

Abstract
We report an approach to the high-throughput screening of asymmetric oxidation catalysts. The strategy is based on application of the one-bead-one-compound library approach, wherein each of our catalyst candidates is based on a peptide scaffold. For this purpose we rely on a recently developed catalytic cycle that employs an acid-peracid shuttle. In order to implement our approach, we developed a compatible linker and demonstrated that the library format is amenable to screening and sequencing of catalysts employing partial Edman degradation and MALDI mass spectrometry analysis. The system was applied to the discovery (and re-discovery) of catalysts for the enantioselective oxidation of a cyclohexene derivative. The system is now poised for application to unprecedented substrate classes for asymmetric oxidation reactions.
Keywords: Asymmetric catalysis, asymmetric epoxidation, peptides
Peptide catalysts are versatile tools for asymmetric synthesis that mediate a wide variety of reactions.1 Among the very early demonstrations of this concept was the application of oligopeptides to the epoxidation of enones, a manifestation of the Juliá-Colonna epoxidation reaction.2 While the Juliá-Colonna reaction relies on a nucleophilic epoxidation mechanism, we subsequently developed an alternative peptide-catalyzed oxidation reaction that relies on electrophilic peracids.3,4 Given that peptides offer the potential for massive catalyst diversity, methods for efficient catalyst discovery remain in demand. Many high-throughput catalyst screening strategies exist,5 but these methods are often reaction-specific. For example, libraries of enzymes for oxidation reactions6 have led to the discovery of remarkable stereoselective enzymatic catalysts using the tools of molecular biology. One-bead-one-compound catalyst libraries based on metal complexes have been evaluated for oxidation catalysis,7 and the principle of bead tagging has been applied to facilitate catalyst deconvolution.8 However, tagging of libraries can itself be labor intensive, cumbersome, and pose stability issues. Our goal in the present study was the development of a high-throughput screening protocol for peptide-based oxidation catalysts. A method free of tagging could in principle employ direct peptide sequencing of catalyst hits, provided the catalyst screening and deconvolution protocols are compatible with the oxidizing conditions of the targeted processes. As we describe below, we found that extant methodology for screening peptide-based catalysts required modification for implementation in the oxidation reactions under study.
The asymmetric oxidation process we recently described depends on the catalytic action of an aspartic acid residue's carboxylic acid, which is transformed to the peracid with a carbodiimide and hydrogen peroxide (Figure 1). Other additives, including the nucleophilic co-catalyst N,N-dimethylaminopyridine (DMAP), or its corresponding N-oxide, were found to facilitate catalytic turnover. The acid catalyst is regenerated upon O-atom transfer from the peracid to the alkene substrate, forming the corresponding epoxides. Furthermore, peptide catalyst 1 was found to be effective for enantioselective epoxidation of 2, yielding 3 in up to 92% ee.
Figure 1.

A. Catalytic cycle of aspartic acid-mediated epoxidations. B. Peptide-catalyzed reaction.
We sought a method to evaluate catalysts quickly using split-and-pool synthesis and one-bead-one-compound (OBOC) type libraries9,10 that are compatible to the oxidizing conditions of the desired transformation. Our lab has previously found such methods successful in conjunction with a fluorescent sensor to quickly distinguish active catalysts for enantioselective acyl transfer reactions.11 The enantioselectivity afforded by the most active catalysts was then determined in a secondary assay for ee using either chiral GC or chiral HPLC. In the present study, we opted for automated analytical HPLC for analysis of reaction outcomes. While lower throughput than most fluorescence-based assays,12 the advantages for the present pilot study include high information content (e.g., simultaneous determination of rate and selectivity) and applicability to a variety of reactions. The focus of this study is thus the adaptation of the OBOC method for the synthesis, screening, and deconvolution (i.e., hit catalyst sequence determination) of a combinatorial library of aspartic acid-based asymmetric epoxidation catalyst candidates.
Our workflow for the proposed catalyst discovery protocol is presented in Figure 2. In the initial stage, the sequence space to be explored in the OBOC library is defined and the library is synthesized using the split-and-pool method. The protocol then calls for sorting the beads into individual reaction vessels (e.g., wells of a multi-well plate) and epoxidation reactions are then performed in the individual wells through the addition of reagents. Reaction contents are then analyzed by chiral HPLC, which allows for simultaneous determination of reaction conversion and enantioselectivity. Hit catalysts are then selected and subjected to single bead sequencing using partial Edman degradation-mass spectrometry (PED/MS),13 which can either be performed on a single bead or a batch of beads to determine the catalyst structure. Hit catalyst sequences may then be resynthesized and evaluated/validated for their performance in epoxidation reactions while in solution or remaining on-bead.
Figure 2.

Schematic diagram of screening approach.
To examine the viability of our approach, we began our investigation by testing a variant of peptide 1 attached directly to polystyrene resin. Our previous studies14 had shown that this substitution of phenylalanine for α-methylbenzylamine in peptide 1 when attached to Merrifield resin affords 3 with reduced, but still significant enantioselectivity (70% ee; Table 1, entry 1). This reaction was scaled down to a single functionalized polystyrene macrobead-catalyzed reaction, and the product was obtained with 57% ee (Table 1, entry 2). A slight background reaction can occur without a bead, perhaps due to oxidation induced by the carbodiimide-hydrogen peroxide adduct.15 Further studies suggest that rate of the background reaction may be attenuated by varying conditions and substrate choice.
Table 1.
Comparison of linkers and reaction conditions.
 |
||||
|---|---|---|---|---|
| Entryd | Linker | Solvent | Avg. ee | |
| 1 | Nonea | DCMb | 70% |
|
| 2 | None | DCMb | 57% | |
| 3 | 4 | DCMb | <15% | |
| 4 | 5 | DCMb | 24% | |
| 5 | None | Tolueneb | 54% | |
| 6 | 5 | Tolueneb | 16% | |
| 7 | None | THF/H2Oc | 50% | |
| 8 | 5 | THF/H2Oc | 35% | |
| 9 | None | MeOHc | 41% | |
| 10 | 5 | MeOHc | 21% | |
|
|
||||
Merrifield resin, many beads, single run.
With DMAP.
With HOBt and DMAP.
Average of three runs.
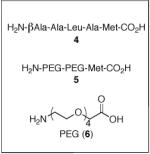
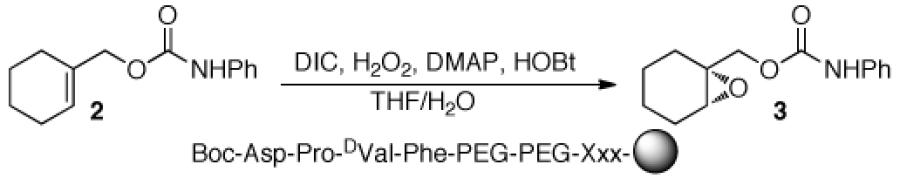
A critical issue proved to be the choice of linker between the peptide catalyst and the polystyrene bead. Sequencing peptides using PED/MS requires a linker between the resin and the peptides to increase the mass of the cleaved peptide fragments, thus avoiding interference from MALDI matrix peaks. In addition, a methionine residue incorporated at the beginning of the linker provides an acid/base-stable moiety that may be cleaved using CNBr with aqueous 70% TFA.16,17 Initially, we examined a tailored peptide linker (4: Table 1, entry 3), containing the βala-Ala-Leu-Ala-Met sequence attached to the Boc-Asp-Pro-DVal-Phe sequence. Unfortunately, single bead reactions employing this catalyst-linker construct provided 3 in <15% ee, which we judged too low for a peptide that will eventually be our positive control. The low selectivity may be attributed to an unfavorable linker conformation, low peptide loading, low solubility, and/or methionine oxidation (vide infra).
We then attempted to address these issues by using a PEG-linker (5) based upon amino acid 6,18 which we reasoned would provide solubility in analogy to PEG-based resins. The peptide with the PEG-linker provides improved selectivity (24% ee; Table 1, entry 4) relative to linker 4, although the results are still markedly less impressive in comparison to single bead reactions performed with beads lacking a linker altogether. Switching reaction solvents from DCM to toluene gives lower enantioselectivity, suggesting that screening should be avoided in toluene as the linker is deleterious to selectivity, possibly because of low solubility (Table 1, entries 5 and 6). However, use of THF/H2O as a solvent increases the selectivity of the peptide attached to linker 5 (35% ee; Table 1, entry 8), though without the linker there is a slight increase (50% ee; Table 1, entry 7). Interestingly, the reaction proceeds in methanol, a nucleophilic solvent that could compete with H2O2 to intercept aspartic acid intermediates in the catalytic cycle or break catalyst-substrate associations by competing for hydrogen bonds (Table 1, entries 9 and 10).
Since peracids can oxidize sulfides to sulfones,19 we hypothesized that the peptide may engage in non-productive oxidation of the methionine sulfide, eroding selectivity by sequestering active catalyst. Furthermore, over-oxidation of the methionine sulfide to a sulfone interferes with our ability to sequence the peptides since the fully reduced sulfide is optimal for cleavage from resin.20 Thus, we studied the effect of methionine on selectivity by making linkers that are co-functionalized with differing amounts of methionine and alanine at the position adjacent to the resin (Table 2). Though reducing the methionine content increases the yield of 3, the selectivity remains constant when the reaction is performed in THF/H2O. In DCM, however, there is an increase in selectivity as the methionine ratio declines, with a maximum of 37% ee for the 1:4 Met:Ala peptide (see Supplementary Information for complete data set). These data suggest that the methionine is indeed deleterious under certain conditions. Lowering the ratio of Met:Ala to 1:4 produces adequate results to proceed and was used in subsequent studies.
Table 2.
Effect of methionine on selectivity. Uncorrected HPLC yield is defined as the ratio of the area under the product peaks to the sum of the total area of the products and starting material.
 | |||||
|---|---|---|---|---|---|
| Xxx | |||||
| Entrya | Met | Ala | Avg. yieldb | Avg. ee | |
| 1 | 1 | : | 0 | 13% | 37% |
| 2 | 1 | : | 1 | 22% | 37% |
| 3 | 1 | : | 4 | 22% | 37% |
| 4 | 0 | : | 1 | 12% | 32% |
| 5c | 0 | : | 0 | 6% | 33% |
| 6 | No Linker | 38% | 50% | ||
Average of three replicates.
Uncorrected, crude HPLC yield.
No Xxx in peptide.
Despite having lower amounts of methionine, the beads recovered from the oxidation reactions of Table 2, entry 3 were successfully sequenced using established PED/MS techniques.21 After analyzing several beads recovered from other reactions, we found that the peptide fragments present in the MALDI-TOF often had two masses for each fragment, one with the expected value and another less ~83 amu. This mass discrepancy is consistent with hydrolysis of the amide of the C-terminal homoserine lactone that results from cleavage of the methionine with CNBr/TFA. While we are uncertain of the mechanism of this loss, treatment of peptide/linker construct on resin with hydrogen peroxide before cleavage results in the reduced mass. However, treatment with hydrogen peroxide followed by treatment with a cocktail of NH4I, DMS, and TFA mostly gives the expected masses upon cleavage from resin. With this knowledge and an expanded set of masses for which to search, we have successfully sequenced some peptides without reducing the methionine. Nonetheless, sequencing of peptides after the oxidation reaction was challenging at times, possibly due to peptide oxidation or other modifications of certain peptide sequences during reactions. It is also possible that the truncation products produced after PED are more hydrophobic and not as easily ionized as with the traditional linker.17 These challenges are often surmountable, but they generally required more data analysis to complete a sequence assignment. The risk of losing a hit bead due to sequencing difficulties is a standard liability in most HTS assays involving bead-based peptide synthesis. That said, as long as “winning” beads may be found, the impact of beads not amenable to sequencing is minimized. In this light, we were confident that we had sufficiently robust methods in place to proceed.
To test the protocol from start to finish, a split-and-pool library with three variable positions was constructed and screened for selective oxidation of 2. The library was designed as shown in Figure 3. The i-position was fixed as Asp. Then i+1, i+2 and i+3 positions were varied with eight residues, creating a library of 512 theoretical peptide sequences for screening. To ensure that a successful catalyst could be identified from the library, one positive control bead (Table 2, entry 3) was added to 49 other beads from the library in a double-blind fashion.
Figure 3.

Reaction yield and selectivity with catalysts from split-and-pool library.
As revealed in Figure 3, two beads from the screen outperformed the rest. These were then identified by PED. Significantly, one of the beads was identified as the positive control (Boc-Asp-Pro-DVal-Phe); the other proved similar to the control, but with Tyr(OtBu) in the i+3 position (Boc-Asp-Pro-DVal-Tyr(OtBu)). Notably, in the OBOC assay, the positive control and its Tyr-variant afforded 3 with 37% and 40% ee, respectively.
The two peptides were then resynthesized as bead-bound peptide catalysts 7 and 8 respectively on polystyrene resin and validated as successful catalysts (Figure 4).22 When reexamined in triplicate in an epoxidation reaction employing single beads, each gave a satisfactory recapitulation of the results from the OBOC assay, with 7 affording 3 with 46% ee, and 8 providing 3 with 50% ee. Each was then synthesized as a C-terminal methyl ester for study in homogeneous reactions to increase selectivity, facilitate purification and allow greater control of the catalyst loading. Under the homogeneous conditions optimized for peptide 1, peptide 9 provides 3 with 64% ee (81% yield), and peptide 10 provides 3 with 65% ee (74% yield). These results are in line with expectations from our earlier study and provide validation for our approach to find selective catalysts.
Figure 4.

Validation of peptides from screen.
With the validation studies of the process presented herein, we are now poised to search for catalysts capable of selective oxidation of more complex molecules in at least a medium throughput, and perhaps a high-throughput mode. The use of general chromatography technologies to analyze complex product mixtures will continue to prove useful in these studies. Of course, advances in the rapid analysis of reactions remains an ongoing challenge for this field. Nonetheless, the present results provide the path forward for the synthesis, testing, and sequence determination of OBOC peptide-based oxidation catalyst libraries that function under conditions of electrophilic oxidation catalysis.
Experimental Procedures
Peptide synthesis for on-bead screening
Polystyrene macrobeads (500-560 μm, Rapp Polymere) were swelled in DMF for 20 min and then coupled twice for 3 h each to amino acid coupling partner using Fmoc-protected amino acid monomer (4 equiv.), O-benzotriazole-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU: 4 equiv.), 1-hydroxybenzotriazole hydrate (HOBt•H2O: 4 equiv.), and iPr2EtN (8 equiv.). After coupling, the resin was washed several times with DMF and DCM. Deprotections commenced with two 20 min treatments of 20% piperidine/DMF and were followed by exhaustive washing with DMF and DCM. The final coupling was performed with Boc-Asp(OFm)-OH. The Asp-side chain was deprotected by treatment with 20% piperidine/DMF four times for 10 min each. Finally, beads were washed exhaustively with DMF, DCM, and MeOH; then dried under N2.
Screening protocol
Polystyrene macrobeads were individually transferred to 200 μL PCR tubes and inspected for uniformity of size. Three solutions were sequentially added to each tube, each addition followed by centrifugation: H2O (1.84 μL); 2.65 μL of a THF solution containing 2 (1 μmol, 1 equiv.), DIC (2 μmol, 2 equiv.), HOBt•H2O (0.1 μmol, 0.1 equiv.), and DMAP (0.1 μmol, 0.1 equiv.); and 0.5 μL of a 30% aq. H2O2 solution (5 equiv.), bringing the total reaction concentration of 2 to ~0.2 M. After the last addition, the tubes were closed, centrifuged, and allowed to stand for 12 h. Reactions were quenched with ~3 drops of sat. aq. Na2SO3, centrifuged, and then 175 μL HPLC grade hexanes was added. The biphasic mixture was vortexed, centrifuged, and the organic layer was carefully removed to an HPLC vial with insert. The samples were concentrated to dryness under a stream of N2, dissolved in 125 μL HPLC hexanes, and resolved by HPLC using a Chiralpak® AD-H column, eluting with 1.0% isopropanol in hexanes.
Bead recovery
High performing beads were recovered from the leftover aqueous layers into BioRad chromatography tubes. These beads were washed several times with H2O, MeOH, and DCM; and then dried. The beads were moved to a glass vessel and treated with anhydrous TFA once for 20 min, and then again for 10 min. The resin was washed with DCM, and then the PED protocol from Thakkar et al. was followed using 10 μL of PITC with 160 μL of 31 mM Fmoc-OSu in pyridine. After three cycles of PED, the beads were washed with H2O, MeOH, DCM, then DMF. They were treated with 20% piperidine in DMF during two 20 min cycles and then rinsed with DMF, DCM, and MeOH. Optional reduction: the beads were treated with about 50 μL of a 1 mL solution of TFA containing 25 mg NH4I (does not dissolve well) and >20 μL DMS. After 20 min, the bead was removed, and washed exhaustively with H2O, DCM, and MeOH.
Peptide cleavage and analysis
After drying in a PCR tube, these beads were treated with a few drops of 20 mg/mL CNBr in 70% TFA (aq.) overnight in the dark for at least 12 h. After drying the beads under vacuum, the resulting white solid was dissolved in 30% H2O/MeCN. 0.5 μL of the peptide solution was mixed with a 0.5 μL 30% H2O/MeCN with either 4.5 mg/mL α-cyano-4-hydroxycinnamic acid or 20 mg/mL sinapinic acid, and dried on a plate for MALDI-TOF analysis.
Supplementary Material
ACKNOWLEDGMENT
This work is supported by National Institutes of Health (R01-GM096403) to SJM. PAL was partially supported by NIH CBI-TG-GM-067543.
Footnotes
Supporting Information Available. Synthetic protocols and characterization of 6, 9, and 10; experimental details of peptide and library validation; and additional oxidation studies of library.
References
- 1.(a) Miller SJ. In Search of Peptide-Based Catalysts for Asymmetric Organic Synthesis. Acc. Chem. Res. 2004;37:601–610. doi: 10.1021/ar030061c. [DOI] [PubMed] [Google Scholar]; (b) Colby Davie EA, Mennen SM, Xu Y, Miller SJ. Asymmetric Catalysis Mediated by Synthetic Peptides. Chem. Rev. 2007;107:5759–5812. doi: 10.1021/cr068377w. [DOI] [PubMed] [Google Scholar]; (c) Revell JD, Wennemers H. Peptidic Catalysts Developed by Combinatorial Screening Methods. Curr. Opin. Chem. Biol. 2007;11:269–278. doi: 10.1016/j.cbpa.2007.05.015. [DOI] [PubMed] [Google Scholar]
- 2.(a) Kelly DR, Roberts SM. Oligopeptides as Catalysts for Asymmetric Epoxidation. Biopolymers. 2006;84:74–89. doi: 10.1002/bip.20373. [DOI] [PubMed] [Google Scholar]; (b) Berkessel A, Koch B, Toniolo C, Rainaldi M, Broxterman QB, Kaptein B. Asymmetric Enone Epoxidation by Short Solid-Phase Bound Peptides: Further Evidence for Catalyst Helicity and Catalytic Activity of Individual Peptide Strands. Biopolymers. 2006;84:90–96. doi: 10.1002/bip.20413. [DOI] [PubMed] [Google Scholar]
- 3.(a) Peris G, Jakobsche CE, Miller SJ. Aspartate-Catalyzed Asymmetric Epoxidation Reactions. J. Am. Chem. Soc. 2007;129:8710–8711. doi: 10.1021/ja073055a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jakobsche CE, Peris G, Miller SJ. Functional Analysis of an Aspartate-Based Epoxidation Catalyst with Amide-to-Alkene Peptidomimetic Catalyst Analogues. Angew. Chem. Int. Ed. 2008;47:6707–6711. doi: 10.1002/anie.200802223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.For selected reviews and recent advances involving catalytic asymmetric epoxidation, see: Johnson RA, Sharpless KB. Addition Reactions with Formation of Carbon-Oxygen Bonds: (ii) Asymmetric Methods of Epoxidation. In: Trost BM, editor. Comprehensive Organic Synthesis. Vol. 7. Pergamon; New York: 1991. p. 389. Jacobsen EN, Wu MH. Epoxidation of Alkenes Other Than Allylic Alcohols. In: Jacobsen EN, Pfaltz A, Yamamoto H, editors. Comprehensive Asymmetric Catalysis II. Springer-Verlag; Berlin: 1999. p. 649. Aggarwal VK, Winn CL. Catalytic, Asymmetric Sulfur Ylide-Mediated Epoxidation of Carbonyl Compounds: Scope, Selectivity, and Applications in Synthesis. Acc. Chem. Res. 2004;37:611–620. doi: 10.1021/ar030045f. Xia QH, Ge HQ, Liu ZM, Su KX. Advances in Homogeneous and Heterogeneous Catalytic Asmmetric Epoxidation. Chem. Rev. 2005;105:1603–1662. doi: 10.1021/cr0406458. Wong OA, Shi Y. Organocatalytic Oxidation. Asymmetric Epoxidation of Olefins Catalyzed by Chiral Ketones and Iminium Salts. Chem. Rev. 2008;108:3958–3987. doi: 10.1021/cr068367v. Li Z, Zhang W, Yamamoto H. Vanadium-Catalyzed Enantioselective Desymmetrization of meso Secondary Allylic Alcohols and Homoallylic Alcohols. Angew. Chem. Int. Ed. 2008;47:7520–7522. doi: 10.1002/anie.200802523.
- 5.For representative reviews of combinatorial catalysis, see: Francis MB, Jamison TF, Jacobsen EN. Combinatorial Libraries of Transition-Metal Complexes, Catalysts and Materials. Curr. Opin. Chem. Biol. 1998;2:422–428. doi: 10.1016/s1367-5931(98)80019-7. Kuntz KW, Snapper ML, Hoveyda AH. Combinatorial Catalyst Discovery. Curr. Opin. Chem. Biol. 1999;3:313–319. doi: 10.1016/S1367-5931(99)80048-9. Jandeleit B, Schaefer DJ, Powers TS, Turner HW, Weinberg WH. Combinatorial Materials Science and Catalysis. Angew. Chem. Int. Ed. 1999;38:2494–2532. Reetz MT. Combinatorial and Evolution-Based Methods in the Creation of Enantioselective Catalysts. Angew. Chem. Int. Ed. 2001;40:284–310. Wennemers H. Combinatorial Methods for the Discovery of Catalysts. In: Schmuck C, Wennemers H, editors. Highlights in Bioorganic Chemistry. Wiley-VCH; Weinheim: 2004. pp. 436–445.
- 6.For a few examples of enantioselective oxidations mediated by enzymes, see: Landwehr M, Hochrein L, Otey CR, Kasravan A, Bäckvall J-E, Arnold FH. Enantioselective aHydroxylation of 2-Arylacetic Acid Derivatives and Buspirone Catalyzed by Engineered Cytochrome P450 BM-3. J. Am. Chem. Soc. 2006;128:6058–6059. doi: 10.1021/ja061261x. Peters MW, Meinhold P, Glieder A, Arnold FJ. Regio- and Enantioselective Alkane Hydroxylation with Engineered Cytochromes P450 BM-3. J. Am Chem. Soc. 2003;125:13442–13450. doi: 10.1021/ja0303790. Kayser MK. ‘Designer Reagents’ Recombinant Microorganisms: New and Powerful Tools for Organic Synthesis. Tetrahedron. 2009;65:947–974. Reetz MT, Wu S. Laboratory Evolution of Robust and Enantioselective Baeyer-Villiger Monooxygenases for Asymmetric Catalysis. J. Am. Chem. Soc. 2010;131:15424–15432. doi: 10.1021/ja906212k. Reetz MT. Laboratory Evolution of Stereoselective Enzymes: A Prolific Source of Catalysts for Asymmetric Reactions. Angew. Chem. Int. Ed. 2011;50:138–174. doi: 10.1002/anie.201000826. For a more general discussion of stereoselectivity in enzyme engineering, see:
- 7.(a) Francis MB, Jacobsen EN. Discovery of Novel Catalysts for Alkene Epoxidation from Metal-Binding Combinatorial Libraries. Angew. Chem. Int. Ed. 1999;38:937–941. doi: 10.1002/(SICI)1521-3773(19990401)38:7<937::AID-ANIE937>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]; (b) Moreira R, Havranek M, Sames D. New Fluorogenic Probes for Oxygen and Carbene Transfer: A Sensitive Assay for Single Bead-Supported Catalysts. J. Am. Chem. Soc. 2001;123:3927–3931. doi: 10.1021/ja003786+. [DOI] [PubMed] [Google Scholar]
- 8.Ohlmeyer MHJ, et al. Complex Synthetic Chemical Libraries Indexed with Molecular Tags. Proc. Natl. Acad. Sci. USA. 1993;90:10922–10926. doi: 10.1073/pnas.90.23.10922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Furka A, Sebestyen F, Asgedom M, Dibo G. General Method for Rapid Synthesis of Multicomponent Peptide Mixtures. Int. J. Pept. Protein Res. 1991;37:487–493. doi: 10.1111/j.1399-3011.1991.tb00765.x. [DOI] [PubMed] [Google Scholar]; (b) Lam KS, Salmon SE, Hersh EM, Hruby VJ, Kazmierski WM, Knapp RJ. A New Type of Synthetic Peptide Library for Identifying Ligand-Binding Activity. Nature. 1991;354:82–84. doi: 10.1038/354082a0. [DOI] [PubMed] [Google Scholar]; (c) Lam KS, Lebl M, Krchnák V. The “One-Bead-One-Compound” Combinatorial Library Method. Chem. Rev. 1997;97:411–448. doi: 10.1021/cr9600114. [DOI] [PubMed] [Google Scholar]
- 10.For several applications of the OBOC approach to catalyst discovery, see: Taylor SJ, Morken JP. Thermographic Selection of Effective Catalysts from an Encoded Polymer-Bound Library. Science. 1998;280:267–270. doi: 10.1126/science.280.5361.267. Krattiger P, McCarthy C, Pfaltz A, Wennemers H. Catalyst-Substrate Coimmobilization: A Strategy for Catalyst Discovery in Split-and-Mix Libraries. Angew. Chem. Int. Ed. Engl. 2003;42:1722–1724. doi: 10.1002/anie.200250422. Krattiger P, Kovasy R, Revell JD, Wennemers H. Using Catalyst-Substrate Coimmobilization for the Discovery of Catalysts for Asymmetric Aldol Reactions in Split-and-Mix Libraries. QSAR Comb. Sci. 2005;24:1158–1163. Upert G, Merten CA, Wennemers H. Nanoliter Plates-Versatile Tools for the Screening of Split-and-Mix Libraries On-Bead and Off-Bead. Chem. Commun. 2010;46:2209–2211. doi: 10.1039/b927017e.
- 11.(a) Copeland GT, Miller SJ. A Chemosensor-Based Approach to Catalyst Discovery in Solution and on Solid Support. J. Am. Chem. Soc. 1999;121:4306–4307. [Google Scholar]; (b) Harris RF, Nation AJ, Copeland GT, Miller SJ. A Polymeric and Fluorescent Gel for Combinatorial Screening of Catalysts. J. Am. Chem. Soc. 2000;122:11270–11271. [Google Scholar]; (c) Copeland GT, Miller SJ. Selection of Enantioselective Acyl Transfer Catalysts from a Pooled Peptide Library through a Fluorescence-Based Activity Assay: An Approach to Kinetic Resolution of Secondary Alcohols of Broad Structural Scope. J. Am. Chem. Soc. 2001;123:6496–6502. doi: 10.1021/ja0108584. [DOI] [PubMed] [Google Scholar]
- 12.Sensor-based approaches for epoxide formation have been reported. See: Moreira R, Havranek M, Sames S. New Fluorogenic Probes for Oxygen and Carbene Transfer: A Sensitive Assay for Single Bead-Supported Catalysts. J. Am. Chem. Soc. 2001;123:3927–3931. doi: 10.1021/ja003786+.
- 13.(a) Chait BT, Wang R, Beavis RC, Kent SBH. Protein Ladder Sequencing. Science. 1993;262:89–92. doi: 10.1126/science.8211132. [DOI] [PubMed] [Google Scholar]; (b) Thakkar A, Wavreille A-S, Pei D. Traceless Capping Agent for Peptide Sequencing by Partial Edman Degradation and Mass Spectrometry. Anal. Chem. 2006;78:5935–5939. doi: 10.1021/ac0607414. [DOI] [PubMed] [Google Scholar]
- 14.Jakobsche CE. Yale University; New Haven, CT: 2009. Ph.D. Dissertation. [Google Scholar]
- 15.(a) Majetich G, Hicks R. Carbodiimide-Promoted Epoxidation of Olefins with Dilute Aqueous Hydrogen Peroxide. Synlett. 1996:649–651. [Google Scholar]; (b) Murray RW, Iyanar K. Olefin Epoxidations Using the Dicyclohexylcarbodiimide-H2O2 System. J. Org. Chem. 1998;63:1730–1731. [Google Scholar]; (c) Majetich G, Hicks R, Sun G, McGill P. Carbodiimide-Promoted Olefin Epoxidation with Aqueous Hydrogen Peroxide. J. Org. Chem. 1998;63:2564–2573. doi: 10.1021/jo972026n. [DOI] [PubMed] [Google Scholar]
- 16.(a) Hancock WS, Marshall GR. Cyanogen Bromide as a Cleavage Procedure in Solid Phase Peptide Synthesis. J. Am. Chem. Soc. 1975;97:7488–7489. doi: 10.1021/ja00859a018. [DOI] [PubMed] [Google Scholar]; (b) Wang P, Arabaci G, Pei D. Rapid Sequencing of Library-Derived Peptides by Partial Edman Degradation and Mass Spectrometry. J. Comb. Chem. 2001;3:251–254. doi: 10.1021/cc000102l. [DOI] [PubMed] [Google Scholar]
- 17.Commonly used linkers also contain Asn and Arg residues which aid in detection and likely help to solubilize peptides in aqueous solutions. However, we avoided these two polar residues in our linker, as we wished to evaluate the peptide catalysts in organic solvents; these residues might also be unstable to our reaction conditions (e.g., via N-H oxidation). See: Youngquist RS, Fuentes GR, Lacey MP, Keough T. Generation and Screening of Combinatorial Peptide Libraries Designed for Rapid Sequencing by Mass Spectrometry. J. Am. Chem. Soc. 1995;117:3900–3906. Sweeney MC, Pei D. An Improved Method for Rapid Sequencing of Support-Bound Peptides by Partial Edman Degradation and Mass Spectrometry. J. Comb. Chem. 2003;5:218–222. doi: 10.1021/cc020113+.
- 18.See Supporting Information for synthesis.
- 19.Skatterbøl L, Boulette B, Solomon S. Thiopyran 1,1,-Dioxide Derivatives from Addition of Amines to Propargyl Sulfone. J. Org. Chem. 1968;33:548–552. [Google Scholar]
- 20.Gross E, Witkop B. Selective Cleavage of the Methionyl Peptide Bonds in Ribonuclease with Cyanogen Bromide. J. Am. Chem. Soc. 1961;83:1510–1511. [Google Scholar]
- 21.Thakkar A, Wavreille A-S, Pei D. Traceless Capping Agent for Peptide Sequencing by Partial Edman Degradation and Mass Spectrometry. Anal. Chem. 2006;78:5935–5939. doi: 10.1021/ac0607414. [DOI] [PubMed] [Google Scholar]
- 22.See Supporting Information for performance data.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.