

Abstract
Aberrant glycosylation of cell membrane and secreted glycoproteins is a hallmark of various disease states, including cancer. The natural lectins currently used in the recognition of these glycoproteins are costly, difficult to produce, and unstable towards rigorous use. Herein we describe the design and synthesis of several boronic acid functionalized peptide-based synthetic lectin (SL) libraries, as well as the optimized methodology for obtaining peptide sequences of these SLs. SL libraries were subsequently used to identify SLs with as high as 5-fold selectivity for various glycoproteins. SLs will inevitably find a role in cancer diagnositics, given that they do not suffer from the drawbacks of natural lectins and that the combinatorial nature of these libraries allows for the identification of an SL for nearly any glycosylated biomolecule.
Keywords: Boronic acids, aberrant glycosylation, synthetic lectins, cancer, sensors
Introduction
Most membrane-bound and secreted proteins possess a variety of glycan structures linked to Ser, Thr, and Asn residues.[1] In healthy cells, these various glycans play important roles in mediating cell-cell interactions, protecting proteins from degradation, and, most importantly, in cell signaling.[1] In cancerous cells, however, specific glycans, e.g. sialyl Lewis X (sLex), are known to be over-, under-, or newly-expressed on particular types of tumor cells, and these changes are known to alter the ability of tumor cells to grow, divide, and metastasize.[1–5] Significantly, these changes in glycan expression can be used as diagnostic predictors of morbidity and mortality.[6] While the aberrant expression of numerous glycans has been associated with advanced stages of cancer, these same saccharides are also present in the initial stages of the disease. Therefore, the opportunity to detect cancer early exists based on the development of compounds (e.g., synthetic lectins) capable of sensing these predictive saccharides.
A handful of diagnostic tests based on aberrant glycosylation events have been developed for the initial detection of malignancy, as well as for monitoring disease progression after the initiation of cancer therapy.[7–10] A prominent example is the immunoassay for CEA (carcinoembryonic antigen). CEA is an aberrantly glycosylated glycoprotein that possesses high levels of sLex and elevated levels of CEA in the blood exhibit a strong, positive correlation with an increased risk of relapse and metastasis.[9] Although the CEA test is used to routinely monitor the post-operative serum levels of CEA in colon cancer patients, it is only effective in 4% of stage I and 25% of stage II cancers, stages where the disease is most treatable.[11] Similarly, the serum levels of sLea, also known as the CA19-9 antigen, are commonly used as a colon cancer diagnostic, although it too suffers from similar drawbacks.[12,13] Although these and other tests have proven valuable, they rely on either antibodies or natural lectins (i.e., glycan binding proteins). These biomolecules suffer from serious drawbacks, including instability towards rigorous use, high cost, and difficulty to produce, as well as the fact that most natural lectins and antibodies for cancer associated glycans have not been identified. The preponderance of false positives and the limitations outlined above for existing tests, coupled with the established value of the targeted saccharides, not only justifies, but necessitates, the development of novel diagnostic approaches targeting aberrant glycosylation that can be used as a screen for cancer.
To this end, we and others have previously described the design and synthesis of synthetic lectins (SLs) that incorporate boronic acids.[14–26] Boronic acids are used because they form covalent, and yet reversible interactions with the 1,2 and 1,3 cis diols present on many saccharides. These boronic acid-based synthetic lectins are advantageous over natural lectins due to their ease of synthesis, inherent stability towards rigorous use, and relatively low expense. Also, the combinatorial nature of SL libraries allows for the identification of SLs that bind to cancer associated biomarkers for which natural lectins have not yet been identified. Previous work from our lab has demonstrated the utility of boronic acid functionalized peptide based SLs in binding to glycoproteins.[27] SLs that bind to a particular analyte were identified from a combinatorial library, utilizing fluorescently tagged analytes. Note that both SLs selective for a particular glycoprotein and those able to bind several different glycoproteins, termed cross-reactive, were identified. The incorporation of both selective and cross-reactive SLs into an array based format, similar to established DNA based arrays,[28–30] will allow for the detection of the number and type of carbohydrates present in sera and tissue samples. Furthermore, we predict that the arrays will be able to identify cancer-specific glycosylation patterns, thereby negating false positives and limiting the need for more invasive procedures.
Herein, we describe our efforts to optimize SL library design, identify SLs that bind to proof-of-concept glycoproteins, and validate that these hits can selectively recognize glycoproteins. Furthermore, the methodology for sequencing these SLs, which has been problematic in the past, has been optimized, utilizing a unique set of reactions to remove the boronic acid moiety prior to sequencing. These studies highlight the intricacies of designing a synthetic lectin library and demonstrate that it is possible to use this approach to identify SLs that recognize a particular glycoprotein with as high as 5-fold selectivity versus another glycoprotein. These results provide definitive proof-of-concept for our approach to discovering SLs that can be used for the diagnosis, and potentially the treatment, of cancer.
Results and Discussion
Previous work from our lab has shown that a phenylboronic acid (PBA) functionalized peptide library, containing only alanine and diamino acids (to attach the PBA moiety), can exhibit selectivity for glycoproteins containing unique glycan structures (e.g., ovalbumin (OVA), bovine submaxillary mucin (BSM), and porcine stomach mucin (PSM)).[27] Termed the Low Diversity Library (LDL), this library demonstrated our ability to design, synthesize, and identify SLs that could bind particular glycoproteins, although the hit rate (i.e., percentage of strongly fluorescent beads within a screening) was somewhat high for this library (~10%). Additionally, the determination of peptide sequences from this library via both Edman and MS/MS based methodologies was problematic. We considered the possibility that the high hit rate was due to non-specific binding, suggesting that the isolated SL “hits” may not have been strong binders. This prompted the design and synthesis of several higher diversity libraries which had significantly lower hit rates, suggesting that the high hit rate for the LDL may be due to non-specific binding.
Given that our original library incorporated only alanine and four diamino acids, i.e., diamino propionic acid (Dpr), diaminobutanoic acid (Dab), ornithine (Orn), and Lys, we considered the possibility that the relatively high hit rate obtained with this library was due to a lack of diversity within the library. To address this possibility, a ‘High’ Diversity Library (HDL) containing 17 of the 20 natural amino acids was synthesized. The sequence of this library is Cbz-A-(X)10-BBRM-resin; where B is beta-alanine and X is one of the 17 natural amino acids used. The Met on the C-terminus is used for orthogonal cleavage from the resin using cyanogen bromide, the Arg aids in MS/MS sequencing of the peptide, and the beta-alanine acts as a spacer to project the SL away from the resin. Cys, Met, and Ile were excluded from the library, due to its high reactivity, its use in cyanogen bromide cleavage, and disambiguation of isomers (Leu versus Ile), respectively. Standard Fmoc protocols were used for library synthesis and acid-labile side-chain protecting groups were cleaved in one step, after incorporation of the boronic acid. Orthogonally protected (ivDde) lysine was used during synthesis because the ivDde group can be selectively removed with hydrazine to allow for the installation of the PBA moiety onto the diamino acid side-chain amine via reductive amination of 2-formylphenylboronic acid (Scheme 1).
Scheme 1.

Dde deprotection and reductive amination to attach the phenyl boronic acid moiety to the peptide backbone.
The library was synthesized using a biased split-and-pool combinatorial approach that on average resulted in the incorporation of 4 boronic acids per peptide. All libraries were synthesized on TentaGel amino resin (320 μm) because it is dynamic in aqueous and organic solvents, stable in acidic solutions, and provides a good balance between loading capacity, which is important for peptide sequencing, and the number of beads per gram, which is important for sequence diversity.
This library was screened against several fluorescently labeled proof-of-concept glycoproteins (i.e., OVA, PSM, and BSM) by incubating a portion of the library (e.g., 5 mg of library resin) with a tagged analyte (e.g., FITC-OVA). Note that screening smaller portions of the library, rather than the entire library at once, improves the signal to noise, thereby increasing the likelihood of identifying hits, or beads with a luminosity at least 30% greater than the library average. For library screens, the library was first soaked briefly in Screening Buffer (100 mM NaH2PO4, 150 mM NaCl, 10% glycerol, pH 7.2) and then pre-blocked with 1 mg/mL bovine serum albumin (BSA) in Screening Buffer to reduce non-specific binding. Glycerol was added to the buffer used for wetting, pre-blocking, and glycoprotein incubation to compete with weak boronic acid binders. The resin was then incubated with, for example, 0.5 mg/mL FITC-OVA in Screening Buffer plus 1 mg/mL BSA for 18 h at 23 °C. After washing the resin three times with PBS (100 mM NaH2PO4, 150 mM NaCl, pH 7.2) to remove unbound analyte, fluorescent beads were visualized using a fluorescent stereoscope. Hits were picked manually, and for the brightest beads, presumably the strongest binders, we attempted to directly determine the sequence by first boiling in 1% SDS for 1 h to remove bound analyte, before cleaving the peptides from the resin using 40 mg/mL cyanogen bromide under acidic conditions. Cleavage reactions were dried in vacuo, and MS and MS/MS data acquired.
Although this library had a decreased hit rate (Table 1), presumably due to its increased diversity, it had relatively high background fluorescence and proved quite difficult to sequence. It was reasoned that by reducing the number of different amino acids used in the library synthesis, we could increase the likelihood of determining peptide sequences. In order to test this theory, a library termed the Neutral ‘Medium’ Diversity Library (NMDL), of the same general sequence as the HDL, i.e. Cbz-A-(X)10-BBRM-resin, was synthesized using the methodology outlined above and incorporating only 11 of the 20 natural amino acids. Excluded from this library were any charged amino acids, as well as Pro, Met, Cys, Ile, and Trp.
Table 1.
Information about the SL libraries synthesized
| fLibrary | General Sequence | Diamino Acid | Randomized Amino Acids | Theoretical Diversity | Hit Rate | Advantages |
|---|---|---|---|---|---|---|
| LDL | Cbz-A(X)10A-resin | Lys, Orn, Dab, Dpr | A, K, Orn, Dab, Dpr | 510 | 10% | |
| HDL | Cbz-A(X)10BBRM-resin | Lys | A, R, N, D, E, Q, G, H, L, K, F, P, S, T, W, Y, V | 1710 | 1% | Increased diversitya |
| NMDL | Cbz-A(X)10BBRM-resin | Lys | A, N, Q, G, L, F, S, T, Y, V | 1110 | 1% | Increased diversitya |
| Dab NMDL | Cbz-A(X)10BBRM-resin | Dab | A, N, Q, G, L, F, S, T, Y, V | 1110 | 0.5% | Increased selectivity and decreased background fluorescenceb |
| Arg MDL | Cbz-A(X)10BBRM-resin | Dab | A, N, Q, G, L, F, S, T, Y, V, R | 1210 | 0.5% | Increased selectivityb |
| FPL | Ac-RXD*(X)3D*XBBRM-resin | Dab | A, N, Q, G, L, F, S, T, Y, V, R | 115 | 1% | Enhanced sequence determinationb |
Versus the Low Diversity Library;
Versus all previous Libraries
This library was screened against FITC-labeled OVA, BSM, and PSM as described above and had a similar hit rate (~1%) to the HDL (Table 1), indicating that even though the theoretical diversity of the NMDL was not as great as that of its predecessor, this library was still diverse enough to generate selective SLs. Nevertheless, this library displayed relatively high background fluorescence, similar to the HDL, and still proved difficult to sequence using MS based techniques (i.e., Partial Edman Degradation and MS/MS sequencing).
Given our observations with previous libraries, we considered the possibility that the high background fluorescence could be due to the use of Lys as the site of PBA incorporation. The long, flexible side-chain in Lys can adopt numerous conformations capable of binding the target analyte, if only weakly, thereby increasing background noise. To evaluate this possibility, the Dab NMDL was synthesized, where Lys was replaced with Dab, because it was expected that the shorter side-chain of Dab (4 methylene units for Lys versus 2 for Dab) would allow for increased pre-organization and enhanced secondary interactions with the peptide backbone, thereby decreasing non-specific binding and increasing selectivity. Note that Edwards et al utilized Dab in their peptidyl library for the same reason.[31] This library incorporates the same amino acids, utilizes the same synthetic methodology, and is of the same general sequence as the NMDL, thereby allowing for direct comparison of the effects of the different diamino acid side-chains.
This library, and the NMDL, which incorporates the PBA moiety via Lys, were screened in parallel against the same set of proof-of-concept glycoproteins. As expected, a slightly lower hit rate was observed for the Dab NMDL (Table 1). This suggests that by increasing library pre-organization, it is possible to identify hits that are more selective for a particular analyte. Also as expected, the incorporation of Dab in place of Lys further decreased non-selective binding, as indicated by the decreased background fluorescence and increased library differentiation. Representative data obtained for the screening of FITC-OVA is depicted in Figure 1. Note that the chart in this figure is a binning chart, in which individual bead luminosities are plotted for each screen. The greater spread in the data obtained for the Dab-containing library, versus the otherwise identical Lys-containing library, is an indication of greater differentiation and selectivity for binding the targeted glycoprotein. Even though selective hit identification was enhanced upon Dab incorporation, it was still difficult to consistently obtain peptide sequences using MS/MS methodologies.
Figure 1.

Fluorescence images of equal portions of the Dab NMDL (A.) and the NMDL (B.) after treatment with FITC-OVA. Note the increased diversity in bead luminosity when the Dab library is used. (C.) A binning chart showing individual bead luminosities from this comparative screening, demonstrating decreased background fluorescence and increased differentiation when Dab is used in a library instead of Lys.
In order to explore the importance of charge within the SL library, a library of the same general design as the NMDL was synthesized, utilizing 12 of the 20 natural amino acids, excluding Pro, Met, Cys, Ile, and Trp, as well as any charged amino acids except for Arg. It was hypothesized that the positively charged guanidinium group of Arg would form a favorable salt bridge with negatively charged sialic acid moieties commonly found on cancer related glycoproteins, as well as on two of our proof-of-concept glycoproteins (i.e., BSM and PSM). This library was synthesized with the general sequence Cbz-A-(X)10-BBRM-resin using the methodology described above. Subsequent screening indicated that the Arg containing library exhibited mildly increased diversity versus the Dab NMDL, allowing us to conclude that the addition of Arg into a library can enhance the binding strength and selectivity of SLs. Although optimization of library design allowed for recognition of selective SLs, the sequencing of these SLs was still problematic.
Initial sequencing efforts focused on utilizing the partial Edman degradation (PED) technique.[32] Unlike traditional Edman peptide sequencing, which immobilizes a resin bound peptide, removes one amino acid at a time, and analyzes it by high-performance liquid chromatography (HPLC), PED uses a mixture of capping and degradation reagents to produce a latter of peptide fragments on the bead, which can be cleaved and analyzed by MALDI-TOF MS (Figure 2).
Figure 2.

The PED procedure employed initially to sequence peptides involves several cycles of treating with a mixture of capping (Fmoc-OSU) and degradation (PITC) reagents, followed by TFA. After generating the peptide library, the Fmoc groups are removed, and the peptides are cleaved from the resin and analyzed by MALDI-TOF MS.
In example, a small portion of beads with free N-termini were treated with a 25:1 mixture of phenyl isothiocyanate (PITC) and Fmoc-O-succinimide (Fmoc-OSU) under basic conditions. Upon subsequent treatment with TFA, those peptides capped with PITC would undergo a degradation reaction to remove one amino acid, while those capped with Fmoc remained intact. This process was repeated several times to generate the peptide ladder before removing the Fmoc groups, cleaving the peptides from the resin, and analyzing by MALDI-TOF MS. Although his technique worked moderately well with known peptide sequences, overall the spectra were quite poor, with low signal-to-noise ratios, making it difficult to identify the peaks corresponding to the laddered peptides.
It was considered that the difficulty obtaining peptide sequences was potentially due to the presence of the boronic acid moiety. To evaluate this possibility, we explored several techniques for removing this moiety from the SL prior to MS-based sequencing. Treatment of the resin bound SL with hydrogen peroxide in water successfully oxidized the phenylboronic acid to a phenol.[33] Upon cleavage from the resin with CNBr and subsequent analysis by MALDI-TOF MS it was observed that the PBA moiety had in fact been completely removed, leaving the free amine (Scheme 2 and Figure 3).
Scheme 2.

Hydrogen peroxide treatment oxidizes the phenyl boronic acid to the phenol and subsequent treatment with CNBr under acidic conditions cleaves the methyl phenol moiety from SLs altogether. It is suggested that this moiety is oxidized to the quinine methide.
Figure 3.

Mass Spectra of (A.) a boronic acid functionalized model SL (exp m/z = 1085.0; obs m/z = 1084.8) and (B.) the same SL after oxidation with H2O2 (exp m/z = 969.1; obs m/z = 968.7). Both sequences were treated with CNBr to cleave the peptide from the resin. Note that oxidation/CNBr treatment removes the entire PBA moiety.
To further explore the oxidation and cleavage reactions that appeared to cause complete removal of the PBA moiety before sequencing, an all Ala SL was synthesized with differing groups appended to Dab. This peptide, of the sequence Ac-AAAD*AAABBRM-resin, was synthesized on amino terminated TentaGel resin using the Fmoc strategy, where D* is modified Dab. Upon removal of the Dde protecting group from Dab, the free amine was reductively aminated with either benzaldehyde, salicylaldehyde, or 2-formylphenylboronic acid, to give Ala-H, Ala-OH, or Ala-BOH respectively (Scheme 3).
Scheme 3.

Reductive amination to complete the synthesis of Ala-H, Ala-OH, and Ala-BOH.
Each compound was subjected to various conditions (Table 2) and analyzed by MALDI-TOF to explore the mechanism of removal of the PBA moiety from an SL. Observed masses and their corresponding structures are summarized in Table 3. In testing Ala-H (Figure 4), no difference was seen between the control (Test A) and oxidized Ala-H (Test B), as both exhibited major peaks at 1058.7 Da and 1080.7 Da, corresponding to the expected m/z for Ala-H (compound 2) and the sodium adduct of Ala H, respectively.
Table 2.
Various tests of Ala model compounds
| Test | Compound | Treatment 1 | Treatment 2 | Treatment 3 | Compound # |
|---|---|---|---|---|---|
| A | Ala-H | CNBr | 2 | ||
| B | Ala-H | H2O2 | CNBr | 2 | |
| C | Ala-BOH | CNBr | 4 | ||
| D | Ala-BOH | H2O2 | CNBr | 1 | |
| E | Ala-BOH | H2O2 | AcOAc | CNBr | 4, 5, 6, & 7 |
| F | Ala-OH | CNBr | 1 & 3 | ||
| G | Ala-OH | H2O2 | CNBr | 1 | |
| H | Ala-OH | AcOAc | CNBr | 6 & 7 |
Table 3.
Observed m/z values and their corresponding structures
| Compound | R′ | m/z |
|---|---|---|
| 1 |
|
968.7 |
| 2 |
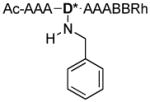
|
1058.7 |
| 3 |
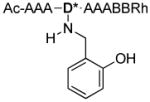
|
1074.7 |
| 4 |
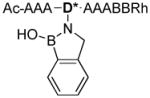
|
1084.8 |
| 5 |
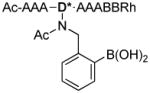
|
1143.8 |
| 6 |
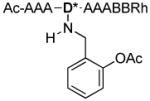
|
1116.8 |
| 7 |
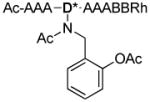
|
1158.8 |
Figure 4.

MALDI-TOF analysis of Ala-H testing.
CNBr cleavage of Ala-BOH (Figure 5) (Test C) resulted in the formation of the dehydrated boronic acid form of Ala-BOH with an m/z of 1084.8 Da (compound 4). Oxidation and subsequent cleavage of Ala-BOH (Test D) resulted in complete removal of the PBA moiety, giving a major m/z of 968.7 Da (compound 1). Interestingly, the peak at 1152.7 Da exhibits a bromine isotope pattern and corresponds to Ala-OH, the predicted oxidation product of Ala-BOH, plus bromine. This peak does not normally show up in spectra of our library SLs and is thought to be a unique adduct for this model compound. It was thought that by following oxidation of Ala-BOH with an acylation step before cleaving it from the resin (Test E), we might be able to trap the predicted phenol intermediate by either limiting the phenol or benzyl amine reactivity. Several peaks are observed in this spectrum, corresponding to dehydrated Ala-BOH (1084.7 Da; compound 4) and hydrated Ala-BOH plus one acyl group (1143.8 Da; compound 5), presumably on the benzyl amine. The other two major peaks in this spectrum correspond to the mono- and di-acylated versions of the oxidized Ala-BOH (Ala-OH) (1116.8 Da and 1158.8 Da; compounds 6 and 7, respectively), presumably on the either the benzyl amine, phenol, or both. This supports the theory that treatment with hydrogen peroxide does convert the PBA moiety to a phenol and that this phenol is an important intermediate in the removal of PBA from SLs.
Figure 5.

MALDI-TOF analysis of Ala-BOH testing.
The last set of tests were performed to explore the reactivity of Ala-OH (Figure 6), the predicted product after Ala-BOH oxidation. Perhaps not surprisingly, simple treatment of this compound with CNBr (Test F) to remove it from the resin results in removal of the phenol moiety to give the free amine (968.7 Da; compound 1). This spectrum is not particularly clean, as some intact Ala-OH is still present (1074.7 Da; compound 3). Interestingly, treatment of this compound with hydrogen peroxide before cleavage (Test G) results in a cleaner spectrum, with nearly all Ala-OH being converted to free amine. Note that again, the unique bromine adduct is observed at 1152.7 Da. Lastly, Ala-OH was first acylated and then treated with CNBr (Test H), resulting in the starting Ala-OH plus either one or two acyl groups (1116.8 Da or 1158.8 Da; compounds 6 and 7, respectively), a similar result to Test E with Ala-BOH.
Figure 6.

MALDI-TOF analysis of Ala-OH testing.
The results of this testing suggest that both the phenol and the secondary benzylic amine are necessary for complete removal of this moiety. Work with compounds of this nature suggests that o-hydroxy benzyl amines are capable of readily oxidizing to a quinine methide at slightly decreased pH (Scheme 2).[34] While removal of the PBA moiety resulted in much cleaner MS/MS spectra, on average, complete sequence information could only be obtained for ~50% of the hits we attempted to sequence. Therefore, it was hypothesized that simplification of the library design may aid in successfully obtaining hit sequences, and as such a library with the general sequence Ac-RXD*XXXD*XBBRM-resin, where the positions of the PBA moieties were fixed was synthesized; D* denotes PBA functionalized Dab, B is beta-alanine, and X is a randomized amino acid. Known as the Fixed Position Library (FPL), this design approach was taken because it was expected that the fixed PBA groups would provide known mass differences that could be used to aid in the interpretation of MS/MS spectra. Although this library has a lower theoretical diversity (~1.6 × 105) than the other libraries tested, the complete diversity space for this library could not be exhausted based on 1 gram of synthesized library (~6.5 × 104), as was the synthetic scale for the higher diversity libraries discussed above. In this regard, considerable work has shown that efficient and effective sensors and ligands have been isolated from combinatorial libraries with similar or even significantly lower theoretical diversities.[35–40] Arginine was included at the N-terminus because the positive charge of this amino acid was expected to aid ionization during MS analyses and provide for favorable interactions with the negative charge present on sialylated glycoproteins.
The FPL was initially screened against FITC–OVA, demonstrating that this library possessed comparable background fluorescence to the ‘medium’ diversity libraries described above (i.e., Lys NMDL, Dab NMDL, and Arg MDL), with only a slightly higher hit rate (Table 1). These data indicate that decreasing the number of PBA moieties from ~4 to 2 did not affect the ability of the library to differentiate different glycoproteins. This library was then screened against FITC-labeled OVA, BSM, and PSM and hits were picked manually. After washing with 1% SDS, sequences were obtained by treating individual beads with hydrogen peroxide followed by cyanogen bromide (to remove the PBA moiety and cleave the peptide from the resin) and subsequent MS/MS analysis. Note that the defined structure of this library allowed for sequencing of greater than 80% of the hits isolated, including SL1 and SL2, which were isolated from a screen selecting for OVA-specific SLs (Table 4).
Table 4.
Sequence information for isolated hits and subsequent mutants from the FPL
| SL Hit | Sequence | Glycoprotein Screen | Glycoprotein Selectivity |
|---|---|---|---|
| SL1 | Ac–RGD*VTFD*R–BBRM–resin | OVA | Cross reactive |
| SL2 | Ac–RTD*RFLD*V–BBRM–resin | OVA | OVA |
| SL2-Ala | Ac–RTARFLAV–BBRM–resin | NA | NA |
SL1 and SL2 were re-synthesized and their selectivity and cross-reactivity evaluated by incubating small sets of the re-synthesized SLs (20 to 50 beads) with fluorescently-tagged analytes at a concentration of 0.1 mg/mL. Note that BSA is not a glycoprotein but was used as a control to assess non-specific hydrophobic protein binding to the SLs. Also note that since there is some natural heterogeneity in glycoproteins, causing variation in the screening data, at least 20 replicates were selected from each screening.
Similar size sets of the fixed position SL library were treated identically to serve as a statistical background/reference. This approach was taken because the different glycoproteins used in these investigations exhibit differing degrees of glycosylation and fluorescent labeling; thus, using non-normalized fluorescence intensities gave misleading information about SL selectivity. Data normalization began by subtracting the fluorescence intensity of the library from the fluorescence intensity of the re-synthesized SL. The obtained difference provided a relative value for the binding affinity of a particular analyte to a specific SL referenced against a statistically constant background control. Dividing this difference by the fluorescence intensity of the library provided the percent change in binding of the SL over the library for any given analyte (Figure 7). This manipulation puts all data on the same linear scale regardless of the extent of labeling and glycosylation for each analyte.
Figure 7.

Plot of the percent change in luminosity (i.e., the percent change in binding of the SL over the library for any given analyte) of SL1, SL2, and SL2-Ala towards 4 different analytes, ovalbumin (OVA), bovine submaxillary mucin (BSM), porcine stomach mucin (PSM) and bovine serum albumin (BSA). Broadly speaking, SL1 is cross-reactive and SL2 is selective for OVA. Substitution of the boronic acid modified Dab moieties for Ala (SL2-Ala) results in essentially no binding, indicating the importance of the boronic acid-carbohydrate interaction for SL binding and selectivity.
A selectivity factor (Table 5) was calculated by dividing the percent increase for each analyte by the percent increase of the weakest binder within an SL screening. To compare the selectivity of an SL between two glycoproteins, simply divide the selectivity factors for each glycoprotein, which affords a relative fold-selectivity.
Table 5.
Selectivity factors for each SL screened against 4 different glycoproteins. Values were calculated by dividing the percent increase for each analyte by the percent increase of the weakest binder within each SL screening series.
| OVA | BSM | PSM | BSA | |
|---|---|---|---|---|
| SL1 | 1.0 | 1.3 | 1.8 | 1.3 |
| SL2 | 5.1 | 1.6 | 1.0 | 2.5 |
The fold-selectivity of an SL for one glycoprotein over another can be obtained by dividing their respective selectivity factors.
SL1, selected from screening the library against OVA, was cross-reactive, showing little selectivity for any of the analytes screened (Tables 5). Also, SL1 shows a relatively high average luminosity when treated with FITC-BSA, indicating that this SL exhibited non-specific protein binding, which may account for the observed cross-reactivity. SL2 exhibited moderate selectivity for OVA, although it did exhibit some non-specific binding to BSA. This was demonstrated by the fact that SL2 bound OVA with ~3 fold selectivity over BSM and ~5 fold selectivity over PSM but only 2-fold selectivity over BSA. These results were particularly gratifying because they demonstrated that this approach is capable of identifying SLs that recognize glycoproteins with moderate selectivity.
To investigate whether the importance of the multivalent, bead based display of SL2 is important for a strong SL2-OVA interaction, a soluble SL2 analogue (sSL2) was synthesized on Rink amide resin and purified by HPLC. This soluble SL was then added to standard SL2-FITC-OVA screenings at varying concentrations, to see at what concentration competition for OVA binding occurred (i.e., when the beads began to look dimmer). The data indicate that millimolar concentrations of sSL2 are required for the fluorescent intensity to begin to diminish (Figure 8). These results indicate that the multivalent nature of the bead based SLs is important for a strong interaction between the SL and the glycoprotein. To further explore SL-glycoprotein binding, and, in particular, the importance of the boronic acid-carbohydrate interaction on the strength of binding, a derivative of SL2 was synthesized where the boronic acid functionalized Dab moieties were substituted with Ala (i.e., SL2-Ala). This SL was screened against OVA, BSM, PSM, and BSA to evaluate its selectivity (Figure 7). Interestingly, this SL exhibited essentially no binding with any of the analytes screened, including OVA; the glycoprotein that boronic acid functionalized SL2 bound to with good affinity. These results indicate that the boronic acid-carbohydrate complex is the major component of a strong SL-glycoprotein interaction, at least with respect to SL2.
Figure 8.

Soluble SL2 (sSL2) competes for FITC-OVA binding at millimolar concentrations, indicating that the multivalent nature of bead based SL2 is important for strong binding.
Conclusions
Herein are described efforts towards developing an SL library that covers a large diversity space and exhibits excellent binding selectivity towards a variety of glycoproteins, as well as the development of methodologies for readily sequencing boronic acid-based SLs using MS/MS techniques. From these efforts, it was determined that the incorporation of PBA moieties onto Dab, rather than Lys, decreases background fluorescence by reducing non-specific binding, likely as a result of increased pre-organization of the SL binding site. Also, a novel series of reactions was identified that completely removes the amine-linked PBA moiety from a SL, allowing for unambiguous sequence determination. Successful high throughput screening of an optimized library, the Fixed Position Library, was demonstrated against proof-of-concept glycoproteins. Subsequently, SLs exhibiting selective binding for specific glycoproteins were identified, sequenced, and their binding characteristics validated. Several selective and cross-reactive SLs have been identified for OVA, including SL2, which exhibits as high as ~5-fold selectivity for this glycoprotein.
Having developed the methodology to synthesize, screen, and sequence bead based SLs, current work in the area of library design is focused on tuning the boronic acid moiety to improve reactivity and selectivity. Also, efforts are underway to identify highly selective, as well as cross-reactive SLs for cancer associated glycans and glycoproteins (e.g., sLex, sLea, and CEA), assemble these into a diagnostic array, and probe the SL-glycoprotein interaction.
Experimental Methods
Chemicals
Glycoproteins and FITC were purchased from Sigma-Aldrich (Milwaukee, WI). Fluorescently labeled glycoproteins were prepared and validated as previously described.[27] Fmoc-protected amino acids and O-Benzotriazole-N,N,N′,N′-tetramethyl-uronium-hexafluoro-phosphate (HBTU) were purchased from Novabiochem Corp (Gibbstown, NJ). TentaGel resin (Cat. No. MB-300-002; loading level 0.25–0.30 mmol/g) was acquired from Rapp Polymere (Tübingen, Germany). All other chemicals were purchased from Acros Organics (Morris Plains, NJ) and used without further purification.
Microscopy
All fluorescent images were taken using a Leica MZ 16F microscope equipped with a GFP filter set (excitation 450–490 nm; emission filter 500–550 nm), and a QImaging MicroPublisher 5.0 RTV digital camera. The images were analyzed with Adobe Photoshop, using the marquee tool to select the entire bead, obtaining luminosity values that represent the fluorescent intensity of individual beads. High average luminosity values correspond to brighter beads that presumably bind strongly to the analyte under study.
General library design
All libraries were synthesized using standard Fmoc/HBTU chemistry on amino terminated TentaGel resin (320 μm) that had first been derivatized with the following sequence: BBRM; B is beta-alanine and was included as a spacer to project the SL away from the resin. The Met and Arg residues were included to permit orthogonal cleavage of the peptide from the resin using cyanogen bromide, and aid ionization during MS/MS sequencing of the peptide, respectively. Randomized amino acids were then introduced using the split-and-pool methodology.[27] Cbz-Ala-OH or alternatively, Arg followed by acylation with acetic anhydride, was used to cap the N-terminus. Dde or ivDDE protected diamino acids were incorporated to provide a side chain handle on which to attach phenyl boronic acid moieties by reductive amination; the Dde or ivDDE protecting group was used because it can be removed, in an orthogonal fashion to the other side chain protecting groups, using hydrazine. The reactive side chains of all other amino acids were protected with acid-labile protecting groups. For all libraries, save the fixed position library, a biased split-and-pool combinatorial method (see below) was used for library synthesis. The procedures for coupling BBRM were the same for all of the libraries.
Synthesis of the BBRM sequence
All reactions were performed at ambient temperature unless otherwise specified. For example, TentaGel resin (200 mg; 0.05 mmol; ~12,800 beads) was swollen in DMF for 10 min. The DMF was removed and Fmoc-Met-OH (4 eq, 0.2 mmol, 74.3 mg), preactivated with HBTU (4 eq, 0.2 mmol, 75.9 mg) for 10 min in 5% N-methylmorpholine in DMF, was then added. After tumbling for 45 min, unreacted amino acid was removed by vacuum filtration, and the beads were then washed with DMF, methanol (MeOH), and DMF. Piperidine (20% in DMF; 10 mL; 10 min; 2x) was then used to remove the Fmoc group. The same procedure was used to subsequently couple arginine, and the two β-alanines.
Synthesis of High Diversity Library
The sequence of this library was Cbz-A-(X)10-BBRM-resin, where X is a randomized amino acid. Amino acids incorporated into the 10 randomized positions included: Ala, Gly, Pro, Val, Leu, Ile, Phe, Tyr, Ser, Thr, Asn, Gln, Lys, Arg, His, Asp, and Glu. Both Boc and ivDde protected Lys were used in this library; Lys-ivDde was used for coupling the PBA moiety and Lys-Boc was used in the randomized portion of the library as a positively charged amino acid. Resin which already contained the BBRM sequence was divided into 18 filtered vials by adding DMF (28 mL) to the beads, agitating them to a homogeneous suspension, and transferring 11 mL (~5000 beads) of the DMF containing beads to one vial and 1 mL (~450 beads) of the mixture to each of the remaining 17 vials. The DMF was then removed by vacuum filtration. The vial containing the 11 equivalents of resin was then treated with Fmoc-Lys(ivDde)-OH (4 eq, 0.08 mmol, 42.6 mg) activated with HBTU (4 eq, 0.08 mmol, 30.3 mg) to couple the ivDde-protected Lys, incorporating a statistical average of 4 PBA moieties per peptide. To the remaining 17 vials, which each contained one equivalent of resin, was added one of the 17 different amino acids (4 eq, 0.007 mmol) and HBTU (4 eq, 0.007 mmol, 2.7 mg). The coupling reactions were allowed to proceed for 45 min, at which point, the reaction solutions were drained, and the beads subsequently washed sequentially with DMF, MeOH, and DMF by adding the wash solution, agitating the resin, and then removing the solution by vacuum filtration. The beads were then combined, randomized, and the next amino acid coupled using the methodology outlined above. After incorporation of the last randomized position, the beads were combined, washed, and the N-terminus capped with Cbz-Ala-OH (4 eq, 0.20 mmol, 44.6 mg).
Incorporation of Phenyl Boronic Acid
The DDE or ivDde protecting group of the side-chain amine on a diamino acid was removed by tumbling the resin in hydrazine monohydrate (10 mL, 2% (v/v) in DMF) for 1 h. 2-formylphenyl boronic acid (8 eq, 0.8 mmol, 119.95 mg) was dissolved in a mixture of 10% methanol in DMF. This solution was added to the resin along with activated 3Å molecular sieves and was tumbled at 37 °C overnight. NaBH4 (8 eq, 0.8 mmol, 30.26 mg) was added, gas was evolved, and the resin was tumbled at 37 °C for 4 h. The beads were then sequentially washed with DMF, MeOH, and DMF. Molecular sieves were then removed by floating off the resin with CH2Cl2, followed by further washing with DMF, MeOH, and DMF. Subsequently, the acid-labile protecting groups were removed by tumbling in 95% trifluoroacetic acid (TFA), 2.5% water, and 2.5% triisopropysilane (TIS) (10 mL) for 1 h. The beads were then washed again with DMF, MeOH, DMF, and MeOH. Resin was dried by aspiration and stored at −20 °C.
Synthesis of Neutral Medium Diversity Library
The sequence of this library is Cbz-A-(X)10-BBRM-resin. Amino acids incorporated into the 10 randomized positions include: Ala, Gly, Val, Leu, Phe Tyr, Ser, Thr, Asn, and Gln. For this library, Lys(ivDde) was used as the site of boronic acid incorporation and a statistical average of 4 PBA moieties per peptide were incorporated. Library synthesis was accomplished by suspending the beads in DMF (17 mL), transferring 7 mL of the DMF/bead mixture into 1 vial and 1 mL of the mixture to 10 smaller vials. The vial containing the larger amount of resin was then coupled to Fmoc-Lys(ivDde)-OH (4 eq, 0.02 mmol, 10.65 mg), activated with HBTU (4 eq, 0.02 mmol, 7.6 mg). The remaining 10 vials were coupled to the 10 different amino acids (4 eq, 0.003 mmol) as described above. The synthesis of the remainder of the library proceeded similarly to the methods outlined above. The N-terminus of this library was also capped with Cbz-Ala-OH, and the PBA moiety incorporated identically.
Synthesis of Dab Neutral Medium Diversity Library
The sequence of this library is Cbz-A- (X)10-BBRM-resin. The only difference between this library and the Neutral Medium Diversity Library is that, for this library, Dde-protected Dab is used as the diamino acid for PBA installation instead of Lys.
Synthesis of Arg-Dab Neutral Medium Diversity Library
Synthesis of this library is similar to that for the Dab Neutral Medium diversity library just described. The only difference is that the randomized amino acids also include Arg.
Synthesis of Fixed Position Library
The general sequence of this library is Ac-RXD*XXXD*XBBRM-resin where D* denotes Dab and X is a randomized amino acid (i.e., Ala, Gly, Val, Leu, Phe Tyr, Ser, Thr, Asn, Arg, and Gln). The BBRM resin was split equally into 11 different tubes by suspending the beads in 11 mL of DMF and transferring 1 mL of this homogeneous solution to 11 different tubes. Subsequently, each portion was coupled with a different amino acid, combined together in a fritted reaction column, washed, the Fmoc group removed, and Dde-protected Dab installed as previously described. The split-and-pool method described above was then used to incorporate three randomized positions, at which point the second Dde-protected Dab moiety was installed. The beads were divided equally to permit the incorporation of the final diversity element. The beads were recombined, and the N-terminal Arg was coupled to the resin. The peptide was then N-terminally acetylated by treating the resin with 10 mL of CH2Cl2 containing 5% acetic anhydride and 5% pyridine. The PBA moiety was incorporated, and the acid labile protecting groups removed, as described above.
General Screening Process
Library beads (2 to 10 mg) were added to a 1.5 mL microcentrifuge tube, and equilibrated by tumbling in Screening Buffer (100 mM NaH2PO4, 150 mM NaCl, 10% glycerol, pH 7.2) for 10 min. Note that 10% glycerol was added to prevent weak binding to the SLs. The Screening Buffer was then removed, and the resin tumbled in 1% BSA in Screening Buffer for 15 min to prevent nonspecific binding. After removal of this solution, the beads were tumbled with a FITC-tagged glycoprotein (0.1 mg/mL) in Screening Buffer containing 1% BSA for ~20 h. Resin was subsequently washed three times with PBS (100 mM NaH2PO4, 150 mM NaCl, pH 7.2) to remove non-specifically bound protein and fluorescent images were obtained using the methodology outlined above. Highly fluorescent beads were manually picked, and stored for peptide sequence determination.
Glycoprotein Removal
To obtain the peptide sequences of SL hits, beads were first treated with alternating acid, base washes to remove the FITC-tagged glycoproteins. This procedure involved tumbling the beads with 70% TFA for 20 min, washing with Screening Buffer for 2 min, then treating them with 0.1 M NaOH for 20 min, followed by a second Screening Buffer wash. This procedure was repeated once, at which point the beads were washed with Screening Buffer three times, and then treated with 1% SDS in water for 1 h at 100 °C.
Partial Edman Degradation
Partial Edman degradation (PED) reactions were performed according to the methods developed by Pei and colleagues.[32] Briefly, a homemade glass reaction vessel measuring 25 mm high with an internal diameter of 10 mm was used for all reactions. One end of the vessel was equipped with a fine frit filter and a Luer tip and the other end with a screw cap. A solution containing Fmoc-O-succinimide (Fmoc-OSU; 1.67 mg; 5.0 μmol), pyridine (160 μL; 1.99 mmol), and phenyl isothiocyanate (PITC; 18 μL; 0.150 mmol) per PED cycle was prepared (30:1 ratio of PITC:Fmoc-OSU). Approximately 5–10 beads were placed in the reaction vessel and washed with an excess of water, 300 μL of pyridine, and 300 μL of 2:1 pyridine in water containing 0.1% triethylamine (TEA). An aspirator with an attached solvent trap was used to evacuate the reaction vessel after each wash. The reaction vessel was removed from suction and the beads were suspended in 160 μL of the 2:1 pyridine in water solution containing 0.1% TEA. To this was added 160 μL of the PITC:Fmoc-OSU solution and the vessel was agitated on a vortex for six minutes. After draining the solution, the beads were washed with 300 μL of pyridine, an excess of dichloromethane (CH2Cl2), and 300 μL of TFA. The vessel was removed from suction and the beads were suspended in 600 μL of TFA and agitated on the vortex for six minutes. The solution was removed and the beads were again suspended in 600 μL of TFA for another six minute agitation. Again the solution was removed and the beads were washed with an excess of CH2Cl2 and 300 μL of pyridine. This procedure was repeated n times to generate the desired peptide ladder. The beads were then treated with 600 μL of 20% piperidine in DMF for five minutes with agitation in order to remove the Fmoc groups. The solution was drained and the beads were washed with 600 μL of DMF. This deprotection step was then repeated.
Beads were resuspended in TFA (0.5 mL) containing dimethyl sulfide (10 μL, 0.135 mmol) and ammonium iodide (12.5 mg, 0.09 mmol) to assure that the methionine was reduced. The beads were then incubated in an ice bath for 20 min, at which point the solution was drained, and the beads were washed three times with water. Individual beads were then transferred to separate microcentrifuge tubes and treated with 50 μL of 40 mg/mL CNBr in 0.1 M HCl for 12–16 h in the dark. The bead was then removed and excess CNBr, TFA, and water were removed under reduced pressure using an AES 1010 SpeedVac system. The cleaved SLs were redissolved in 10 μL of 0.1% TFA in water. For MS analyses, 1 μL of matrix containing saturated α–cyano-4-hydroxycinnamic acid (CHCA) in 50:50:0.1 acetonitrile/water/TFA was applied on to a MALDI plate, and then mixed with 2 μL of the cleaved SL solution. Samples were then analyzed by MALDI-TOF MS.
PBA Removal and MS/MS analysis
To afford the removal of the boronic acid, the beads were incubated with 100 mM H2O2 in water at 50 °C for 1 h. After washing with water three times, the peptide was cleaved from the resin as described previously and analyzed by MALDI-TOF MS and MS/MS techniques. Sequences were identified manually by comparing the mass differences between two adjacent MS/MS peaks.
Dab Fixed Position Library Hit Validation
To examine selectivity trends, hits were synthesized manually using Fmoc strategies as described above. Four individual portions of the beads (2 mg) were equilibrated in Screening Buffer for 10 min, and then tumbled with 1% BSA in Screening Buffer. Library beads (2 mg), used to normalize fluorescence intensity, were treated identically. Each of the four tubes was then individually incubated with 1.0 mL of 0.1 mg/ml FITC-OVA, BSM, PSM, or BSA for 20 h at 23 °C, at which point the beads were washed three times with PBS. Images of the resin were taken with a fluorescent microscope and data analyzed, as described above.
Screening of sSL2
Portions of SL2 were screened as previously described against 0.1 mg/mL FITC-OVA in Screening Buffer containing varying concentrations of sSL2 (i.e., 0, 1, 50, 100, 500, 1000, 5000, 10000 μM). Images were analyzed as previously described and the luminosity of resin incubated with FITC-OVA and sSL2 divided by the luminosity of resin only treated with FITC-OVA to provide a value for fraction bound.
Data Analysis
Percent Change in luminosity was calculated using eq 2.1,
| eq 2.1 |
where A equals the average luminosity of the library and B equals the average luminosity of resynthesized SLs (n=20). A selectivity factor was then determined by dividing each percent increase by the lowest percent increase for a particular SL.
Acknowledgments
This work was supported by funds provided from NIH COBRE grant P20RR17698. This work is dedicated to the memory of Dr. Marc S. Maynor.
References
- 1.Dube DH, Bertozzi CR. Glycans in cancer and inflammation--potential for therapeutics and diagnostics. Nat Rev Drug Discov. 2005;4:477–488. doi: 10.1038/nrd1751. [DOI] [PubMed] [Google Scholar]
- 2.Baldus SE, Zirbes TK, Monig SP, Engel S, Monaca E, Rafiqpoor K, Hanisch FG, Hanski C, Thiele J, Pichlmaier H, Dienes HP. Histopathological subtypes and prognosis of gastric cancer are correlated with the expression of mucin-associated sialylated antigens: Sialosyl-Lewis(a), Sialosyl-Lewis(x) and sialosyl-Tn. Tumour Biol. 1998;19:445–453. doi: 10.1159/000030036. [DOI] [PubMed] [Google Scholar]
- 3.Fuster MM, Brown JR, Wang L, Esko JD. A disaccharide precursor of sialyl Lewis X inhibits metastatic potential of tumor cells. Cancer Res. 2003;63:2775–2781. [PubMed] [Google Scholar]
- 4.Ogawa J-i, Sano A, Koide S, Shohtsu A. Relation between recurrence and expression of proliferating cell nuclear antigen, sialyl Lewis X, and sialyl Lewis a in lung cancer. J Thorac Cardiovasc Surg. 1994;108:329–336. [PubMed] [Google Scholar]
- 5.Nakamori S, Kameyama M, Imaoka S, Furukawa H, Ishikawa O, Sasaki Y, Izumi Y, Irimura T. Involvement of carbohydrate antigen sialyl Lewis(x) in colorectal cancer metastasis. Dis Colon Rectum. 1997;40:420–431. doi: 10.1007/BF02258386. [DOI] [PubMed] [Google Scholar]
- 6.Dube DH, Bertozzi CR. Glycans in cancer and inflammation--potential for therapeutics and diagnostics. Nat Rev Drug Discov. 2005;4:477–488. doi: 10.1038/nrd1751. [DOI] [PubMed] [Google Scholar]
- 7.Hollingsworth MA, Swanson BJ. Mucins in cancer: protection and control of the cell surface. Nat Rev Cancer. 2004;4:45–60. doi: 10.1038/nrc1251. [DOI] [PubMed] [Google Scholar]
- 8.Magnani JL. The discovery, biology, and drug development of sialyl Lea and sialyl Lex. Arch Biochem Biophys. 2004;426:122–131. doi: 10.1016/j.abb.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 9.Nakagoe T, Sawai T, Tsuji T, Jibiki MA, Nanashima A, Yamaguchi H, Yasutake T, Ayabe H, Arisawa K. Preoperative serum level of CA19-9 predicts recurrence after curative surgery in node-negative colorectal cancer patients. Hepatogastroenterology. 2003;50:696–699. [PubMed] [Google Scholar]
- 10.Wang WS, Lin JK, Lin TC, Chiou TJ, Liu JH, Yen CC, Chen WS, Jiang JK, Yang SH, Wang HS, Chen PM. Tumor marker CEA in monitoring of response to tegafur-uracil and folinic acid in patients with metastatic colorectal cancer. Hepatogastroenterology. 2002;49:388–392. [PubMed] [Google Scholar]
- 11.Fakih MG, Aruna P. CEA monitoring in colorectal cancer. What you should know. Oncology. 2006;20:579–587. [PubMed] [Google Scholar]
- 12.Magnani JL, Steplewski Z, Koprowski H, Ginsburg V. Identification of the gastrointestinal and pancreatic cancer-associated antigen detected by monoclonal antibody 19-9 in the sera of patients as a mucin. Cancer Res. 1983;43:5489–5492. [PubMed] [Google Scholar]
- 13.Koprowski H, Herlyn M, Steplewski Z, Sears HF. Specific antigen in serum of patients with colon carcinoma. Science. 1981;212:53–55. doi: 10.1126/science.6163212. [DOI] [PubMed] [Google Scholar]
- 14.Walker DB, Joshi G, Davis AP. Progress in biomimetic carbohydrate recognition. Cell Mol Life Sci. 2009;66:3177–3191. doi: 10.1007/s00018-009-0081-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jin S, Cheng Y, Reid S, Li M, Wang B. Carbohydrate recognition by boronolectins, small molecules, and lectins. Med Res Rev. 2010;30:171–257. doi: 10.1002/med.20155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.James TD, Samankumara KRAS, Shinkai S. Saccharide Sensing with Molecular Receptors Based on Boronic Acid. Angew Chem, Int Ed Engl. 1997;35:1911–1922. [Google Scholar]
- 17.James TD, Shinkai S. Artificial Receptors as Chemosensors for Carbohydrates. Top Curr Chem. 2002;218:159–200. [Google Scholar]
- 18.Lavigne JJ, Anslyn EV. Teaching Old Indicators New Tricks: A Colorimetric Chemosensing Ensemble for Tartrate/Malate in Beverages. Angew Chem Int Ed. 1999;38:3666–3669. [PubMed] [Google Scholar]
- 19.Li M, Lin N, Huang Z, Du L, Altier C, Fang H, Wang B. Selecting aptamers for a glycoprotein through the incorporation of the boronic acid moiety. J Am Chem Soc. 2008;130:12636–12638. doi: 10.1021/ja801510d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamamoto M, Takeuchi M, Shinkai S. Molecular Design of a PET-based Chemosensor for Uronic Acids and Sialic Acids Utilizing a Cooperative Action of Boronic Acid and Metal Chelate. Tetrahedron. 1998;54:3125–3140. [Google Scholar]
- 21.Yang W, Fan H, Gao XGS, Karnati VVRNW, Hooks WB, Carson J, Weston B, Wang B. The First Fluorescent Diboronic Acid Sensor Specific for Hepatocellular Carcinoma Cells Expressing Sialyl Lewis X. Chem Biol. 2004;11:439–448. doi: 10.1016/j.chembiol.2004.03.021. [DOI] [PubMed] [Google Scholar]
- 22.James TD, Phillips MD, Shinkai S. Boronic acids in saccharide recognition. Royal Society of Chemistry; Cambridge, UK: 2006. [Google Scholar]
- 23.Jin S, Cheng Y, Reid S, Li M, Wang B. Carbohydrate recognition by boronolectins, small molecules, and lectins. Med Res Rev. 2010;30:171–257. doi: 10.1002/med.20155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duggan PJ, Offermann DA. Remarkably selective saccharide recognition by solid-supported peptide boronic acids. Tetrahedron. 2009;65:109–114. [Google Scholar]
- 25.Pal A, Berube M, Hall DG. Design, synthesis, and screening of a library of peptidyl bis(boroxoles) as oligosaccharide receptors in water: identification of a receptor for the tumor marker TF-antigen disaccharide. Angew Chem Int Ed Engl. 2010;49:1492–1495. doi: 10.1002/anie.200906620. [DOI] [PubMed] [Google Scholar]
- 26.James TD, Shinmori H, Shinkai S. Novel fluorescence sensor for ‘small’ saccharides. Chem Commun. 1997:71–72. [Google Scholar]
- 27.Zou Y, Broughton DL, Thompson PR, Lavigne JJ. Peptide Borono-Lectins (PBLs): A New Tool for Glycomics and Cancer Diagnostics. Chem BioChem. 2007;8:2048–2051. doi: 10.1002/cbic.200700221. [DOI] [PubMed] [Google Scholar]
- 28.Kranaster R, Marx A. Taking fingerprints of DNA polymerases: Multiplex enzyme profiling of DNA arrays. Angew Chem, Int Ed. 2009;48:4625–4628. doi: 10.1002/anie.200900953. [DOI] [PubMed] [Google Scholar]
- 29.Lin C, Katilius E, Liu Y, Zhang J, Yan H. Self-assembled signaling aptamer DNA arrays for protein detection. Angew Chem, Int Ed. 2006;45:5296–5301. doi: 10.1002/anie.200600438. [DOI] [PubMed] [Google Scholar]
- 30.Marx J. DNA arrays reveal cancer in its many forms. Science. 2000;289:1670–1672. doi: 10.1126/science.289.5485.1670. [DOI] [PubMed] [Google Scholar]
- 31.Edwards NY, Sager TW, McDevitt JT, Anslyn E. Boronic acid based peptidic receptors for pattern-based saccharide sensing in neutral aqueous media, an application in real-life samples. J Am Chem Soc. 2007;129:13575–13583. doi: 10.1021/ja073939u. [DOI] [PubMed] [Google Scholar]
- 32.Thakkar A, Wavreille AS, Pei D. Traceless capping agent for peptide sequencing by partial edman degradation and mass spectrometry. Anal Chem. 2006;78:5935–5939. doi: 10.1021/ac0607414. [DOI] [PubMed] [Google Scholar]
- 33.Simon J, Salzbrunn S, Prakash GKS, Petasis NA, Olah GA. Regioselective conversion of arylboronic acids to phenols and subsequent coupling to symmetrical diaryl ethers. J Org Chem. 2001;66:633–634. doi: 10.1021/jo0015873. [DOI] [PubMed] [Google Scholar]
- 34.Antonio MD, Doria F, Richter SN, Bertipaglia C, Mella M, Sissi C, Palumbo M, Freccero M. Quinone methides tethered to naphthalene diimides as selective G-quadruplex alkylating agents. J Am Chem Soc. 2009;131:13132–13141. doi: 10.1021/ja904876q. [DOI] [PubMed] [Google Scholar]
- 35.Schneider SE, O’Neil SN, Anslyn EV. Coupling rational design with libraries leads to the production of an ATP selective chemosensor. J Am Chem Soc. 2000;122:542–543. [Google Scholar]
- 36.Simpson LS, Burdine L, Dutta AK, Feranchak AP, Kodadek T. Selective toxin sequestrants for the treatment of bacterial infections. J Am Chem Soc. 2009;131:5760–5762. doi: 10.1021/ja900852k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Udugamasooriya DG, Dineen SP, Brekken RA, Kodadek T. A peptoid “antibody surrogate” that antagonizes VEGF Receptor 2 activity. J Am Chem Soc. 2008;130:5744–5752. doi: 10.1021/ja711193x. [DOI] [PubMed] [Google Scholar]
- 38.Manku S, Hall DG. Synthesis, decoding, and preliminary screening of a bead-supported split-pool library of triboronic acid receptors for complex oligosaccharides. Aust J Chem. 2007;60:824–828. [Google Scholar]
- 39.Gareiss PC, Sobczak K, McNaughton BR, Palde PB, Thornton CA, Miller BL. Dynamic combinatorial selection of molecules capable of inhibiting the (CUG) repeat RNA-MBNL1 interaction in vitro: Discovery of lead compounds targeting myotonic dystrophy (DM1) J Am Chem Soc. 2008;130:16254–16261. doi: 10.1021/ja804398y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McNaughton BR, Gareiss PC, Miller BL. Identification of a selective small-molecule ligand for HIV-1 frameshift-inducing stem-loop RNA from a 11,325 member resin bound dynamic combinatorial library. J Am Chem Soc. 2007;129:11306–11307. doi: 10.1021/ja072114h. [DOI] [PubMed] [Google Scholar]