

Abstract
Background
During the transition to alcohol and drug addiction, neuromodulator systems in the extended amygdala are recruited to mediate aspects of withdrawal and relapse via convergence on inhibitory GABA neurons in central amygdala (CeA).
Methods
This study investigated the role of neuropeptide Y (NPY) in excessive alcohol drinking by making rats dependent on alcohol via alcohol vapor inhalation. This study also utilized intracellular and whole-cell recording techniques to determine the effects of NPY on GABAergic inhibitory transmission in CeA, synaptic mechanisms involved in these NPY effects, and NPY interactions with alcohol in the CeA of alcohol-naïve and alcohol-dependent rats.
Results
Chronic NPY treatment blocked excessive operant alcohol-reinforced responding associated with alcohol dependence, as well as gradual increases in alcohol responding by intermittently tested non-dependent controls. NPY decreased baseline GABAergic transmission and reversed alcohol-induced enhancement of inhibitory transmission in CeA by suppressing GABA release via actions at presynaptic Y2 receptors.
Conclusions
These results highlight NPY modulation of GABAergic signaling in central amygdala as a promising pharmacotheraputic target for the treatment of alcoholism. GABA neurons in the CeA likely constitute a major point of convergence for neuromodulator systems recruited during the transition to alcohol dependence.
Keywords: Central amygdala, Y2 receptor, Y1 receptor, BIIE0246, BIBP3226, Negative reinforcement
INTRODUCTION
Alcoholism, or alcohol dependence, is a progressive and chronically relapsing disorder. The development of alcoholism is characterized by frequent episodes of intoxication, preoccupation with alcohol and the use of alcohol despite adverse consequences, compulsive alcohol-seeking behavior, loss of control limiting intake, and a negative emotional state in the absence of alcohol (1). A major goal of basic research on alcoholism is to understand the neural underpinnings of alcohol use and the pathological progression to alcohol dependence (2).
The transition from casual drinking to alcohol dependence involves numerous neuroadaptive changes in brain reward and stress systems. The recruitment of brain stress systems contributes to the aversive aspects of alcohol withdrawal and, along with dysregulation of brain reward systems, promotes alcohol-seeking behaviors and relapse (3). Brain stress systems in the “extended amygdala” (4) appear to be especially important in mediating motivational deficits associated with alcohol withdrawal and excessive alcohol drinking. The extended amygdala contains the central nucleus of the amygdala (CeA), the lateral portion of the bed nucleus of the stria terminalis (BNST), and a transition area in the nucleus accumbens (NAc) shell. These three regions are heavily interconnected and have high concentrations of stress-related neuropeptides.
Several neuropeptides have prominent roles in the aversive aspects of alcohol withdrawal and relapse via their actions in the CeA. Neuropeptide Y (NPY) is an inhibitory peptide produced in abundance in the hypothalamus and phylogenetically conserved across species (5). NPY is highly co-localized with GABA in the amygdala (6), which is important because NPY reduces anxiety (7) via actions in the amygdala (8,9). Recently, it has been shown that NPY suppresses alcohol drinking in rats (10) via its actions in CeA (11-14). More specifically, NPY microinjection into the CeA exhibits an enhanced ability to suppress alcohol drinking in certain subpopulations of drinkers, including rats that are made dependent on alcohol via vapor inhalation.
In vitro electrophysiological studies have revealed that acute alcohol facilitates spontaneous and evoked GABAergic transmission in the CeA via presynaptic and postsynaptic mechanisms (15). In CeA neurons from rats exposed to chronic alcohol vapor, baseline GABAergic transmission is increased and CeA neurons do not exhibit tolerance to the facilitatory effects of acute alcohol (16). Recent data have highlighted the effects of various neuropeptides on alcohol-induced facilitation of inhibitory transmission in the CeA of rats. For example, CRF facilitates (17) and nociceptin opposes (18) GABAergic transmission in the CeA.
In the present study, we tested the hypothesis that NPY has a key role in the development of excessive alcohol drinking during the transition to alcohol dependence. We also examined the effects of NPY on baseline GABAA-mediated inhibitory transmission in the CeA and the interactions of NPY and acute alcohol in alcohol-naïve and alcohol-dependent rats, as well as the synaptic mechanisms and NPY receptor subtypes responsible for these effects. We report that NPY administration blocks the development of excessive alcohol drinking associated with the transition to alcohol dependence, and that NPY opposes alcohol effects on GABAergic transmission in CeA, likely via activation of presynaptic Y2 receptors.
METHODS AND MATERIALS
Behavioral Studies
Animals
We used forty-two adult male Wistar rats (Charles River, Kingston, NY). The average body weight before vapor exposure was 541.7+21.4 grams. Animals were single-housed in standard plastic cages with wood chip bedding under a 12 hr light/12 hr dark cycle (lights off at 8 AM) with ad libitum access to food and water throughout except during experimental drinking sessions. All procedures were conducted in the dark cycle and met the guidelines of the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Operant Alcohol Self-administration Training
Operant boxes and training procedures have been described previously (19). Briefly, we trained Wistar rats to respond on a continuous fixed ratio-1 (FR1) schedule for 0.1 ml deliveries of “supersaccharin” (3% glucose and 0.125% saccharin; see (20) versus water in a concurrent, two-lever, free-choice contingency during 30-min sessions. Following these operant training sessions, 10% (w/v) ethanol was added and sweeteners gradually removed from the solution. Upon completion of this fading procedure, we allowed rats 25 operant responding sessions for 10% (w/v) ethanol versus water to stabilize operant responding. Rats were stereotaxically implanted with cannulae, and subsequently divided into four groups based on mean intakes during four post-surgery pre-vapor operant self-administration sessions: 1) chronic alcohol vapor exposure and NPY infusion (dependent-NPY, n=7); 2) chronic alcohol vapor exposure and vehicle infusion (dependent-aCSF; n=8); 3) chronic air vapor exposure and NPY infusion (non-dependent-NPY; n=7); and 4) chronic air vapor exposure and vehicle infusion (non-dependent-aCSF; n=8).
Stereotaxic Surgeries
We surgically implanted intracerebroventricular cannulae using aseptic procedures as previously described (11), after isoflurane anesthesia (Abbott Laboratories, North Chicago, IL), A guide cannula (22 gauge) was unilaterally implanted according to the appropriate stereotaxic coordinates (21), with a dummy cannula (28 gauge) in the guide cannula at all times except during infusions. We monitored rats for a one-week recovery period to determine that animals resumed normal activity.
Microinfusions
We used a Harvard 33 microinfusion pump for all infusions into the lateral ventricles (ICV) at a rate of 2.5μl/minute for two minutes, and left the injection cannula in place one additional minute to allow for adequate diffusion. We delivered infusions via polyethylene tubing (PE 20) connected to a Hamilton 25 μl syringe.
Operant Tests During Alcohol Vapor Exposure
We infused dependent and non-dependent rats with NPY or vehicle and tested them for operant behavior at the 6-hr withdrawal time point, on multiple days during the first 15 days of chronic intermittent alcohol vapor exposure (CIE; see Supplement). Rats in the dependent groups were exposed to alcohol vapor 14 hrs/day (vapor off at 8 a.m.); rats in the non-dependent groups were exposed to air vapor 24 hrs/day. Operant tests and NPY infusions never occurred on the same day. On days 2, 4, 6, 8, 10, 12, and 14 of CIE), rats received either 0.0 μg or 10.0 μg NPY ICV (Sigma-Aldrich, St. Louis, MO) in 5.0 μl aCSF. On days 3, 7, 11, and 15 of CIE, we tested rats for operant alcohol responding. On day 16 of CIE, rats were sacrificed and cannula placements histologically verified. The 10.0 μg NPY dose was chosen based on prior studies that showed that acute ICV infusion of this dose reliably suppresses ethanol drinking in rats (10, 12).
Statistical Analysis
We analyzed operant responses, ethanol consumption (g ethanol/kg body weight) and ethanol preference (ethanol consumed/total fluid consumed) using 3-way repeated-measures analyses of variance (RM ANOVAs), where between-subjects factors were vapor exposure (alcohol vapor vs. air vapor) and NPY dose (0.0μg vs. 10.0μg), and day was the within-subjects factor (baseline vs. operant tests on vapor days 3, 7, 11, and 15). Because a priori differences were not expected early in alcohol vapor exposure, we also analyzed operant test days individually to determine when vapor effects and/or NPY effects manifested during the transition to alcohol dependence. We made post-hoc comparisons using the Student Newman-Keuls test and set statistical significance at p<0.05.
Electrophysiological Studies
Slice Preparation
Following ≥2 weeks of CIE (14 hrs/day) or air vapor exposure (see Supplement), we prepared CeA slices as previously described (14,15). Briefly, male Sprague-Dawley rats (120-300 g; 4-7 weeks old, n=57) were removed from vapor chambers, anesthetized with halothane (3%), and decapitated. We cut 400 μm coronal slices on a Vibratome Series 3000 (Technical Products International), incubated them in an interface configuration for 30 min, then completely submerged and continuously superfused (flow rate of 2-4 ml/min) them with warm (31° C), gassed artificial cerebrospinal fluid (aCSF) of the following composition in mM: NaCl, 130; KCl, 3.5; NaH2PO4, 1.25; MgSO4•7H2O, 1.5; CaCl2, 2.0; NaHCO3, 24; glucose, 10. Drugs were added to the aCSF from stock solutions to obtain known concentrations in the superfusate.
Intracellular Recording of IPSPs in CeA Neurons
We recorded in alcohol-free aCSF from CeA neurons (n=64) of alcohol-naïve or alcohol-dependent rats for 2-8 hours after cutting, as previously described (15). We recorded with sharp micropipettes (3M KCl) using current-clamp mode, holding potentials near resting membrane potential (mean Vm = −77 mV). Data were acquired with an Axoclamp-2A preamplifier and pClamp software (Axon Instruments). We used a bipolar stimulating electrode to evoke pharmacologically-isolated GABAA receptor-mediated inhibitory postsynaptic potentials (IPSPs) while superfusing the slices with the glutamate receptor blockers 6,7-dinitroquinoxaline-2,3-dione (DNQX; 10 μM) and DL-2-amino-5-phosphonovalerate (APV; 30 μM), and the GABAB receptor antagonist CGP 55845A (1 μM). To determine the IPSP response parameters for each cell, we performed an input-output (I-O) protocol (14,15). The stimulus strengths were maintained throughout the duration of the experiment. We normalized three stimulus intensities of equal steps (threshold, half-maximal and maximal) as 1-3X.
We examined paired-pulse facilitation (PPF) using paired stimuli at 50 msec inter-stimulus intervals (15). The amplitude of the first IPSP was 50% of the maximal stimulus strength, as determined from the I-O relationship. We calculated the PPF ratio as the amplitude of the second IPSP divided by that of the first IPSP. All measures were taken before the first drug superfusion (control), during superfusion (15-20 min per drug), and following washout (~15 min). To avoid tachyphylaxis, we superfused each drug only once onto a single cell.
Whole-Cell Patch-Clamp Recording of mIPSCs in CeA Neurons
We visualized CeA neurons in brain slices (350-400μm) using infrared differential interference contrast (IR-DIC) optics and CCD camera (EXi Aqua, QImaging). We used a w60 water immersion objective (Olympus) for identifying and approaching CeA neurons. Whole-cell voltage-clamp recordings were made with a Multiclamp 700B amplifier (Molecular Devices), low-pass filtered at 2-5kHz, digitized (Digidata 1440A; Molecular Devices), and stored on a PC using pClamp 10 software (Axon Instruments). Patch pipettes (4-8MΩ) were pulled from borosilicate glass (Warner Instruments) and filled with an internal solution composed of (in mM): potassium chloride (KCl) 145; EGTA 0.5; MgCl2 2; HEPES 10; Na-ATP 2; Na-GTP 0.2. Series resistance (<10 MΩ) was continuously monitored with a 10mV hyperpolarizing pulse. Drugs were constituted in aCSF and applied by bath superfusion. Recordings were performed in the presence of DNQX (10μM), APV (30μM), CGP55845A (1μM) and tetrodotoxin (1μM) to isolate GABAA receptor mIPSCs. All cells were clamped at −60mV for the duration of the recording.
Statistical Analysis
Data were analyzed with 1-way and 2-way between-subjects ANOVA or repeated-measures (RM) ANOVA and, when appropriate, with the Student Newman-Keuls post hoc test, with p<0.05 considered statistically significant. Because there were unequal error variances between groups and there was a significant positive correlation between means and variance of I/O data, we performed a log-transformation on all I/O data prior to analysis. In some cases, we used independent-samples or paired-samples t-tests for comparing individual pairs of means. Means reported in figures represent measurements at the end of particular drug infusion periods.
RESULTS
Behavioral Studies
Effects of Alcohol Vapor Exposure on Operant Behavior
To track the effects of alcohol vapor on operant behavior over time (i.e., the transition to alcohol dependence), we analyzed data only from vapor-exposed and air-exposed aCSF-infused rats. Two-way vapor history × day RM ANOVAs indicated that over time, there was a global increase in alcohol responding, F(3,57)=3.62, p<0.05, and alcohol consumption (g/kg), F(3,57)=4.32, p<0.01, which was attributable to the gradual increase in responding and consumption by CIE vapor-exposed rats (Table S1 in the Supplement). CIE-exposed rats consumed more ethanol (g/kg) than air-exposed rats across days, F(1,19)=4.45, p<0.05. The ethanol response rates and consumption quantities observed following 11 and 15 days of CIE exposure (in aCSF controls) reliably produce BALs of approximately 100 mg/dl in alcohol-dependent animals (19). There were no effects of dependence history or day on water responding in aCSF-treated rats (p>0.05).
Effects of NPY on Operant Behavior
Figure 1A shows operant alcohol responding by CIE- and air-exposed rats infused chronically with NPY or aCSF and tested across the first 15 days of vapor exposure. A 3-way (vapor exposure × NPY dose × day) RM ANOVA revealed a significant suppression of operant alcohol responding across days by chronic NPY infusions, F(1,38)=6.23, p=0.017. There was no 3-way interaction effect (p>0.05), but there was a significant dependence × day interaction effect on operant alcohol responding, F(3,114)=3.60, p=0.016. Two-way (vapor exposure × NPY dose) ANOVAs for each of the 4 test days (vapor days 3, 7, 11, 15) revealed that NPY suppressed operant alcohol responding on day 15 of vapor exposure, F(1,38)=5.84, p<0.05, and that there was a tendency toward a suppressive NPY effect on alcohol responding on days 3 (p=0.06) and 11 (p=0.07) of vapor exposure.
Figure 1.
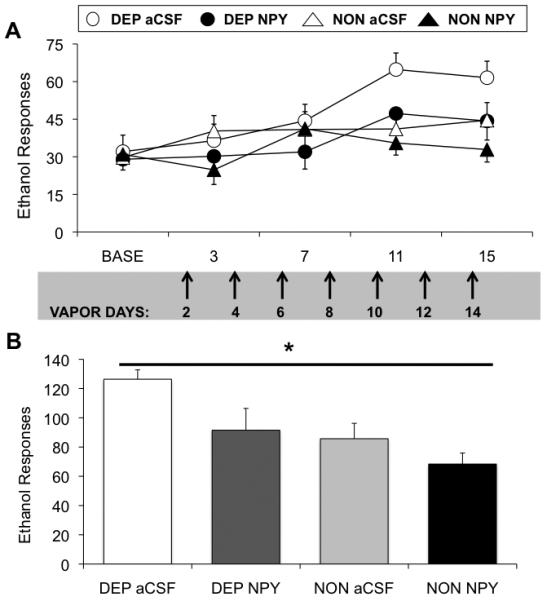
Chronic NPY administration in a clinically relevant treatment regimen blocks gradual and cumulative elevations in alcohol drinking similarly in alcohol-dependent and –non-dependent rats. A: Chronic NPY treatment blocks the development of alcohol dependence-induced increases in alcohol responding (n=11/vapor-exposed group), and also blocks moderate increases in alcohol drinking by non-dependent rats (n=10/air-exposed group) over time. Rats were infused in the ventricles with NPY (10μg) or vehicle on even-numbered days of vapor exposure (indicated by arrows), and tested for responding at 6-8-hr withdrawal on days 3, 7, 11, and 15 of vapor exposure. B: Chronic NPY treatment suppresses cumulative alcohol responding across operant test sessions on vapor days 11 and 15. * indicates significant (p<0.05) suppression by NPY relative to aCSF vehicle regardless of vapor history (main effect of NPY).
A separate 2-way (vapor exposure × NPY dose) RM ANOVA for cumulative alcohol responding on days 11 and 15 of vapor exposure (Figure 1B) indicated a significant suppression of operant alcohol responding by NPY, F(1,38)=5.84, p=0.021. Finally, a 3-way (vapor exposure × NPY dose × day) RM ANOVA revealed that alcohol-dependent rats responded less for water across days, F(1,38)=7.82, p=0.008, but water responding was not affected by chronic NPY infusions (p>0.05). Two-way (vapor exposure × NPY dose) ANOVAs for each of the 4 test days (vapor days 3, 7, 11, 15) revealed no significant effects of NPY on water responding (p>0.05 in all cases).
Electrophysiology Studies
NPY & Ethanol Effects on IPSPs
As previously described (15), baseline GABAA-IPSP input-output curves were higher (data not shown) and basal PPF ratio of IPSPs was lower in slices from dependent rats relative to naïve control rats, suggesting an increased GABAergic tone via GABA release (statistics below and in figure captions).
We first applied ethanol alone, then concomitantly applied NPY to CeA slices from vapor-exposed rats and naïve controls (time course in Figure 2A). As previously demonstrated (14), acute ethanol (44 mM) produced robust 35-45% (p<0.001) increases in IPSP amplitudes (Figures 3A and 3B). Subsequent application of NPY (0.5 μM) returned IPSPs to baseline levels (p=0.025 difference from ethanol; p>0.05 no difference from baseline), suggesting that NPY reverses ethanol-induced enhancement of inhibitory transmission in CeA.
Figure 2.
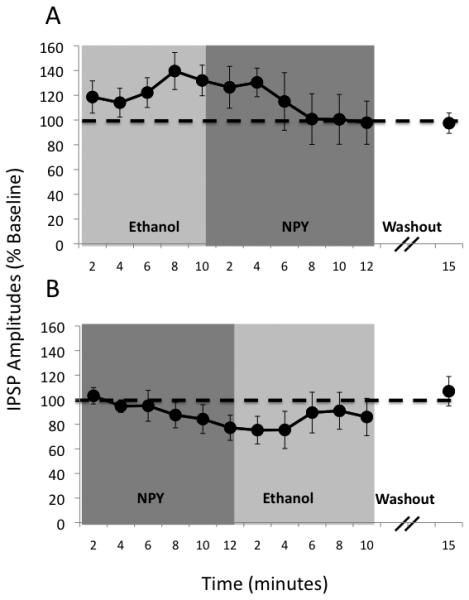
Time course of mean ± SEM IPSP amplitudes (% change from baseline) in CeA of alcohol-naïve rats during ethanol application followed by NPY application (A; n=8) and also during NPY application followed by ethanol application (B; n=8). NPY reversed (A) and prevented (B) ethanol-induced facilitation of inhibitory transmission in CeA.
Figure 3.

NPY prevents and reverses ethanol-induced increases in GABAergic transmission via presynaptic release. A: In CeA neurons from alcohol-naïve rats, acute ethanol increases GABAergic transmission in CeA neurons, and subsequent NPY reverses this effect. Top Panel: Representative evoked IPSPs in a CeA neuron. Bottom Panel: In CeA neurons, ethanol alone (n=9) significantly (p<0.05) increases mean IPSP amplitudes to 136% of control (at stimulus intensity = ½ maximal IPSP amplitude). NPY in the presence of ethanol abolishes the ethanol-induced facilitation of evoked IPSP amplitude. B: Top Panel: Representative evoked IPSPs in a CeA neuron from an alcohol-dependent rat. Bottom Panel: Ethanol alone (n=10) increases mean IPSP amplitudes to 145% of control. Subsequent NPY reverses this ethanol effect. C: Ethanol significantly reduces the mean PPF ratio (at 50 msec interval) of IPSPs in CeA of naïve and chronic vapor-exposed rats (p<0.05) suggesting increased GABA release (i.e., changes in PPF ratio are inversely related to transmitter release). NPY completely blocks this ethanol effect on PPF, (p>0.05 relative to baseline), with recovery on washout. Note that baseline PPF in chronic vapor-exposed rats is significantly (# p<0.05) lower than baseline PPF in alcohol-naïve rats, (p<0.05). D: NPY slightly decreases basal GABAergic transmission in CeA and blocks ethanol-induced enhancement of IPSPs. Top panel: Evoked IPSPs in a CeA neuron from a naïve rat. Bottom panel: In CeA neurons, NPY alone (n=8) decreases the mean amplitudes of evoked IPSPs to 90% of control and prevents the enhancement of IPSPs induced by subsequent ethanol (compare to panel A). E: Top panel: Representative evoked IPSPs in a CeA neuron from an alcohol -dependent rat. Bottom panel: NPY alone (n=7) decreases the mean IPSP amplitudes to ~85% of control and prevents the enhancement of IPSPs induced by subsequent ethanol (compare to panel B). F: NPY does not significantly affect the 50-ms PPF ratio (p>0.05) relative to baseline. Ethanol added to NPY does not affect the 50-ms PPF ratio (p>0.05) relative to baseline. Note that baseline PPF in alcohol-dependent rats is significantly (# p< 0.05) lower than PPF in alcohol-naïve rats.
We applied NPY alone, followed by concurrent application of ethanol, (time course in Figure 2B). NPY alone did not significantly affect IPSPs (p>0.05), but blocked the ethanol-induced augmentation of IPSP amplitudes (p>0.05 relative to baseline; see Figures 3D & 3E), and these effects were similar in vapor-exposed rats and naïve controls. Longer (22 min) application of NPY alone produced a persistent decrease in IPSP amplitudes (Figure S2 in the Supplement) that returned to baseline levels 20 minutes into washout. These results suggest that NPY blocks ethanol-induced enhancement of inhibitory transmission in CeA.
NPY & Ethanol Effects on PPF of IPSPs
In CeA slices from both vapor-exposed rats and naïve controls, acute ethanol decreased the PPF ratio by 10-20% (p=0.04; Figure 3C) (15). NPY application reversed this ethanol-induced decrease in PPF ratio (p>0.05; Figure 3C) to baseline levels. Vapor-exposed rats exhibited significantly lower baseline PPF relative to naïve rats (p=0.02), suggesting increased baseline GABA release in vapor-exposed rats (Figures 3C & 3F). These results confirm that acute ethanol augments CeA GABA transmission via increased GABA release (and a lack of tolerance to this effect in alcohol vapor-exposed animals), and suggest that NPY reverses acute ethanol effects via a presynaptic mechanism.
In both vapor-exposed rats and naïve controls, NPY alone did not affect PPF (p>0.05; Figure 3F) but blocked the ethanol effect on PPF (p>0.05; Figure 3F), suggesting that NPY blocked the ability of ethanol to increase GABA release in CeA. These results again suggest that NPY blocks ethanol effects via a presynaptic mechanism.
Y1- and Y2-receptor Antagonist Effects on IPSPs in CeA from Naïve Rats
To investigate the NPY receptor subtypes responsible for NPY blockade of ethanol effects on GABAergic transmission in CeA, we performed experiments using Y1 and Y2 receptor antagonists.
We applied the Y2 receptor antagonist, BIIE0246 (0.5 μM) – followed by co-application with ethanol – to slices from naïve rats (Figure 4A). Superfusion of BIIE0246 alone produced ~10% increases in CeA IPSP amplitudes relative to baseline (p=0.046). Subsequent application of ethanol produced a further increase in IPSP amplitudes that was 50% greater than baseline (p<0.001) and was also significantly greater than BIIE0246 alone (p=0.009). These results suggest a tonic inhibitory action of NPY on basal and post-ethanol GABAergic transmission that is mediated by Y2 receptors. Furthermore, the fact that ethanol altered IPSPs in the presence of BIIE0246 suggests that ethanol does not produce its effect via direct actions at Y2 receptors.
Figure 4.

A Y2-receptor antagonist, but not a Y1-receptor antagonist, blocks NPY inhibition of ethanol-induced IPSP increase. A: Representative traces and cumulative histograms showing that BIIE0246, a Y2-receptor antagonist, significantly (# p<0.05 relative to baseline) increases the IPSP amplitudes in CeA neurons (n=6) from naïve rats. Ethanol applied in presence of BIIE0246 further increases the amplitudes of IPSPs (* p<0.05 relative to BIIE0246 alone), with recovery during washout. Note on the right the increase in IPSPs produced by acute ethanol alone. B: Representative traces and cumulative histograms showing that in CeA neurons (n=8) from slices pretreated with BIIE0246, NPY slightly decreases IPSP amplitudes and does not block the ethanol-induced IPSP increase (* p<0.05 relative to baseline). Note on the right that NPY blocks ethanol-induced increase of IPSPs in the absence of BIIE0246. C: Representative traces and cumulative histograms showing that BIBP3226, a Y1-receptor antagonist, does not affect IPSP amplitudes in CeA neurons (n=5) from naïve rats. Ethanol applied in presence of BIBP3226 increases the amplitudes of IPSPs (* p<0.05 relative to BIBP3226 alone), with recovery during washout. Note on the right the increase in IPSPs produced by acute ethanol alone. D: Representative traces and cumulative histograms showing that in CeA neurons (n=9) from slices pre-treated with BIBP3226, NPY decreases the amplitude of IPSPs (* p<0.05 relative to baseline) and blocks ethanol-induced increase of IPSPs (p>0.05). Note on the right the increase in IPSPs produced by acute ethanol alone.
In slices from naïve rats pretreated with BIIE0246, ethanol applied in the presence of NPY produced a 50% increase in IPSP amplitude relative to the NPY condition (p=0.01), whereas there was no such effect of ethanol in control slices that did not receive BIIE0246 pretreatment (p>0.05) (Figure 4B). Furthermore, amplitudes of baseline IPSPs (p=0.006) and post-ethanol IPSPs (p<0.001) were larger in slices pretreated with BIIE0246 relative to controls. In the presence of BIIE0246, NPY did not alter the PPF of IPSPs and did not block ethanol-induced decrease in PPF of IPSPs (Figure S1 in the Supplement). These results suggest that the ability of NPY to block ethanol-induced increases in inhibitory transmission in CeA is mediated by Y2 receptors and support the notion that NPY blockade of ethanol effects occurs at presynaptic sites, because Y2 receptors primarily function as presynaptic autoreceptors (22).
We applied the Y1 receptor antagonist, BIBP3226 (0.5 μM) – followed by co-application with ethanol to slices from naïve rats (Figure 4C). Superfusion of BIBP3226 did not (p>0.05) affect baseline IPSP amplitudes and did not (p<0.05) block the ethanol-induced increase in IPSP amplitudes relative to BIBP3226 alone.
In CeA slices from naïve rats pretreated with BIBP3226, NPY alone significantly decreased IPSP amplitudes (p=0.042) (Figure 4D) and blocked the ethanol-induced increase of IPSP amplitudes. In the presence of BIBP3226, NPY also increased by 10% the PPF of IPSPs and blocked ethanol-induced decrease in PPF of IPSPs (Figure S1 in the Supplement). These results suggest that NPY decreases tonic inhibitory transmission in CeA, probably via Y2 receptors, and that this tonic effect is buffered by NPY activity at Y1 receptors. Also, these data suggest that NPY blockade of ethanol effects does not occur at postsynaptic Y1 receptors.
NPY & Ethanol Effects on mIPSCs
To confirm the synaptic site of action for NPY blockade of ethanol effects on GABAergic transmission, we performed whole-cell voltage-clamp recording of mIPSCs in the CeA of naïve rats (Figure 5). In CeA slices from naïve rats, NPY did not (p>0.05) alter either frequency or amplitude of mIPSCs but completely blocked the ethanol-induced increase in mIPSC frequencies. As previously demonstrated (15) ethanol alone significantly (p<0.05) increased mIPSC frequencies but not amplitudes. These data suggest that NPY blocks ethanol-induced facilitation of presynaptic GABA release.
Figure 5.
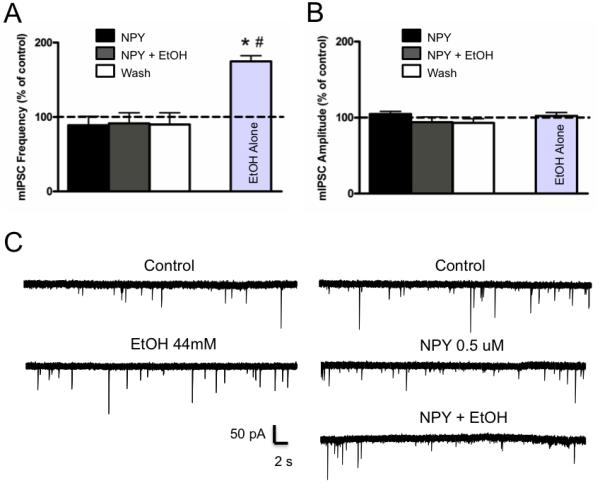
NPY blocks ethanol-induced increase in mIPSC frequency, but neither NPY nor ethanol affect mIPSC amplitude. Mean ± SEM percent change in mIPSC frequency (A) and amplitude (B) following superfusion of NPY (0.5μM), NPY + EtOH (44mM), and wash (n=6) as compared to superfusion of EtOH alone (n=6). NPY blocks ethanol-induced increase in mIPSC frequency relative to baseline condition (* p<0.05) and NPY + EtOH condition (# p<0.05). C: Whole-cell voltage-clamp recordings from a representative CeA neuron depicting mIPSCs during baseline and following superfusion of 44mM EtOH (left panel). Whole-cell voltage-clamp recording from a representative CeA neuron depicting mIPSCs during baseline and following superfusion of 0.5μM NPY and NPY + 44mM EtOH (right).
DISCUSSION
We found that chronic NPY treatment blocks excessive alcohol drinking associated with the development of alcohol dependence, and also suppresses modest increases in alcohol drinking associated with intermittent testing in non-dependent animals. A key aspect of these findings is that rats chronically injected with NPY during the transition to alcohol dependence exhibited a moderate alcohol-drinking phenotype similar to that of vehicle-treated non-dependent control rats. NPY also blocked alcohol-induced facilitation of inhibitory transmission in CeA neurons via actions at presynaptic receptors. These findings identify a locus and mechanism for the involvement of brain NPY systems in pathological and excessive alcohol consumption.
Three additional aspects of our behavioral findings with chronic NPY treatment are worth noting. First, repeated NPY administration not only blocked the development of excessive alcohol consumption in the course of dependence induction, but also tempered the moderate increase in alcohol consumption following periods of abstinence in non-dependent rats (10, 12). Thus, like chronic administration of CRF1R antagonists (17), prolonged NPY administration may prevent excessive alcohol consumption under a variety of behavioral and physiological conditions. However, the fact that chronic NPY also suppressed alcohol drinking by non-dependent rats does not detract from the importance of its ability to abolish excessive alcohol drinking by dependent animals. Chronic infusion of a lower dose may have revealed potential increased sensitivity to the behavioral effects of NPY in dependent rats. Second, NPY exhibits long-term efficacy in suppressing alcohol self-administration, highlighting the potential utility of treatments that target brain NPY systems in a prolonged treatment protocol likely in a clinical setting. Finally, the behavioral effects of ICV NPY persist for 24h post-injection, consistent with previously observed NPY effects (10). This latter finding may be explained by the slow receptor dissociation rate of NPY from Y2 receptors (20% after 8h), a property not shared with Y1 receptors (23).
The ability of NPY to suppress alcohol drinking in rats is mediated by its actions in the CeA. Acute injection of NPY into the CeA suppresses excessive alcohol consumption by alcohol-dependent rats (11) and alcohol-deprived rats (12), and viral vector-induced amygdalar NPY overexpression suppresses alcohol consumption by alcohol-deprived (14) and innately “anxious” rats (13). It has been suggested that anti-anxiety NPY systems are recruited to counteract pro-anxiety CRF effects during the transition to alcohol dependence (24). This is especially interesting because both peptides are abundant in the CeA and their local actions are responsible for mediating effects on anxiety-like behavior.
We previously demonstrated that acute alcohol enhances GABAergic transmission at both pre- and postsynaptic sites in rat CeA slices (15), and dependent rats exhibit increased basal GABA release and no tolerance to acute alcohol effects relative to naïve rats (16). We also showed that CRF increases GABAergic transmission in rat CeA via a presynaptic mechanism (17). Furthermore, CRF1R antagonists block acute alcohol-induced increases in GABAergic transmission in CeA, suggesting a critical role for these receptors in alcohol effects. Here, we sought to identify the role and site of action for NPY effects on GABAergic transmission in rat CeA. NPY slightly decreased basal GABAergic transmission, and also blocked and reversed acute alcohol-induced increases in IPSP amplitude in both naïve and vapor-exposed rats. This finding correlates well with the ability of chronic NPY to suppress excessive alcohol drinking during the transition to dependence as well as moderate increases in alcohol drinking by non-dependent animals during intermittent testing.
Consistent with prior studies (15,16, 17), these data show that alcohol increases IPSP amplitudes and decreases PPF of IPSPs in CeA neurons, suggesting that alcohol increases GABAergic transmission by increasing GABA release. NPY alone did not affect PPF ratio or mIPSC frequency in alcohol-naïve rats, but did reverse acute alcohol-induced decreases in PPF ratio, suggesting that NPY opposes acute alcohol-induced GABA release. Furthermore, acute alcohol applied in the presence of NPY does not affect PPF ratio or mIPSC frequency, again suggesting that NPY prevention/reversal of alcohol-induced increases in GABAergic transmission in CeA occurs at a presynaptic site (see Figure 6). Importantly, NPY normalized alcohol dependence-induced decreases in PPF ratio, but did not affect basal PPF ratios in alcohol-naïve rats, suggesting that alcohol dependence produces neuroadaptations in amygdalar NPY systems that modulate inhibitory transmission in that brain region.
Figure 6.
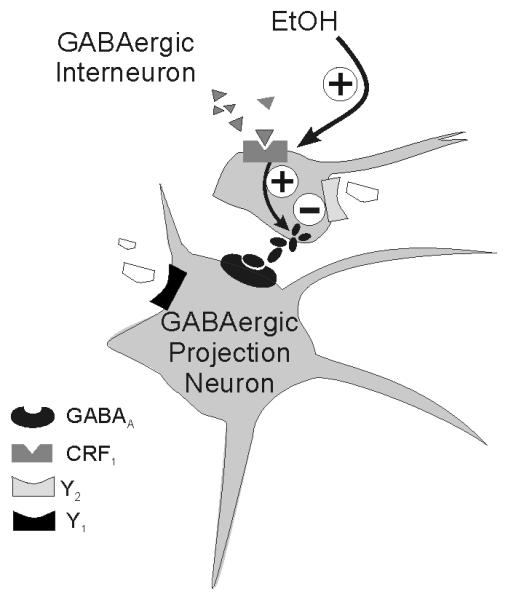
Simplified schematic of rodent CeA circuitry and hypothetical sites of ethanol and NPY action on GABAergic synapses. Most neurons in the CeA are GABAergic inhibitory projection neurons or interneurons that co-transmit GABA and one of several neuromodulators, including NPY and CRF. Ethanol may enhance the release of GABA from GABAergic afferent neurons via direct activation of CRF1 receptors on those afferents (17), but regardless of the mechanism of action, CRF and ethanol both likely inhibit the activity of GABAergic neurons projecting out of CeA. Conversely, activation of presynaptic Y2 receptors by NPY reduces inhibition of GABAergic neurons projecting out of CeA, thereby facilitating the release of GABA onto downstream targets. These results are important because the interplay of CRF and NPY actions in CeA was previously identified to be important for the co-regulation of emotionality and anxiety (42). Therefore, recorded increases in GABAergic transmission (e.g., by CRF and ethanol) likely reflect a disinhibition of downstream target regions (e.g., BNST, hypothalamus, periaqueductal gray), whereas recorded decreases in GABAergic transmission (e.g., by NPY) reflect a net inhibition of downstream target regions. This hypothesis is supported not only by the behavioral effects of NPY in CeA (e.g., decreases in anxiety and decreases in alcohol drinking; 7, 10, 11), but also by recent data regarding CRF (17), nociceptin (18), vasopressin (43), and endocannabinoid (44) effects on cellular function in the rat CeA. Further details of this circuitry remain to be elucidated.
To further assess the mechanism and site of action for NPY, we tested the effects of BIIE0246, a presynaptic Y2Rs antagonist, and BIBP3226, a postsynaptic Y1Rs antagonist, on NPY- and alcohol-induced alterations in CeA GABAergic transmission. BIIE0246 modestly increased evoked IPSP amplitudes, and acute alcohol further increased GABAergic transmission. Pre-treatment with BIIE0246 prevented NPY from blocking alcohol-induced increases in IPSPs. Conversely, pre-treatment with BIBP3226 revealed a tonic suppressive effect of NPY on IPSP amplitude, but did not prevent NPY from blocking acute alcohol effects. In the presence of the Y1R, but not the Y2R antagonist, NPY produced a 10% increase in the PPF of IPSPs, further suggesting a presynaptic Y2R site of NPY action (see Figure S1 in the Supplement). These results agree with findings that NPY suppresses GABAergic transmission in BNST by suppressing GABA release via actions at presynaptic Y2Rs (25), and that presynaptic Y2Rs can function as heteroceptors that modulate the release of other neurotransmitters (26). The compound increase in GABAergic transmission produced by BIIE0246 and alcohol suggest that Y2Rs modulate, but do not directly mediate, alcohol effects. Furthermore, NPY actions at postsynaptic Y1Rs may serve as a “brake” on Y2R effects, perhaps explaining the lack of NPY effects on PPF ratio and mIPSC frequency in alcohol-naïve rats, and suggesting that blockade of Y1Rs unmasks a tonic NPY presynaptic site of action.
Antagonism of Y1Rs in the amygdala suppresses operant alcohol responding in rats (27). Our results provide a possible mechanism for this effect, suggesting that Y1R blockade eliminates the brake on tonic NPY action at Y2Rs, potentially resulting in suppression of alcohol consumption. A wealth of evidence suggests a role for Y2Rs in alcohol-drinking behavior. Intra-ventricular administration of BIIE0246 suppresses alcohol consumption by rats (28), and alcohol-dependent rats exhibit increased sensitivity to the suppressive effects of BIIE0246 on alcohol drinking (29). Y2R KO mice also consume significantly less alcohol than wild-type controls (30). Furthermore, Y2Rs bind ligands in an apparently irreversible manner, and NPY dissociates from Y2Rs much more slowly than from Y1Rs (23), suggesting a possible Y2R mechanism for prolonged NPY effects on alcohol drinking (10). Indeed, single nucleotide polymorphisms in the gene encoding for Y2Rs are associated with alcohol dependence and alcohol withdrawal symptoms in humans (31), and site-specific ablation of the Y2R gene in CeA and BLA affects anxiety-like and depression-like behaviors in mice, presumably via alteration of GABAergic transmission (32). Although prior studies have shown that NPY and Y2R antagonists both suppress alcohol drinking by rats, those findings do not necessarily contradict the opposite effects of NPY and BIIE0246 reported here on CeA GABAergic transmission. The suppressive effects of NPY on alcohol drinking have been localized to the CeA, but the rodent behavioral studies described above antagonized Y2R function in whole brain. The present results predict that site-specific antagonism of Y2Rs in the CeA would actually increase drinking by alcohol-dependent rats and that antagonism of Y1Rs constitute a promising pharmacotherapeutic target for lowering excessive alcohol drinking in the alcohol-dependent organism.
Many studies suggest a prominent role for amygdalar NPY in alcohol-drinking and anxiety-like behaviors. Alcohol-preferring rats exhibit deficiencies in CeA NPY mRNA and protein that are restored by voluntary alcohol consumption (33), perhaps via intracellular PKA pathways (34). Rats exhibit increases in anxiety-like behavior and decreased amygdalar NPY 24 hrs into alcohol withdrawal, effects that may be due to reduced histone acetylation in the CeA (35, 36). Restoration of histone acetylation restores depleted NPY levels in amygdala and in turn normalizes alcohol withdrawal-induced increases in anxiety-like behavior (36). Withdrawal-induced decreases in CeA NPY may contribute to the increase in GABAergic tone observed in alcohol-dependent animals. This hypothesis is supported by the fact that NPY normalizes the increased GABA release observed in alcohol-dependent animals (Figure 3F). Consistent with this hypothesis, it has been proposed that inhibitory neuronal populations in CeA mediate the ability of ICV NPY to block stress-induced reinstatement of alcohol-seeking behavior (37).
We showed that chronic NPY administration during prior alcohol withdrawals blocks increases in alcohol self-administration during subsequent withdrawals, a hallmark behavioral feature of the transition to alcohol dependence. Other labs have shown that treatment with various pharmacological compounds during prior alcohol withdrawals prevents increases in anxiety-like behavior during subsequent withdrawals (38), raising the possibility that chronic NPY treatment in our study suppressed alcohol drinking by relieving the negative affect produced by prior withdrawals. Indeed, chronic NPY administration into the BLA produces long-term decreases in stress-induced anxiety-like behavior (39). Furthermore, NPY in BLA blocks CRF-induced increases in anxiety-like behavior (40), suggesting an interaction between amygdalar CRF and NPY systems, perhaps via convergence on GABA neurons. In the BLA, NPY appears to suppress anxiety-like behavior via Y1R activation (41) and promote anxiety-like behavior via Y2R activation (9), suggesting complex roles of NPY receptor subtypes in anxiety-related behaviors that may vary between amygdaloid subnuclei.
Chronic NPY administration blocks excessive alcohol consumption during the transition to alcohol dependence, perhaps via actions in the CeA. NPY may suppress alcohol drinking in rats via tonic activation of pre-synaptic Y2Rs in CeA, and this effect may be buffered by NPY actions at postsynaptic Y1Rs in the same region (see Figure 6). NPY also blocks and reverses acute- and chronic-alcohol-induced increases in GABAergic transmission in CeA, likely via actions at pre-synaptic Y2Rs. CRF1Rs remain a likely locus for direct effects of alcohol on GABAergic transmission in CeA (17), but NPY receptors appear to play an important regulatory role in alcohol-related behaviors. Based on the treatment regimen presented here, manipulation of NPY receptors in the CeA may show future promise as a target for pharmacological management of excessive alcohol drinking, especially as new NPY receptor ligands are developed that cross the blood-brain barrier.
Supplementary Material
ACKNOWLEDGMENTS
The authors acknowledge Dr. George R. Siggins for his helpful comments and Mike Arends for his excellent editorial assistance. This is manuscript number 20659 from The Scripps Research Institute. This work was supported by the Pearson Center for Alcoholism and Addiction Research and NIAAA grants AA016436, AA016985, AA015566, AA06420, AA08459, AA012602.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors report no biomedical financial interests or potential conflicts of interest.
REFERENCES
- 1.American Psychiatric Association . Diagnostic and Statistical Manual of Mental Disorders. American Psychiatric Press; Washington, D.C.: 1998. [Google Scholar]
- 2.National Institute on Alcohol Abuse and Alcoholism . Five-year strategic plan FY08-13. National Institute on Alcohol Abuse and Alcoholism; Washington, D.C.: 2007. [Google Scholar]
- 3.Koob GF. Alcoholism : Allostasis and beyond. Alcohol Clin Exp Res. 2003;27:232–43. doi: 10.1097/01.ALC.0000057122.36127.C2. [DOI] [PubMed] [Google Scholar]
- 4.Heimer L, Alheid GF. Piecing together the puzzle of basal forebrain anatomy. Adv Exp Med Biol. 1991;295:1–42. doi: 10.1007/978-1-4757-0145-6_1. [DOI] [PubMed] [Google Scholar]
- 5.Allen J, Novotny J, Martin J, Heinrich G. Molecular structure of mammalian neuropeptide Y: analysis by molecular cloning and computer-aided comparison with crystal structure of avian homologue. Proc Natl Acad Sci USA. 1987;84:2532–2536. doi: 10.1073/pnas.84.8.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McDonald AJ, Pearson JC. Coexistence of GABA and peptide immunoreactivity in non-pyramidal neurons of the basolateral amygdala. Neurosci Lett. 1989;100:53–8. doi: 10.1016/0304-3940(89)90659-9. [DOI] [PubMed] [Google Scholar]
- 7.Heilig M, Söderpalm B, Engel JA, Widerlöv E. Centrally administered neuropeptide Y (NPY) produces anxiolytic-like effects in animal anxiety models. Psychopharmacol. 1989;98:524–9. doi: 10.1007/BF00441953. [DOI] [PubMed] [Google Scholar]
- 8.Heilig M, McLeod S, Brot M, Heinrichs SC, Menzaghi F, Koob GF, Britton KT. Anxiolytic-like action of neuropeptide Y: mediation by Y1 receptors in amygdala, and dissociation from food intake effects. Neuropsychopharmacol. 1993;8:357–63. doi: 10.1038/npp.1993.35. [DOI] [PubMed] [Google Scholar]
- 9.Sajdyk TJ, Schober DA, Gehlert DR. Neuropeptide Y receptor subtypes in the basolateral nucleus of the amygdala modulate anxiogenic responses in rats. Neuropharmacol. 2002;43:1165–72. doi: 10.1016/s0028-3908(02)00234-4. [DOI] [PubMed] [Google Scholar]
- 10.Gilpin NW, Stewart RB, Murphy JM, Li T-K, Badia-Elder NE. Neuropeptide Y reduces oral ethanol intake in alcohol-preferring (P) rats following a period of imposed ethanol abstinence. Alcohol Clin Exp Res. 2003;27:787–94. doi: 10.1097/01.ALC.0000065723.93234.1D. [DOI] [PubMed] [Google Scholar]
- 11.Gilpin NW, Misra K, Koob GF. Neuropeptide Y in the central nucleus of the amygdala suppresses dependence-induced decreases in alcohol drinking. Pharmacol Biochem Behav. 2008;90:475–80. doi: 10.1016/j.pbb.2008.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gilpin NW, Stewart RB, Badia-Elder NE. Neuropeptide Y suppresses ethanol drinking in ethanol-abstinent, but not non-ethanol-abstinent, Wistar rats. Alcohol. 2008;42:541–51. doi: 10.1016/j.alcohol.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Primeaux SD, Wilson SP, Bray GA, York DA, Wilson MA. Overexpression of neuropeptide Y in the central nucleus of the amygdala decreases ethanol self administration in “anxious” rats. Alcohol Clin Exp Res. 2006;30:791–801. doi: 10.1111/j.1530-0277.2006.00092.x. [DOI] [PubMed] [Google Scholar]
- 14.Thorsell A, Repunte-Canonigo V, O'Dell LE, Chen SA, King AR, Lekic D, et al. Viral vector-induced amygdala NPY overexpression reverses increased alcohol intake caused by repeated deprivations in Wistar rats. Brain. 2007;130:1330–7. doi: 10.1093/brain/awm033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roberto M, Madamba SG, Moore SD, Tallent MK, Siggins GR. Ethanol increases GABAergic transmission at both pre- and postsynaptic sites in rat central amygdala neurons. Proc Natl Acad Sci USA. 2003;100:2053–8. doi: 10.1073/pnas.0437926100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roberto M, Madamba SG, Stouffer DG, Parsons LH, Siggins GR. Increased GABA release in the central amygdala of ethanol-dependent rats. J Neurosci. 2004;24:10159–66. doi: 10.1523/JNEUROSCI.3004-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roberto M, Cruz MT, Gilpin NW, Sabino V, Schweitzer P, Bajo M, et al. Corticotropin releasing factor-induced amygdala gamma-aminobutyric acid release plays a key role in alcohol dependence. Biol Psychiatry. 2010;67:831–9. doi: 10.1016/j.biopsych.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roberto M, Siggins GR. Nociceptin/orphanin FQ presynaptically decreases GABAergic transmission and blocks the ethanol-induced increase of GABA release in central amygdala. Proc Natl Acad Sci USA. 2006;103:9715–20. doi: 10.1073/pnas.0601899103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gilpin NW, Smith AD, Cole M, Weiss F, Koob GF, Richardson HN. Operant behavior and alcohol levels in blood and brain of alcohol-dependent rats. Alcohol Clin Exp Res. 2009;33:2113–23. doi: 10.1111/j.1530-0277.2009.01051.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Valenstein ES, Cox VC, Kakolewski JW. Polydipsia elicited by the synergistic action of a saccharin and glucose solution. Science. 1967;157:552–4. doi: 10.1126/science.157.3788.552. [DOI] [PubMed] [Google Scholar]
- 21.Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. Academic Press; San Diego: 1998. [Google Scholar]
- 22.Chen X, DiMaggio DA, Han SP, Westfall TC. Autoreceptor-induced inhibition of neuropeptide Y release from PC-12 cells is mediated by Y2 receptors. J Am Physiol. 1997;273:H1737–44. doi: 10.1152/ajpheart.1997.273.4.H1737. [DOI] [PubMed] [Google Scholar]
- 23.Dautzenberg FM, Neysari S. Irreversible binding kinetics of neuropeptide Y ligands to Y2 but not to Y1 and Y5 receptors. Pharmacol. 2005;75:21–9. doi: 10.1159/000085897. [DOI] [PubMed] [Google Scholar]
- 24.Valdez GR, Koob GF. Allostasis and dysregulation of corticotropin-releasing factor and neuropeptide Y systems: implications for the development of alcoholism. Pharmacol Biochem Behav. 2004;79:671–89. doi: 10.1016/j.pbb.2004.09.020. [DOI] [PubMed] [Google Scholar]
- 25.Kash TL, Winder DG. Neuropeptide Y and corticotropin-releasing factor bi-directionally modulate inhibitory synaptic transmission in the bed nucleus of the stria terminalis. Neuropharm. 2006;51:1013–22. doi: 10.1016/j.neuropharm.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 26.Greber S, Schwarzer C, Sperk G. Neuropeptide Y inhibits potassium-stimulated glutamate release through Y2 receptors in rat hippocampal slices in vitro. Br J Pharmacol. 1994;113:737–40. doi: 10.1111/j.1476-5381.1994.tb17055.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schroeder JP, Olive F, Koenig H, Hodge CW. Intra-amygdala infusion of the NPY Y1 receptor antagonist BIBP 3226 attenuates operant ethanol self-administration. Alcohol Clin Exp Res. 2003;27:1884–91. doi: 10.1097/01.ALC.0000098875.95923.69. [DOI] [PubMed] [Google Scholar]
- 28.Thorsell A, Rimondini R, Heilig M. Blockade of central neuropeptide Y (NPY) Y2 receptors reduces ethanol self-administration in rats. Neurosci Lett. 2002;332:1–4. doi: 10.1016/s0304-3940(02)00904-7. [DOI] [PubMed] [Google Scholar]
- 29.Rimondini R, Thorsell A, Heilig M. Suppression of ethanol self-administration by the neuropeptide Y (NPY) Y2 receptor antagonist BIIE0246: evidence for sensitization in rats with a history of dependence. Neurosci Lett. 2005;375:129–33. doi: 10.1016/j.neulet.2004.10.084. [DOI] [PubMed] [Google Scholar]
- 30.Thiele TE, Naveilhan P, Ernfors P. Assessment of ethanol consumption and water drinking by NPY Y2 receptor knockout mice. Peptides. 2004;25:975–83. doi: 10.1016/j.peptides.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 31.Wetherill L, Schuckit MA, Hesselbrock V, Xuei X, Liang T, Dick DM, et al. Neuropeptide Y receptor genes are associated with alcohol dependence, alcohol withdrawal phenotypes, and cocaine dependence. Alcohol Clin Exp Res. 2008;32:2031–40. doi: 10.1111/j.1530-0277.2008.00790.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tasan RO, Nguyen NK, Weger S, Sartori SB, Singewald N, Heilbronn R, et al. The central and basolateral amygdala are critical sites of neuropeptide Y/Y2 receptor-mediated regulation of anxiety and depression. J Neurosci. 2010;30:6282–90. doi: 10.1523/JNEUROSCI.0430-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pandey SC, Zhang H, Roy A, Xu T. Deficits in amygdaloid cAMP-responsive element-binding protein signaling play a role in genetic predisposition to anxiety and alcoholism. J Clin Invest. 2005;115:2762–73. doi: 10.1172/JCI24381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang H, Pandey SC. Effects of PKA modulation on the expression of neuropeptide Y in rat amygdaloid structures during ethanol withdrawal. Peptides. 2003;24:1397–1402. doi: 10.1016/j.peptides.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 35.Roy A, Pandey SC. The decreased cellular expression of neuropeptide Y protein in rat brain structures during ethanol withdrawal after chronic ethanol exposure. Alcohol Clin Exp Res. 2002;26:796–803. [PubMed] [Google Scholar]
- 36.Pandey SC, Ugale R, Zhang H, Tang L, Prakash A. Brain chromatin remodeling: a novel mechanism of alcoholism. J Neurosci. 2008;28:3729–37. doi: 10.1523/JNEUROSCI.5731-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cippitelli A, Damadzic R, Hansson AC, Singley E, Sommer WH, Eskay R, et al. Neuropeptide Y (NPY) suppresses yohimbine-induced reinstatement of alcohol seeking. Psychopharmacol. 2010;208:417–26. doi: 10.1007/s00213-009-1741-y. [DOI] [PubMed] [Google Scholar]
- 38.Breese GR, Overstreet DH, Knapp DJ. Conceptual framework for the etiology of alcoholism: a “kindling”/stress hypothesis. Psychopharmacol. 2005;178:367–80. doi: 10.1007/s00213-004-2016-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sajdyk TJ, Johnson PL, Leitermann RJ, Fitz SD, Dietrich A, Morin M, et al. Neuropeptide Y in the amygdala induces long-term resilience to stress-induced reductions in social responses but not hypothalamic–adrenal–pituitary axis activity or hyperthermia. J Neurosci. 2008;28:893–903. doi: 10.1523/JNEUROSCI.0659-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sajdyk TJ, Fitz SD, Shekhar A. The role of neuropeptide Y in the amygdala on corticotropin-releasing factor receptor-mediated behavioral stress responses in the rat. Stress. 2006;9:21–8. doi: 10.1080/10253890600557315. [DOI] [PubMed] [Google Scholar]
- 41.Sajdyk TJ, Vandergriff MG, Gehlert DR. Amygdalar neuropeptide Y Y1 receptors mediate the anxiolytic-like actions of neuropeptide Y in the social interaction test. Eur J Pharmacol. 1999;368:143–7. doi: 10.1016/s0014-2999(99)00018-7. [DOI] [PubMed] [Google Scholar]
- 42.Heilig M, Koob GF, Ekman R, Britton KT. Corticotropin-releasing factor and neuropeptide Y : role in emotional intégration. Trends Neurosci. 1994;17:80–5. doi: 10.1016/0166-2236(94)90079-5. [DOI] [PubMed] [Google Scholar]
- 43.Huber D, Veinante P, Stoop R. Vasopressin and oxytocin excite distinct neuronal populations in the central amygdala. Science. 2005;308:245–8. doi: 10.1126/science.1105636. [DOI] [PubMed] [Google Scholar]
- 44.Roberto M, Cruz M, Bajo M, Siggins GR, Parsons LH, Schweitzer P. The endocannabinoid system tonically regulates inhibitory transmission and depresses the effect of ethanol in central amygdala. Neuropsychopharmacol. 2010;35:1962–72. doi: 10.1038/npp.2010.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.