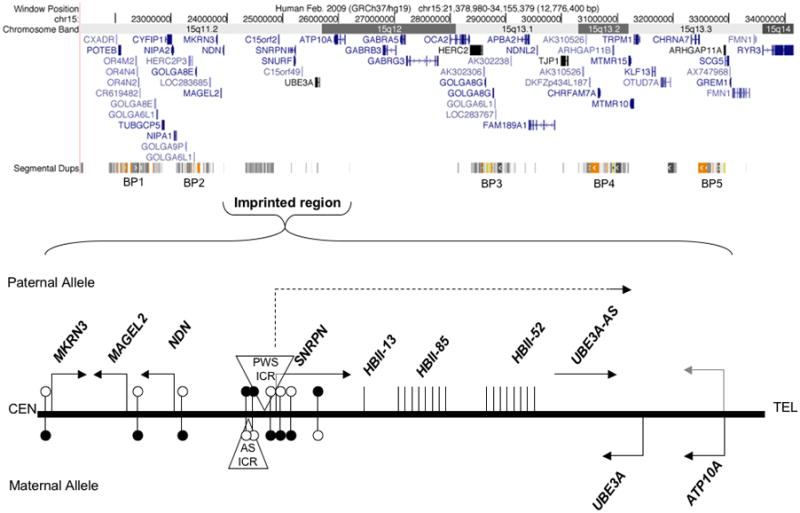
The proximal long arm of chromosome 15, referred to as 15q11-13, spans approximately 13 Mb. Five major clusters of segmental duplications predispose the locus to chromosomal rearrangements observed in Prader-Willi, Angelman, chromosome 15q duplication syndrome and small copy number variants associated with autism, epilepsy, and schizophrenia. The region of the 15q11-13 locus containing imprinted transcripts is expanded in the bottom panel to show the parental preference and direction of transcription, differentially methylated regions (filled circles represent methylated sites, unfilled represent unmethylated), and the positions of the bipartite imprinting control regions (AS-ICR and PWS-ICR, triangles).
The portion of 15q11-13 containing genes with defined parentally imprinted expression is limited roughly to a 3 Mb region extending from MKRN3 to ATP10A (Figure 1). While MKRN3, MAGEL2, NDN and SNURF/SNRPN encode proteins, there are also noncoding RNAs that display paternal allele-specific expression. Of particular interest are the several types of C/D box small nucleolar RNAs (snoRNAs) including clusters of HBII-85 (SNORD116) and HBII-52 (SNORD115) snoRNAs that are contained within a large noncoding host transcript that that also contains the antisense to UBE3A (UBE3A-AS). UBE3A is biallelically expressed in most tissues, but becomes maternally expressed, paternally imprinted, in mature neurons of the brain [Albrecht et al., 1997; Rougeulle et al., 1997; Vu and Hoffman, 1997]. ATP10A appears to be a variable border for the paternal imprint, as recent studies have demonstrated non-imprinted expression in mouse and human brain samples [DuBose et al., 2010; Hogart et al., 2008; Jiang et al., 2010].
The master regulator of the15q11-13 imprinted region is a bi-partite imprinting center (IC) containing a differentially methylated region (DMR) that controls imprinted gene expression over the entire locus [Sutcliffe et al., 1994]. The IC has been defined as the smallest region of overlap in AS and PWS deletion patients (AS-IC on the maternal allele and PWS-IC on the paternal allele). The PWS-IC is contained within the SNURF/SNRPN promoter and methylation of the maternal PWS-IC not only silences SNRPN but is believed to initiate the spread of methylation to the promoters of maternally silenced NDN, MAGEL2 and MKRN3 upstream of the IC [Bielinska et al., 2000]. The PWS-IC is hypomethylated on the paternal allele which is highly active both up and downstream of the IC. The mouse syntenic counterpart, 7qC, also contains a PWS-IC, deletion of which is sufficient to cause a neonatal lethal phenotype [Yang et al., 1998]. Interestingly, mouse models of the PWS-IC have shown that the neonatal lethality can be rescued on different genetic backgrounds and that a human PWS-IC sequence inserted in the mouse locus does not get maternally imprinted, resulting in an AS imprinting mutation mouse model [Chamberlain et al., 2004; Johnstone et al., 2005].
The genes within 15q11-13 function in a variety of biological processes and are regulated by a multitude of epigenetic processes. These include differential DNA methylation, histone modifications and DNAse hypersensitivity sites at the imprinting center (IC). Bioinformatics and experimental approaches have identified possible SP1, NRF1, E2F1, CTCF and YY1 transcription factor binding sites within the PWS-IC [Rodriguez-Jato et al., 2005]. Chromatin immunoprecipitation has also shown that MeCP2 (methyl CpG binding protein 2) binds to the maternal PWS-IC, as well as at other loci throughout 15q11-13, and could act to organize the chromatin in this region [Thatcher et al., 2005; Yasui et al., 2007]. Epigenetic regulation of 15q11-q13 gene function also involves fairly unique features such as the complex and very long paternal transcript, dramatic chromatin decondensation [Leung et al., 2009], tandemly repeated snoRNA expression [Cavaille et al., 2000] and homologous trans interactions [Thatcher et al., 2005].
The very large paternal lncRNA transcript, stretching from SNRPN to UBE3A, covers almost 1 Mb in mouse and over 600 Kb in humans, and is comprised of 148 exons and introns encompassing protein coding, non-coding, and antisense RNAs [Le Meur et al., 2005]. The paternal promoter of this transcript is highly active in multiple tissues but generally only the SNURF/SNRPN coding exons and the first cluster of C/D box snoRNAs are ubiquitously transcribed [Runte et al., 2001]. In contrast, neurons selectively upregulate transcription on the paternal allele to allow complete transcription through the clusters of snoRNAs and ultimately the UBE3A antisense region in certain subsets of neurons. This “extreme” transcriptional spreading corresponds to the highest levels of chromatin decondensation ever observed in an interphase nucleus [Leung et al., 2009]. Decondensation from the paternal allele is neuron specific and developmentally regulated. The paternal allele expands into euchromatic space and decondenses with neuronal maturation, marked by increased nuclear and nucleolar size, and decreased nucleolar number, while the maternal allele remains compact and near or within heterochromatic foci. The paternal allele reaches the level of compaction similar to a 30 nm fiber in many neurons. Yet this degree of chromatin decondensation is not a general feature of highly expressed genes in neurons, as decondensation, as detected by fluorescence in situ hybridization, appears to occur only in the two mammalian loci containing imprinted snoRNA clusters, but not other imprinted, ncRNA encoding, or highly expressed genes in the mouse genome [Leung et al., 2009].
The neuronally expressed long paternal transcript contains three primary components: the SNURF/SNRPN transcript which is processed into a mature mRNA and exported for translation, two embedded clusters of tandemly repeated snoRNAs (24 copies of SNORD115/HBII-52, 44 copies of SNORD116/HBII-85 [Castle et al., 2010]), and the UBE3A-AS that is hypothesized to mediate repression of the paternal UBE3A allele in neurons.
Classic C/D box snoRNAs mediate the post-transcriptional modification of ribosomal RNAs via 2′-O-methylation [Kiss et al., 2006]. Individual C/D box snoRNA genes are located throughout the genome, generally within introns of RNA Polymerase II (Pol II) transcripts. Biogenesis of the majority of snoRNAs is believed to occur at the site of transcription during which the snoRNA containing introns are spliced from the intervening exons, forming a branched lariat which undergoes exonucleolytic degradation to form the final stem loop structures. Processed snoRNAs are then subsequently shuttled to the nucleolus. snoRNAs from the 15q11-13 region show important differences with the canonical snoRNAs. SNORD115 and SNORD116 as well as SNORD109A, SNORD109B show minimal homology to rRNA [Sridhar et al., 2008] and have no known function in rRNA biogenesis. Despite the minimal homology of these “orphan” snoRNAs to rRNA, 15q11-q13 snoRNAs still appear to primarily localize to the nucleolus, suggesting a potential function in rRNA modification or nucleolar maturation. In both mouse and human, neuronal deficiency in these snoRNAs results in smaller nucleoli, while neurons with higher levels of these snoRNAs have larger and more numerous nucleoli [Leung et al., 2009].
The functional ncRNAs within the lncRNA transcript do not appear to be limited to the snoRNAs, however. The exons flanking the snoRNA imbedded introns are non-coding, yet they are spliced to form a lncRNA along with UBE3A-AS [Le Meur et al., 2005; Runte et al., 2001] and a recent study by the Cavaille group suggests a functional role for these exons in neurons. An RNA FISH probe against the non-coding exons, flanking the snoRNAs, revealed a stable nuclear-retained ncRNA cloud-like structure in hippocampal neurons [Vitali et al., 2010]. Treatment of these neurons with a transcriptional inhibitor did not remove the RNA cloud, indicating a remarkably stable nuclear structure encoded from the snoRNA host transcript with possible biological function, perhaps related to gene regulation at this locus
A role for ncRNA in mammalian chromatin organization and structure could involve heterochromatin formation, chromatin remodeling complex recruitment and creation of repressive chromatin compartments [Chaumeil et al., 2006; Khalil et al., 2009; Maison et al., 2002; Redrup et al., 2009]. Recent genomic studies have revealed as many as 4,500 lncRNAs, defined as larger than 200 bp, expressed in the human genome [Khalil et al., 2009]. The high levels of conservation and cell type specificity of these lncRNAs points to biological function rather than stochastic expression [Guttman et al., 2009; Ponjavic et al., 2007].
Recently published reports have suggested that lncRNAs, including the one at 15q11-13, have strong nuclear retention and are frequently localized to their site of transcription. The best-described example of lncRNA localization is Xist, the long ncRNA that is transcribed from and coats the inactive X chromosome in female mammals. Xist forms a RNA cloud into which silent genes are translocated when they are inactivated [Chaumeil et al., 2006; Clemson et al., 2006]. Xist coating of the inactive X chromosome also coincides with repressive histone marks, an absence of RNA Pol II and chromatin compaction [de Napoles et al., 2007; Kohlmaier et al., 2004; Ohhata et al., 2008]. Two other lncRNAs, Air and Kcqn1ot1, have also been shown to form RNA clouds that encase repressed genes in cis [Nagano et al., 2008; Redrup et al., 2009]. Additionally, many of these lncRNAs have been shown to recruit chromatin remodeling complex components, such as PRC2, coREST, SMX, G9a and LSDI which mediate repressive histone marks, directly to their own genomic locus and other loci [Khalil et al., 2009; Nagano et al., 2008; Tsai et al., 2010]. One study has also revealed lncRNAs recruiting active chromatin modifiers during embryonic stem cell differentiation [Dinger et al., 2008]. In the majority of these studies the chromatin modifiers associating with lncRNAs are histone methyltransferases which, depending on the histone residue modified, will confer either an “active” (such as H4K3me) or “silencing” (such as H3K27me) histone mark on the neighboring chromatin.
There are likely to be important differences between the regulation of Xist and the chromosome 15q11-q13 lncRNA. Most importantly, while X-inactivation occurs in early development in the context of rapid cell division, the 15q11-q13 lncRNA displays neuron-specific expression in an apparently post-mitotic context [Chamberlain et al.]. This would suggest that the regulation by the 15q11-q13 lncRNA is unlikely to be mediated by the epigenetic mechanisms associated with dividing cells and observed in the cases of Xist and Igf2r/Air. One example of brain-specific silencing via an antisense transcription is the Commd1/U2af1-rs1 locus. In this case, interference via the RNA polymerase complex engaged by the paternal U2af1-rs1 antisense is hypothesized to mediate paternal-specific Commd1 repression [Joh et al., 2009].
Given the extensive number of lncRNAs being discovered and the realization that we are only just starting to understand their biological roles, the possibilities for their role in chromatin organization appears bountiful. The Lander group has hypothesized that up-regulation of a tissue-specific transcription factor may initiate transcription of specific subsets of lncRNAs which can then guide chromatin remodeling complexes to specific sites throughout the genome in order to organize chromatin specifically for that cell type [Khalil et al., 2009].
A model for neuronal chromatin dynamics at 15q11-13 – one sequence, two distinct modes of regulation
Dynamic and specific changes in transcription, chromatin organization and localization of 15q11-13, and in particular the region transcribing the lncRNA, Snrpn-Ube3a, occur throughout neuronal maturation. While differential methylation of the PWS-IC is what drives imprinting in the germline and its maintenance throughout development, the neuronal-specific manifestations of imprinting involve additional epigenetic processes. The mature neuron treats the maternal and paternal alleles of SNRPN-UBE3A as distinct loci by differential DNA methylation, histone marks, matrix attachment sites, chromatin compaction levels, nuclear localization, and presumably transcription machinery and chromatin remodeling complexes [Buiting et al., 1995; Greally et al., 1999; Perk et al., 2002; Rodriguez-Jato et al., 2005].
A speculative model of the dynamic changes occurring at Snrpn-Ube3a during neuronal development and maturation is proposed in Figure 2. In the early embryonic central nervous system characterized by small heterochromatic nuclei, the structure of the paternal Snrpn-Ube3a locus is similar to the maternal allele in compact chromatin. Yet even at this early stage, the paternal allele is transcriptionally active at Snrpn from the stably unmethylated PWS-IC at relatively high levels.
Figure 2. Model of lncRNA expression and chromatin organization of 15q11-13 during neuronal maturation.
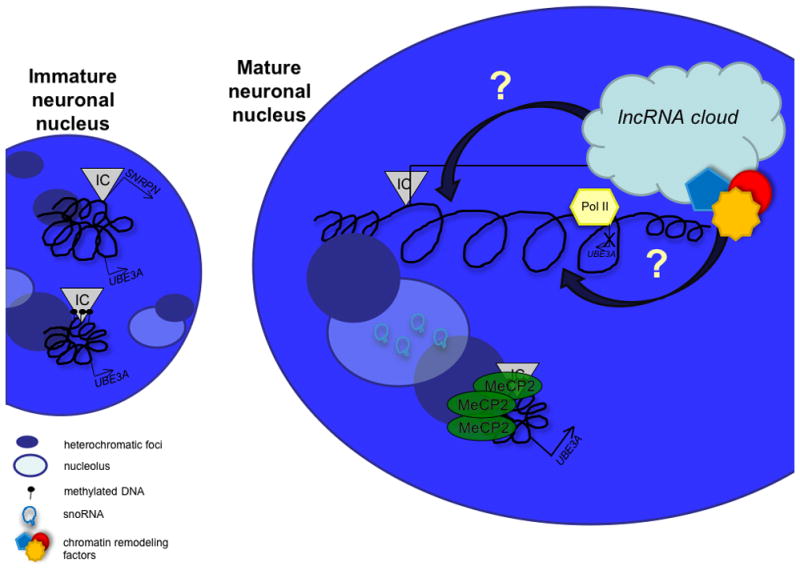
In the immature neuron with multiple nucleoli and heterochromatic foci, both alleles are similarly compacted, the un-methylated paternal IC expresses Snrpn, while both alleles express Ube3a. During maturation the maternal allele remains compact, while the paternal allele decondenses. MeCP2, expressed at higher levels in neurons, co-localizes with heterochromatin and binds to the methylated maternal IC, localizing the maternal allele to heterochromatin and positioning it properly for increased Ube3a expression. At the same time, the paternal allele becomes highly active, expressing not only Snprn, but the snoRNA clusters and the antisense to Ube3a, blocking paternal Ube3a expression from the opposing strand. The snoRNAs are spliced from introns and processed into stem loops. The snoRNA transcript forms a lncRNA cloud possibly localized at the paternal allele which may recruit chromatin remodeling factors to the locus to drive paternal allele decondensation. The snoRNAs are ultimately transported to the nucleolus, increasing nucleolar size contributing to a mature neuronal nuclear phenotype of a single large nucleolus.
By embryonic day 10–11, the snoRNAs are already being expressed at low levels in the developing central nervous system [Le Meur et al., 2005]. The snoRNAs whose biogenesis is believed to occur at the site of transcription, may recruit chromatin remodeling factors, such as Tip48 and Tip49, known to be involved in C/D box snoRNA biogenesis [McKeegan et al., 2009]. We speculate that the RNA cloud of lncRNA formed from host gene exons could be recruited in cis to the active paternal locus, enveloping the paternally expressed genes and enhancing their continued transcription. Recruitment of the lncRNA to the site of active transcription could potentially occur through RNA-DNA hybrids of repetitive regions within the decondensed chromatin, through recruitment by proteins bound to the paternal locus, or perhaps by recruitment of proteins involved in snoRNA biogenesis. This accumulation of activating chromatin remodeling factors within the RNA cloud may serve to aid the chromatin decondensation at the paternal allele. As a neuron matures, nuclear heterochromatin is reorganized into one or two heterochromatic foci surrounding one large central nucleolus [Martou and De Boni, 2000]. This may enable a global euchromatic environment outside of the perinucleolar heterochromatin that allows the paternal allele of Snrpn-Ube3a to further decondense. This high level of decondensation and low level of nucleosome occupancy would presumably mediate RNA Pol II elongation through the locus, extending transcription through Ube3a. Complete Ube3a antisense transcription would be expected to block Pol II from transcribing the sense transcript, resulting in paternally imprinted Ube3a only in mature neurons exhibiting full decondensation of Snrpn-Ube3a. Cellular heterogeneity and variable processivity of Pol II beyond the 3′ end of Ube3a on the paternal allele may also explain the variable imprinting status of Atp10a [Hogart et al., 2008; Kashiwagi et al., 2003; Kayashima et al., 2003a; Kayashima et al., 2003b].
The maternal Snrpn-Ube3a allele, containing a hypermethylated PWS-IC, is compact and associated with heterochromatin in the nuclei of embryonic neurons. Ube3a is expressed from the maternal allele of embryonic neurons, but at relatively low levels [Yamasaki et al., 2003]. As the neurons mature, global MeCP2 protein levels increase, presumably increasing the occupancy of MeCP2 at the methylated maternal PWS-IC [Thatcher et al., 2005]. MeCP2, known to associate with heterochromatin, may be required for localization of the maternal allele to the edge of the large perinucleolar chromocenter forming in the maturing neuron. MeCP2 may help anchor Ube3a to a transcriptionally prime location for its eventual up-regulation that coincides with paternal imprinting and paradoxically the highest cellular levels of Ube3a in mature neurons [Gustin et al., 2010].
In humans, where homologous pairing of 15q11-13 alleles occurs during neuronal maturation [Thatcher et al., 2005], there appears to be an additional regulation of transcriptional optimization. Homologous pairing between the alleles may facilitate transcriptional up-regulation of maternal UBE3A by positioning it within the transcriptionally favorable RNA chromatin cloud of the paternal allele. Multiple layers of epigenetic regulation may be required for full decondensation of the paternal allele and proper transcriptional output on both alleles, reminiscent of X chromosome inactivation which involves multiple, almost redundant, layers of epigenetic regulation to set-up and maintain silencing of the inactive X, including a lncRNA, DNA methylation, histone modifications and homologous pairing [Chaumeil et al., 2006; Csankovszki et al., 2001; Xu et al., 2006]
This multi-layered model of imprinted gene regulation at 15q11-13 is proposed to occur dynamically over a period of weeks in mouse brain and months in human brain during the long and complex process of neuronal maturation. There are several important questions that remain to be addressed in order to determine how the unique 15q11-q13 chromatin modifications determine imprinted gene expression status in brain: What affect does this decondensed chromatin structure have on genes within the 15q11-13 cluster and neighboring chromatin domains? How can decondensed chromatin stretching over an entire quadrant of the nucleus affect inter- and intra-chromosomal interactions between transcription factories and other nuclear bodies? The chromatin dynamics of the SNRPN-UBE3A locus seems to push the boundaries of current thinking as to how chromatin can be altered and localized in post-mitotic interphase nuclei. These studies have also strikingly emphasized the epigenetic principle that identical DNA sequence segments can be read very differently depending on cell type and developmental stage.
Human neuronal cell culture models for improved understanding of neurodevelopmental transcriptional complexities at 15q11-13 and other imprinted loci
Differences in the developmental timing and/or tissue specificity of gene expression between mice and humans make it imperative to develop human neuron culture models to study 15q11-q13 imprinting disorders. Thus far, our understanding of chromatin regulation across the region in neurons versus other tissues is based on mouse models, which show brain-specific expression of all imprinted genes within the syntenic region, except for the ubiquitiously expressed Snrpn gene. Humans, on the other hand, have a broader and more complex pattern of gene expression across the 15q11-q13 region. NDN and the proximal portion of the lncRNA transcribed from the PWS-IC are expressed in multiple tissues, while MKRN3, MAGEL2, and the distal portion of the lncRNA are nearly brain-specific.
Mouse models of 15q imprinting disorders do not perfectly phenocopy the human disorders. For instance, although PWS-IC deletion mice show neonatal failure to thrive [Yang et al., 1998], they do not show hypotonia, hyperphagia, or obesity, despite lacking expression from the same genes in brain. Deletion of Snord116 in mouse resembles PWS is neonatal growth impairments and hyperphagia, but the mice are underweight for their food consumption [Ding et al., 2008]. Even more surprising, a murine model of 15q duplication shows social behavioral deficits only upon paternal transmission of the duplicated allele, rather than the predominant maternal transmission observed in humans [Nakatani et al., 2009]. Whether the species-specific differences in gene regulation within the PWS/AS locus reflect genetic differences in intergenic sequences, such as the primate-specific 15q11-13 segmental duplications and perhaps account for some of the phenotypic differences in cognition between mice and humans is not known due to the lack of appropriate live human tissue for study.
Induced pluripotent stem cells (iPSCs) provide one such source of live human tissue. iPSCs are indefinitely self-renewing and functionally equivalent to human embryonic stem cells, except that they are generated by reprogramming somatic cells, which can be from patients with a genetic disorder. iPSCs generated from individuals with PWS and AS maintain the proper methylation imprint following reprogramming and can be differentiated in vitro into functional neurons [Chamberlain et al., 2010]. These iPSCs may prove to be invaluable tools to study specific changes in gene expression and chromatin configuration during neural development. Furthermore, the availability of PWS and AS iPSCs enables the tracking of the maternal and paternal 15q11-q13 chromatin independently.
Whether the methylation imprints at other imprinted loci will be maintained in iPSCs is not known. Data from normal iPSCs and murine iPSCs suggest that not all methylation imprints are correctly maintained [Pick et al., 2009; Stadtfeld et al.]. Therefore, iPSCs may not be suitable for studying chromatin structure in all imprinting disorders. Due to the stochastic and clonal nature of methylation marks, independently derived clones of iPSCs may show differences in maintenance of methylation, even when derived from donors with identical genetic mutations.
The ability to derive neurons from iPSCs from normal individuals as well as patients with chromosome 15q11-q13 abnormalities also offers unique perspectives for the study of changes to nuclear architecture and chromatin structure as a function of neuronal development. The unique and complex epigenetic mechanisms that establish brain-specific transcription within chromosome 15q11-q13 follow a developmental timeline in brain that can be recapitulated to some extent in vitro through iPSC technology. This in vitro window into human development should allow the study of key processes of 15q11-q13 imprinting as a function of defined stages of neuronal differentiation. In particular, the nature of the lncRNA interaction with UBE3A and the remarkable chromatin decondensation of 15q11-13 can be investigated in normal and abnormal human cells.
References
- Albrecht U, Sutcliffe JS, Cattanach BM, Beechey CV, Armstrong D, Eichele G, Beaudet AL. Imprinted expression of the murine Angelman syndrome gene, Ube3a, in hippocampal and Purkinje neurons [In Process Citation] Nat Genet. 1997;17:75–8. doi: 10.1038/ng0997-75. [DOI] [PubMed] [Google Scholar]
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl- CpG-binding protein 2. Nat Genet. 1999;23:185–8. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Ben-Shachar S, Lanpher B, German JR, Qasaymeh M, Potocki L, Nagamani S, Franco LM, Malphrus A, Bottenfield GW, Spence JE, Amato S, Rousseau JA, Moghaddam B, Skinner C, Skinner SA, Bernes S, Armstrong N, Shinawi M, Stankiewicz P, Patel A, Cheung SW, Lupski JR, Beaudet AL, Sahoo T. Microdeletion 15q13.3: A locus with incomplete penetrance for autism, mental retardation, and psychiatric disorders. J Med Genet. 2009 doi: 10.1136/jmg.2008.064378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird AP, Wolffe AP. Methylation-induced repression--belts, braces, and chromatin. Cell. 1999;99:451–4. doi: 10.1016/s0092-8674(00)81532-9. [DOI] [PubMed] [Google Scholar]
- Brown CJ, Hendrich BD, Rupert JL, Lafreniere RG, Xing Y, Lawrence J, Willard HF. The human XIST gene: analysis of a 17 kb inactive X-specific RNA that contains conserved repeats and is highly localized within the nucleus. Cell. 1992;71:527–42. doi: 10.1016/0092-8674(92)90520-m. [DOI] [PubMed] [Google Scholar]
- Castle JC, Armour CD, Lower M, Haynor D, Biery M, Bouzek H, Chen R, Jackson S, Johnson JM, Rohl CA, Raymond CK. Digital genome-wide ncRNA expression, including SnoRNAs, across 11 human tissues using polyA-neutral amplification. PLoS One. 5:e11779. doi: 10.1371/journal.pone.0011779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavaille J, Buiting K, Kiefmann M, Lalande M, Brannan CI, Horsthemke B, Bachellerie JP, Brosius J, Huttenhofer A. Identification of brain-specific and imprinted small nucleolar RNA genes exhibiting an unusual genomic organization. Proc Natl Acad Sci U S A. 2000;97:14311–6. doi: 10.1073/pnas.250426397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–9. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain SJ, Chen PF, Ng KY, Bourgois-Rocha F, Lemtiri-Chlieh F, Levine ES, Lalande M. Induced pluripotent stem cell models of the genomic imprinting disorders Angelman and Prader-Willi syndromes. Proc Natl Acad Sci U S A. 107:17668–73. doi: 10.1073/pnas.1004487107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton-Smith J, Laan L. Angelman syndrome: a review of the clinical and genetic aspects. J Med Genet. 2003;40:87–95. doi: 10.1136/jmg.40.2.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook EH, Jr, Lindgren V, Leventhal BL, Courchesne R, Lincoln A, Shulman C, Lord C, Courchesne E. Autism or atypical autism in maternally but not paternally derived proximal 15q duplication. Am J Hum Genet. 1997;60:928–34. [PMC free article] [PubMed] [Google Scholar]
- Cook EH, Jr, Scherer SW. Copy-number variations associated with neuropsychiatric conditions. Nature. 2008;455:919–23. doi: 10.1038/nature07458. [DOI] [PubMed] [Google Scholar]
- Cremer T, Cremer M, Dietzel S, Muller S, Solovei I, Fakan S. Chromosome territories--a functional nuclear landscape. Curr Opin Cell Biol. 2006;18:307–16. doi: 10.1016/j.ceb.2006.04.007. [DOI] [PubMed] [Google Scholar]
- Ding F, Li HH, Zhang S, Solomon NM, Camper SA, Cohen P, Francke U. SnoRNA Snord116 (Pwcr1/MBII-85) deletion causes growth deficiency and hyperphagia in mice. PLoS ONE. 2008;3:e1709. doi: 10.1371/journal.pone.0001709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuBose AJ, Johnstone KA, Smith EY, Hallett RA, Resnick JL. Atp10a, a gene adjacent to the PWS/AS gene cluster, is not imprinted in mouse and is insensitive to the PWS-IC. Neurogenetics. 11:145–51. doi: 10.1007/s10048-009-0226-9. [DOI] [PubMed] [Google Scholar]
- Gregg C, Zhang J, Butler JE, Haig D, Dulac C. Sex-specific parent-of-origin allelic expression in the mouse brain. Science. 329:682–5. doi: 10.1126/science.1190831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustin RM, Bichell TJ, Bubser M, Daily J, Filonova I, Mrelashvili D, Deutch AY, Colbran RJ, Weeber EJ, Haas KF. Tissue-specific variation of Ube3a protein expression in rodents and in a mouse model of Angelman syndrome. Neurobiol Dis. 39:283–91. doi: 10.1016/j.nbd.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helbig I, Mefford HC, Sharp AJ, Guipponi M, Fichera M, Franke A, Muhle H, de Kovel C, Baker C, von Spiczak S, Kron KL, Steinich I, Kleefuss-Lie AA, Leu C, Gaus V, Schmitz B, Klein KM, Reif PS, Rosenow F, Weber Y, Lerche H, Zimprich F, Urak L, Fuchs K, Feucht M, Genton P, Thomas P, Visscher F, de Haan GJ, Moller RS, Hjalgrim H, Luciano D, Wittig M, Nothnagel M, Elger CE, Nurnberg P, Romano C, Malafosse A, Koeleman BP, Lindhout D, Stephani U, Schreiber S, Eichler EE, Sander T. 15q13.3 microdeletions increase risk of idiopathic generalized epilepsy. Nat Genet. 2009;41:160–2. doi: 10.1038/ng.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogart A, Patzel KA, Lasalle JM. Gender influences monoallelic expression of ATP10A in human brain. Hum Genet. 2008 doi: 10.1007/s00439-008-0546-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogart A, Wu D, LaSalle JM, Schanen NC. The comorbidity of autism with the genomic disorders of chromosome 15q11.2-q13. Neurobiol Dis. 38:181–91. doi: 10.1016/j.nbd.2008.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang YH, Pan Y, Zhu L, Landa L, Yoo J, Spencer C, Lorenzo I, Brilliant M, Noebels J, Beaudet AL. Altered ultrasonic vocalization and impaired learning and memory in angelman syndrome mouse model with a large maternal deletion from ube3a to gabrb3. PLoS One. :5. doi: 10.1371/journal.pone.0012278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nat Rev Genet. 2007;8:253–62. doi: 10.1038/nrg2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joh K, Yatsuki H, Higashimoto K, Mukai T, Soejima H. Antisense transcription occurs at the promoter of a mouse imprinted gene, commd1, on the repressed paternal allele. J Biochem. 2009;146:771–4. doi: 10.1093/jb/mvp147. [DOI] [PubMed] [Google Scholar]
- Le Meur E, Watrin F, Landers M, Sturny R, Lalande M, Muscatelli F. Dynamic developmental regulation of the large non-coding RNA associated with the mouse 7C imprinted chromosomal region. Dev Biol. 2005;286:587–600. doi: 10.1016/j.ydbio.2005.07.030. [DOI] [PubMed] [Google Scholar]
- Lewis A, Reik W. How imprinting centres work. Cytogenet Genome Res. 2006;113:81–9. doi: 10.1159/000090818. [DOI] [PubMed] [Google Scholar]
- Martou G, De Boni U. Nuclear topology of murine, cerebellar Purkinje neurons: changes as a function of development. Exp Cell Res. 2000;256:131–9. doi: 10.1006/excr.1999.4793. [DOI] [PubMed] [Google Scholar]
- Nakatani J, Tamada K, Hatanaka F, Ise S, Ohta H, Inoue K, Tomonaga S, Watanabe Y, Chung YJ, Banerjee R, Iwamoto K, Kato T, Okazawa M, Yamauchi K, Tanda K, Takao K, Miyakawa T, Bradley A, Takumi T. Abnormal behavior in a chromosome-engineered mouse model for human 15q11-13 duplication seen in autism. Cell. 2009;137:1235–46. doi: 10.1016/j.cell.2009.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pick M, Stelzer Y, Bar-Nur O, Mayshar Y, Eden A, Benvenisty N. Clone- and gene-specific aberrations of parental imprinting in human induced pluripotent stem cells. Stem Cells. 2009;27:2686–90. doi: 10.1002/stem.205. [DOI] [PubMed] [Google Scholar]
- Rougeulle C, Glatt H, Lalande M. The Angelman syndrome candidate gene, UBE3A/E6-AP, is imprinted in brain [letter] [In Process Citation] Nat Genet. 1997;17:14–5. doi: 10.1038/ng0997-14. [DOI] [PubMed] [Google Scholar]
- Schanen NC. Epigenetics of autism spectrum disorders. Hum Mol Genet. 2006;15(Spec No 2):R138–50. doi: 10.1093/hmg/ddl213. [DOI] [PubMed] [Google Scholar]
- Skene PJ, Illingworth RS, Webb S, Kerr AR, James KD, Turner DJ, Andrews R, Bird AP. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol Cell. 37:457–68. doi: 10.1016/j.molcel.2010.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridhar P, Gan HH, Schlick T. A computational screen for C/D box snoRNAs in the human genomic region associated with Prader-Willi and Angelman syndromes. J Biomed Sci. 2008;15:697–705. doi: 10.1007/s11373-008-9271-x. [DOI] [PubMed] [Google Scholar]
- Stadtfeld M, Apostolou E, Akutsu H, Fukuda A, Follett P, Natesan S, Kono T, Shioda T, Hochedlinger K. Aberrant silencing of imprinted genes on chromosome 12qF1 in mouse induced pluripotent stem cells. Nature. 465:175–81. doi: 10.1038/nature09017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefansson H, Rujescu D, Cichon S, Pietilainen OP, Ingason A, Steinberg S, Fossdal R, Sigurdsson E, Sigmundsson T, Buizer-Voskamp JE, Hansen T, Jakobsen KD, Muglia P, Francks C, Matthews PM, Gylfason A, Halldorsson BV, Gudbjartsson D, Thorgeirsson TE, Sigurdsson A, Jonasdottir A, Jonasdottir A, Bjornsson A, Mattiasdottir S, Blondal T, Haraldsson M, Magnusdottir BB, Giegling I, Moller HJ, Hartmann A, Shianna KV, Ge D, Need AC, Crombie C, Fraser G, Walker N, Lonnqvist J, Suvisaari J, Tuulio-Henriksson A, Paunio T, Toulopoulou T, Bramon E, Di Forti M, Murray R, Ruggeri M, Vassos E, Tosato S, Walshe M, Li T, Vasilescu C, Muhleisen TW, Wang AG, Ullum H, Djurovic S, Melle I, Olesen J, Kiemeney LA, Franke B, Sabatti C, Freimer NB, Gulcher JR, Thorsteinsdottir U, Kong A, Andreassen OA, Ophoff RA, Georgi A, Rietschel M, Werge T, Petursson H, Goldstein DB, Nothen MM, Peltonen L, Collier DA, St Clair D, Stefansson K. Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–6. doi: 10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutcliffe JS, Nakao M, Christian S, Orstavik KH, Tommerup N, Ledbetter DH, Beaudet AL. Deletions of a differentially methylated CpG island at the SNRPN gene define a putative imprinting control region. Nat Genet. 1994;8:52–58. doi: 10.1038/ng0994-52. [DOI] [PubMed] [Google Scholar]
- Thatcher K, Peddada S, Yasui D, LaSalle JM. Homologous pairing of 15q11-13 imprinted domains in brain is developmentally regulated but deficient in Rett and autism samples. Hum Mol Genet. 2005;14:785–797. doi: 10.1093/hmg/ddi073. [DOI] [PubMed] [Google Scholar]
- Veenstra-Vanderweele J, Christian SL, Cook EH., Jr Autism as a paradigmatic complex genetic disorder. Annu Rev Genomics Hum Genet. 2004;5:379–405. doi: 10.1146/annurev.genom.5.061903.180050. [DOI] [PubMed] [Google Scholar]
- Vu T, Hoffman A. Imprinting of the Angelman syndrome gene, UBE3A, is restricted to brain. Nature Genet. 1997;17:12–13. doi: 10.1038/ng0997-12. [DOI] [PubMed] [Google Scholar]
- Williams CA, Zori RT, Stone JW, Gray BA, Cantu ES, Ostrer H. Maternal origin of 15q11-13 deletions in Angelman syndrome suggests a role for genomic imprinting [see comments] Am J Med Genet. 1990;35:350–3. doi: 10.1002/ajmg.1320350308. [DOI] [PubMed] [Google Scholar]
- Yamasaki K, Joh K, Ohta T, Masuzaki H, Ishimaru T, Mukai T, Niikawa N, Ogawa M, Wagstaff J, Kishino T. Neurons but not glial cells show reciprocal imprinting of sense and antisense transcripts of Ube3a. Hum Mol Genet. 2003;12:837–47. doi: 10.1093/hmg/ddg106. [DOI] [PubMed] [Google Scholar]
- Yang T, Adamson T, Resnick J, Leff S, Wevrick R, Franke U, Jenkins N, Copeland N, Brannan C. A mouse model for Prader-Willi syndrome imprinting-centre mutations. Nature Genet. 1998;19:25–31. doi: 10.1038/ng0598-25. [DOI] [PubMed] [Google Scholar]
- Yasui DH, Peddada S, Bieda MC, Vallero RO, Hogart A, Nagarajan RP, Thatcher KN, Farnham PJ, Lasalle JM. Integrated epigenomic analyses of neuronal MeCP2 reveal a role for long-range interaction with active genes. Proc Natl Acad Sci U S A. 2007;104:19416–21. doi: 10.1073/pnas.0707442104. [DOI] [PMC free article] [PubMed] [Google Scholar]