

Abstract
Conventional stable isotope labeling with amino acids in cell culture (SILAC) requires extensive metabolic labeling of proteins, and therefore is difficult to apply to cells that do not divide or are unstable in SILAC culture. Using two different sets of heavy amino acids for labeling allows for straightforward SILAC quantitation using partially labeled cells because the two cell populations are always equally labeled. Here we report the application of this labeling strategy to primary cultured neurons. We demonstrated that protein quantitation was not compromised by incomplete labeling of the neuronal proteins. We used this method to study neurotrophin-3 (NT-3) signaling in primary cultured neurons. Surprisingly our results indicate TrkB signaling is a major component of the signaling network induced by NT-3 in cortical neurons. In addition, involvement of proteins such as VAMP2, Scamp1 and Scamp3 suggest NT-3 may lead to enhanced exocytosis of synaptic vesicles.
Keywords: SILAC, mass spectrometry, proteomics, NT-3, neurotrophin 3, quantitation, tyrosine phosphorylation, immunoprecipitation
Introduction
Stable isotope labeling with amino acids in cell culture (SILAC) has proven to be a powerful tool for quantitative proteomics.1 SILAC involves cell culture in media containing “light” (natural) or “heavy” isotope-containing amino acids. The isotopes are incorporated into proteins during protein synthesis in the cells. After labeling, all proteins in the different samples are encoded with either light or heavy versions of the labeling amino acid, allowing for relative quantitation with mass spectrometry. It is important to obtain a high degree of label incorporation because incomplete labeling will skew the SILAC ratio in favor of the light protein. To ensure nearly complete labeling, it is generally required to maintain cells in SILAC media for at least five cell divisions so that even proteins with zero turnover rate will be highly labeled (>97%) merely by dilution.2 However, a variety of cells, for example, postmitotic primary cells, do not divide in culture. These cells are often more biologically relevant for studies of cell signaling than immortalized cell lines, but the application of SILAC to these cells has been greatly limited because of the issue of incomplete labeling. Even for cells that can divide, sometimes SILAC labeling can be difficult. Some cell types are unstable in culture (for example, stem cells), thus are difficult to be kept in SILAC culture for long times. Moreover, quite often cells require supplements of biological sources to maintain their growth or properties. These supplements may contain free amino acids that can cause incomplete labeling. These amino acids can often be removed by dialysis. For example, it has become a general practice to use dialyzed fetal bovine serum instead of conventional serum in SILAC culture 1. However, after dialysis some key components of the supplements can be lost and growth or maintenance of the cell can be compromised. Therefore for each cell type, careful characterization has to be performed to ensure the cells are not affected after they are adapted to SILAC culture. For this reason, it is not trivial to adapt new cell types to SILAC culture for complete labeling.3
In non-dividing cells, the labeling efficiency is dependent on the protein synthesis/turnover rate, which can vary significantly from protein to protein. Primary neurons are widely used as a very important model in neuroscience. Because the neurons do not divide in culture, the application of SILAC has been limited. To allow SILAC analysis of partially labeled neurons, we4 and others5 devised a method in which the SILAC ratio is corrected for incomplete labeling by monitoring the label incorporation of every protein. However, this strategy has several disadvantages. First, each SILAC analysis requires a parallel analysis to measure the label incorporation for each protein quantified in the SILAC analysis. In addition to the extra cost and effort, it is difficult to measure the label incorporation for every protein because it requires the protein be identified and quantified in two analyses. A considerable proportion of the SILAC protein ratios cannot be corrected because for most complex protein mixtures, only 2/3 – 3/4 of the protein identifications overlap for two repetitive liquid chromatography-tandem mass spectrometry (LC-MS/MS) analyses.6 Moreover, the correction step introduces additional random error into the quantitation, compromising the high accuracy of SILAC.
To circumvent these problems, here we report the use of a multiplex SILAC labeling strategy on primary neurons (Figure 1). Instead of using light and heavy labeling amino acids to distinguish the two experimental conditions, we use two different sets of heavy amino acids, D4-lysine/13C6-arginine (Lys4/Arg6) and 13C6-15N2-lysine/13C6-15N4-arginine (Lys8/Arg10). Because the different heavy amino acids are incorporated into the cells at the same rate, the two cell populations are always equally labeled. SILAC quantitation is done using the signals of the medium (Lys4/Arg6) and heavy (Lys8/Arg10) labeled peptides, and the unlabeled peptides can be ignored. This allows for straightforward and accurate SILAC quantitation using partially labeled cells. We implemented the multiplex SILAC approach to circumvent the challenge of correcting for partial labeling of proteins when using SILAC with primary (non-dividing) cell cultures, because it is not possible to achieve complete or nearly complete labeling of all proteins in these non-dividing cells. One group has reported a multiplex SILAC labeling strategy to study rapid protein translation regulation in dividing cells in which cells were pulse labeled by two different heavy isotope amino acids to allow quantitation of newly synthesized proteins,7 while another group has shown that multiplex SILAC can be used with cells grown in non-dialyzed serum. 8
Figure 1.

Schematic of multiplex (AH) SILAC labeling. Unlike conventional SILAC in which light and heavy versions of amino acids are used to label cells representing two different conditions, multiplex SILAC uses two different versions of heavy amino acids (medium and heavy) for labeling. multiplex SILAC does not require complete labeling because the medium and heavy amino acids are incorporated at the same rate.
In this study, we characterized the applicability of multiplex SILAC to primary cultured neurons. We further demonstrated the performance of the labeling method with a quantitative analysis of phosphotyrosine (pY)-associated signaling events initiated by NT-3 in these neurons. NT-3 is a member of the neurotrophin family. It was identified by sequence conservation with NGF and BDNF.9 Compared to other neurotrophins, NT-3 exhibits the most widespread distribution in nonneuronal tissues, including many targets of sympathetic and sensory innervations.10, 11 In vivo studies of gene targeted animals deficient in NT-3 or its receptor TrkC also indicate important functions in peripheral and central nervous system development.12, 13 However, the downstream proteomic targets of NT-3 have not been studied. NT-3 primarily binds to and activates TrkC, a member of the neurotrophin receptor family. In some cellular contexts, NT-3 also is able to activate TrkA and TrkB with much less efficiency.14 To assess the tyrosine phosphorylation dependent signaling events in NT-3-Trk pathway, we performed a SILAC analysis of pY immunoprecipitations (IP) from combined lysates of NT-3 treated and untreated neurons. The analysis identified a number of potential novel effectors which implies several previously unknown aspects of NT-3 signaling in neurons.
Experimental procedures
Primary Neuronal Culture
Pregnant rats were anesthetized and decapitated. Eighteen day old fetuses were collected in ice cold Ca-Mg-free Hanks’ balanced salt solution (HBS) (Invitrogen, Carlsbad, CA) and rapidly decapitated. After removal of meninges and white matter, brain cortices were collected in HBS and mechanically fragmented. Fragments were transferred to conical tubes and treated with 0.25% trypsin in HBS for 15 min at 37°C, washed with HBS, and dissociated by repeated passage through a constricted Pasteur pipette. About 20 million cells were added to each 0.1 mg/mL poly-D-lysine coated 15 cm culture dish (Fisher Scientific, Pittsburg, PA) in Neurobasal Media (Invitrogen) supplemented with B27 (Invitrogen) and 0.5 mM L-glutamine (Invitrogen). 5-fluoro-2-deoxyuridine was added to prevent survival of contaminant dividing cells such as glia. For SILAC labeling, media containing either Lys4/Arg6 or Lys8/Arg10 (Cambridge Isotope Laboratories, Andover, MA) were used. Neurons were maintained in the SILAC media for 10 days before one of the cell populations was stimulated with 25 ng/mL NT-3 ligand for 15 min. Culture media were refreshed every three days by removing half of the volume present on each plate and replacing it with fresh medium. For each SILAC experiment, approximately 108 neurons were used per condition. Cells were lysed in lysis buffer containing 1% Triton X-100, 150 mM NaCl, 20 mM Tris, pH8, 0.2mM EDTA, 2 mM Na3VO4, 2mM NaF, and protease inhibitors (Complete tablet; Roche, Mannheim, Germany).
Immunoprecipitation (IP) and SDS-PAGE
The “medium” and “heavy” lysates were mixed in a 1:1 ratio (v:v) and incubated with agarose-conjugated anti-pY antibody PY99 (Santa Cruz Biotechnology, Santa Cruz, CA) for 4 h, and the beads were washed 4 times with lysis buffer. Precipitated proteins were eluted with a low pH buffer (pH2) containing 0.2% TFA/1% SDS. The eluates were neutralized with 1M NH4HCO3 and separated by SDS-PAGE on a 7.5% Tris-HCl gel (Bio-rad) to remove detergents and to fractionate proteins prior to mass spectrometry. The gel was stained with Coomassie Blue and the gel lanes were cut horizontally into 9 sections for in-gel tryptic digestion.
In-gel Digestion
Gel bands were cut into small pieces and destained in 25 mM NH4HCO3/50% acetonitrile, dehydrated with acetonitrile and dried. Then the gel pieces were rehydrated with 12.5 ng/μl trypsin solution in 25 mM NH4HCO3 and incubated overnight at 37 °C. Peptides were extracted twice with 5% formic acid/50% acetonitrile followed by a final extraction with acetonitrile. Samples were concentrated by vacuum centrifugation to dryness and redissolved with 2% acetonitrile in 0.1% formic acid before further analysis.
LC-MS/MS
For all LC-MS/MS analysis, an LTQ-Orbitrap hybrid mass spectrometer (Thermo Fisher Scientific) equipped with a nano-electrospray ionization source (Jamie Hill Instrument Services) was used. An Eksigent nanoLC system (Eksigent Technologies) equipped with a self packed 100-μm × 12-cm reverse phase column (Reprosil C18, 3 μm, Dr. Maisch GmbH, Germany) was coupled directly to the mass spectrometer. Peptides were eluted by a gradient of 3–40% acetonitrile in 0.1% formic acid over 110 min. Mass spectra were acquired in data-dependent mode with one 60,000 resolution MS survey scan by the Orbitrap and up to eight MS/MS scans concurrent with the Orbitrap survey scan in the LTQ for the most intense peaks selected from each survey scan. Automatic gain control target value was set to 500,000 for Orbitrap survey scans and 10,000 for LTQ MS/MS scans. Survey scans were acquired in profile mode and MS/MS scans were acquired in centroid mode. Mascot generic format files were generated from the raw data using DTASuperCharge (version 1.19) and Bioworks (version 3.3, Thermo Fisher Scientific) for database searching.
Protein Identification and Quantitation
Mascot software (version 2.2.1, Matrix Science, London, UK) was used for database searching. An IPI database containing mouse and rat protein sequences (downloaded 07/01/08) was used. Peptide mass tolerance was 10 ppm, fragment mass tolerance was 0.6 Da, trypsin specificity was required with a maximum of one missed cleavage, and variable modifications were D4-lysine, 13C6-arginine, 13C6-15N2-lysine, 13C6-15N4-arginine, and oxidation of methionine. To estimate the false positive rate for protein identification, a decoy database was created by reversing the protein sequences of the original database. Based on the decoy database search results, three filters for protein identification were applied: (1) Peptide score threshold was 20. (2) Protein score threshold was 40. (3) Each protein was identified based on at least two unique peptide sequences. The estimated false positive rate based on the decoy database search was 1%.
The identified proteins were clustered based on their gene names. Proteins identified based on the same set of peptides were grouped and reported as a single protein match. Proteins that were likely introduced during sample preparation were excluded from the reported protein list. These proteins included keratins and trypsin (from in-gel digestion), immunoglobins (from the PY99 antibody) and serum albumin (from cell culture media). The average emPAI (Exponentially Modified Protein Abundance Index) values for each of these classes of proteins were 0.19, 0.27, and 0.32, respectively, compared to 0.40 for proteins with changed ratios. This suggests that the biologically interesting proteins were more abundant that the contaminants.
SILAC quantitation was carried out using the open source software MSQuant (version 1.4.3a74) developed by Peter Mortensen and Matthias Mann at the University of Southern Denmark. The SILAC ratios of proteins were calculated by comparing the summed MS intensities of all matched medium peptides with those of the heavy peptides. The SILAC results were verified by manual inspection of the MS spectra. MS scans were excluded from quantitation if any of the following occurred: the peptide signal had interfering peaks from coeluting peptides or other isobaric background peaks; the signal was too weak to maintain a proper isotopic distribution; the peptide LC elution profile was abnormal; inconsistent SILAC ratios between different charge states of the same peptide. About 10% of MS spectra were excluded from quantitation after applying the above criteria. As a loading control, a small volume of the combined lysates was subjected to in-gel digestion, LC-MS/MS analysis and the identified proteins were also quantified. The average ratio for all quantified proteins was used as a correction for ratios of proteins identified from the IPs.
Western Blotting
Proteins were separated by SDS-PAGE and transferred to PVDF membranes. Membranes were blocked with Tris buffered saline with Tween 20 containing 2% bovine serum albumin, incubated with the corresponding primary and horseradish peroxidase (HRP)-conjugated secondary antibodies (Santa Cruz Biotechnology), and visualized with ECL (Pierce Biotechnology, Rockford, IL). After the Western blot was performed, the PVDF membrane was stained with Coomassie Blue to confirm that the sample loading was equal in all lanes. For Western blots after IPs (Figure 7), equal amounts of input protein based on Bradford assays were used.
Figure 7. Western blotting of selected proteins.

Neurons were stimulated with NT-3 or BDNF for the indicated times. Anti-pY IPs were probed with the indicated antibodies. a. Time course validation of TrkB, TrkC and VAMP2 involvement in NT-3 signaling. b. Comparison of NT-3 and BDNF as ligands for TrkB and TrkC.
Imaging
Cortical neurons were cultured between 7 and 9 days in vitro, stimulated with 25 ng/ml NT-3, then fixed in 4% paraformaldehyde, 20% sucrose in PBS for 10 min at room temperature (RT). The fixative was quenched in 50 mM NH4Cl in PBS for 5 min, cells were permeabilized with 0.1% TritonX- 100 in PBS for 3 min, blocked with 10% goat serum, 2% BSA, 0.25 % fish skin gelatin in TBS for 30 min and then incubated with anti-TrkB, 1:2000 (Millipore; cross-reacts with TrkC in immunofluorescence) and anti-VAMP2, 1:2000 (clone 69.1, Synaptic Systems) for 30 min at RT in blocking solution. Cells were washed in TBS, 0.25% fish skin gelatin, incubated with Alexa-coupled secondary antibodies (Invitrogen) in blocking solution, washed again and mounted in Mowiol488. Images were taken on a LSM 510 laser scanning confocal microscope equipped with a 40X, Plan Neofluor NA 1.3 DIC oil immersion objective using LSM510 software (Carl Zeiss Microimaging, Thornwood, NY). Images (single z planes) were exported and scale bars added using ImageJ (NIH).
For the recycling assay, cells were incubated with anti-synaptotagmin antibody (1:300, Synaptic Systems) and anti-TrkB (1:1000) for 20 min on ice, then shifted into pre-warmed medium (37°C) for 5 min in presence or absence of 50 mM KCl before washing and fixation. As a control, a separate set of cells was incubated with mouse and rabbit IgGs instead of indicated antibodies. After fixation, cells were stained with secondary antibodies, mounted in Mowiol488 and imaged as described above.
Results and Discussion
Labeling of neurons with multiplex SILAC
Because primary cultured neurons do not divide in culture, conventional SILAC labeling is difficult and requires careful incorporation control analysis 4. To characterize the applicability of multiplex SILAC to primary cultured neurons, cortical neurons from E18 rat embryos were cultured for 2, 6 or 10 days in the two multiplex SILAC media before the lysates of the medium and heavy isotope labeled neurons were mixed, separated by SDS-PAGE, digested with trypsin in-gel and analyzed by LC-MS/MS. Incorporation of the heavy amino acids was measured together with the SILAC ratios. We identified and quantified 746, 844 and 862 proteins from the 2, 6 and 10 day samples respectively. A total of 448 proteins were identified in all three samples. Figure 2 shows label incorporation and heavy/medium SILAC ratios after 2, 6, and 10 days in culture.
Figure 2.

Label incorporation and the heavy/medium SILAC ratios after SILAC labeling of primary cultured neurons after two, six and ten days in culture. a. MS spectra of a representative peptide (LLASLVK from IPI00763269 Trim28). b. Incorporation and multiplex SILAC ratios of all measured proteins at each time point.
Several observations were made based on this experiment. First, with the multiplex SILAC strategy, the heavy/medium SILAC protein ratios remained very nearly 1:1 for proteins from all the three samples, indicating the accuracy of quantitation was not compromised by incomplete labeling. Second, the majority of the identified proteins were labeled to a significant degree and thus quantifiable by multiplex SILAC after the cells were labeled for only two days (median incorporation over 60%, Figure 3), which is a considerably shorter labeling period than in most conventional SILAC studies. This suggests that it is possible to use multiplex SILAC to study young neurons. This result is consistent with our previous study in which the incorporation time courses of 12 proteins were measured. However, our result is considerably different from that in another publication 5, which showed that only around 50% label incorporation was achieved after 14 days in culture. This discrepancy could be attributed to the different culture protocols used, which resulted in different neuron growth rates. Although our result shows that the multiplex labeling method enabled SILAC analysis after only two days of labeling, if circumstances permit, longer labeling times are more likely preferred to increase the amount of protein material for analysis. Third, different proteins were labeled at dramatically different rates. This phenomenon is especially significant for short labeling times. Although most proteins can be extensively labeled by using prolonged cell culture time, some proteins remained poorly labeled even after 10 days in culture, when most proteins were >90% labeled. For example, histone was only 30% labeled (spectrum not shown). Therefore, even when studying cells after many days in culture (relatively mature neurons), the multiplex labeling strategy is extremely useful for correcting for incomplete labeling of individual proteins.
Figure 3. SILAC label incorporation of proteins in primary cultured neurons after two, six and ten days in culture.

In the box plots, the outliers are defined as the data points that are more than 1.5 fold of the interquartile range (length of the box) away from the box body.
The quantified proteins were grouped into subcellular components based on their gene ontology annotations. Proteins from different components displayed different labeling efficiencies (Figure 3). In general, proteins from plasma membrane, cytoskeleton and Golgi apparatus were labeled faster than average proteins. This is probably because embryonic neurons grow many neurites while in culture, and proteins that are required to build the cytoskeleton and membranes are produced at higher rates than proteins from organelles such as ribosomes and mitochondria, which do not undergo significant increase in number. This suggests that proteins required to form neurites may have higher labeling efficiencies in SILAC experiments.
SILAC analysis of NT-3 signaling in neurons
Next we used multiplex SILAC to screen for effector proteins in NT-3 triggered signaling in cultured primary neurons. Upon binding to NT-3, the Trk receptor is activated and triggers a downstream signaling cascade that is largely tyrosine phosphorylation dependent. Anti-pY Western blotting using whole cell lysates and anti-pY IPs of E18 cortical neurons showed some increase in protein phosphorylation at the approximate MW expected for Trk receptors (i.e. approximately 95K and 145K), but did not reveal significant overall changes after the neurons were stimulated with NT-3 (Figure 4). This result suggests the changes in level of tyrosine phosphorylation for most proteins were fairly subtle compared to basal levels.
Figure 4. Tyrosine phosphorylation detected by anti-pY Western blotting of proteins in neuron lysates and pY IPs after NT-3 treatment.
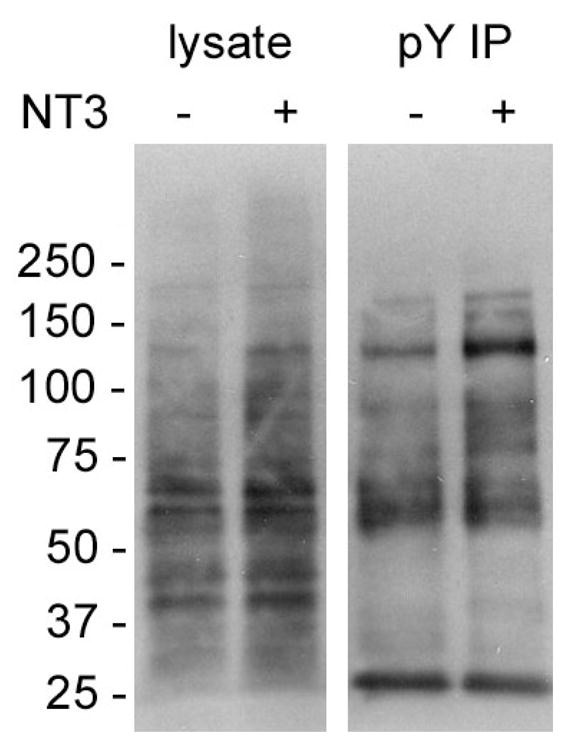
Neurons were treated with or without NT-3 as indicated in the Materials and Methods. Total cell lysates and pY IPs were probed with PY99-HRP. Numbers to the left of the gel show MW × 10−3 corresponding to protein MW standards.
E18 cortical neurons were maintained in Lys4/Arg6 or Lys8/Arg10 medium for 10 days. Then one of the cell populations was treated with NT-3 for 15 min, the treated and untreated cells were lysed and the lysates were mixed for anti-pY IP. The immunoprecipitated proteins were fractionated using 1-D SDS-PAGE, digested in-gel with trypsin and analyzed by LC-MS/MS. Proteins that changed in abundance in the pY IPs are either proteins that are tyrosine phosphorylated/dephosphorylated upon NT-3 stimulation, or binding partners of tyrosine phosphorylated proteins and thus identified as effectors in the pathway. The SILAC analyses were performed twice, and labeling conditions were reversed between replicates. In total 597 proteins were identified and 562 of them quantified (421 and 384 in one experiment respectively and 243 in both). Figure 5 shows good agreement between the SILAC ratios from the two replicates. Supporting Information Table 1 lists all the identified proteins with their SILAC ratios and extent of label incorporation. Most of the SILAC ratios are clustered tightly around 1 (Figure 6). Twenty-seven proteins exhibited stimulated/unstimulated ratios greater than 1.5 and four showed ratios less than 0.67 upon NT-3 stimulation (Table 1). Most of these proteins (21 out of 31) are known effectors in Trk signaling, demonstrating the reliability of the SILAC analysis. The known effectors identified in the SILAC analysis are involved in various aspects of Trk signaling including regulation of cell survival, differentiation and synaptic plasticity. Note that some known effectors had only slightly changed SILAC ratios, suggesting that the multiplex SILAC method is able to detect subtle but biologically significant changes in intracellular signaling. A known Trk signaling effector, Camk2b, had an insignificant SILAC ratio in our analysis. It is known that Camk2b is activated by Trk through phosphorylation on Thr rather than of Tyr sites.15 It is likely that the tyrosine phosphorylation status of this protein did not change in our experiment.
Figure 5.

SILAC protein ratios (stimulated/control) from two SILAC experiments.
Figure 6. SILAC ratios and label incorporation of proteins identified from two replicates of the NT3 SILAC experiment.
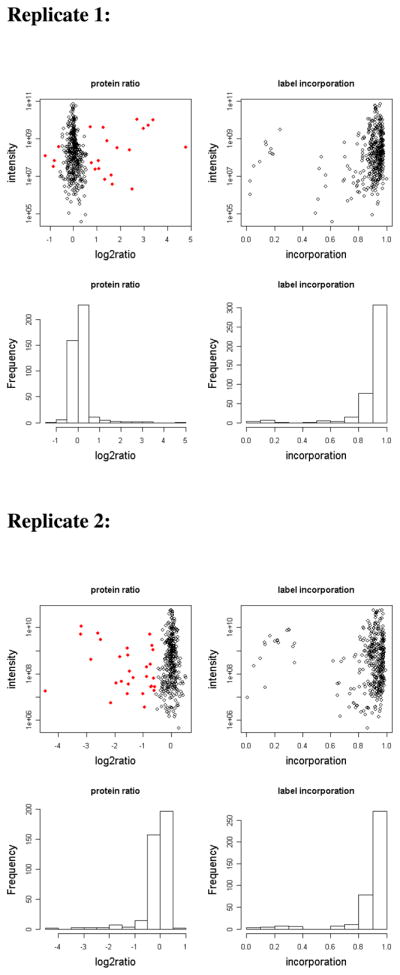
Protein ratios were calculated as heavy/medium SILAC ratios. In replicate 1, the heavy labeled sample was stimulated and the medium labeled sample was used as a control. The labeling of the stimulated and control cells was reversed in the second replicate. Red dots represent proteins with SILAC ratios showing greater than 1.5-fold change in abundance in the pY IP after stimulation.
Table 1.
proteins with changed SILAC ratios after NT-3 stimulation
| accession number | protein name | mean ratio (sti/control) | known ? |
|---|---|---|---|
| IPI00324794 | Isoform p66 of SHC-transforming protein 3 | 17.2 | Yes |
| IPI00870679 | phosphoinositide-3-kinase, class 2, beta polypeptide | 13.8 | Yes |
| IPI00210944 | Isoform GP145-TrkB of BDNF/NT-3 growth factors receptor precursor | 9.8 | Yes |
| IPI00231029 | Isoform TRKC of NT-3 growth factor receptor precursor | 9.2 | Yes |
| IPI00206172 | Isoform 1 of Mitogen-activated protein kinase 3 | 6.8 | Yes |
| IPI00119663 | Mitogen-activated protein kinase 1 | 6.3 | Yes |
| IPI00194572 | SH2B adapter protein 2 | 4.4 | Yes |
| IPI00364933 | signal transducing adaptor molecule (SH3 domain and ITAM motif) 1 | 4.0 | Yes |
| IPI00212420 | 148 kDa protein | 3.8 | Yes |
| IPI00121215 | Brain-derived neurotrophic factor precursor | 3.6 | Yes |
| IPI00206037 | similar to Secretory carrier-associated membrane protein 3 | 3.4 | No |
| IPI00119058 | Isoform 1 of Growth factor receptor-bound protein 2 | 3.0 | Yes |
| IPI00116554 | Isoform 1 of Tyrosine-protein phosphatase non-receptor type 11 | 2.8 | Yes |
| IPI00201027 | Isoform 1 of Hepatocyte growth factor-regulated tyrosine kinase substrate | 2.8 | Yes |
| IPI00212552 | fibroblast growth factor receptor substrate 2 | 2.5 | Yes |
| IPI00194958 | Isoform 2 of Phosphatidylinositol-binding clathrin assembly protein | 2.5 | Yes |
| IPI00763245 | similar to Rho GTPase-activating protein | 2.2 | Yes |
| IPI00193983 | Clathrin heavy chain 1 | 2.0 | Yes |
| IPI00208759 | syntaxin 12 | 2.0 | Yes |
| IPI00194179 | Secretory carrier-associated membrane protein 1 | 1.9 | No |
| IPI00207305 | Isoform Brain of Clathrin light chain A | 1.8 | Yes |
| IPI00466699 | Isoform 3 of Glutamate receptor 2 precursor | 1.8 | Yes |
| IPI00128454 | Isoform 2 of Seizure 6-like protein 2 precursor | 1.7 | No |
| IPI00204447 | Vesicle associated membrane protein 2B | 1.7 | No |
| IPI00750889 | Ubc protein | 1.6 | Yes |
| IPI00121131 | Neuron-specific protein family member 1 | 1.6 | No |
| IPI00364450 | Nedd4 family interacting protein 2 | 1.5 | No |
| IPI00212014 | Transitional endoplasmic reticulum ATPase | 0.7 | No |
| IPI00331865 | Isoform 2 of Drebrin-like protein | 0.6 | No |
| IPI00372478 | NCK interacting protein with SH3 domain | 0.6 | No |
| IPI00326236 | Hepatocyte growth factor receptor precursor | 0.6 | No |
TrkB is a major effector in NT-3 signaling
In addition to known effectors, the multiplex SILAC experiments revealed several interesting but previously unreported potential features of NT-3 signaling. Our SILAC results showed that TrkB was significantly activated upon NT-3 stimulation, with a ratio of 9.8 in stimulated/unstimulated in pY IPs.
The number of identified TrkB peptides was more than that of TrkC, suggesting that the amount of phosphorylated TrkB was most likely slightly greater than or at least comparable to that of TrkC (19 TrkB and 14 TrkC peptides from replicate 1 and 22 TrkB and 14 TrkC peptides from replicate 2, 125 TrkB and 75 TrkC MS/MS spectra from replicate 1 and 211 TrkB and 104 TrkC MS/MS spectra from replicate 2) and hence TrkB signaling is a major component of the signaling network induced by NT-3 in cortical neurons. This was not expected because it is generally believed that NT-3 primarily activates TrkC but not TrkB.16 We further verified this result using Western blotting. As shown in Figure 7a, both TrkB and TrkC were activated after only 1 min of NT-3 treatment. To compare the ability of NT-3 with BDNF, the primary ligand for TrkB, to activate TrkB and TrkC, neurons were stimulated with the two ligands respectively and probed for TrkB and TrkC after pY IP (Figure 7b). The result shows that while NT-3 activates both receptors in our system, BDNF specifically activates TrkB, without affecting TrkC. NT-3 and BDNF appears to be able to activate TrkB with comparable efficiencies.
Validation of Vamp2 as a downstream target of NT-3 signaling
We also verified a potential novel downstream target of NT-3 signaling found in our multiplex SILAC experiments, VAMP2, using Western blotting (Figure 7a). VAMP2 is a SNARE protein that promotes synaptic vesicle exocytosis.17 Unlike TrkB and TrkC, VAMP2 was only found in the pY IP at a later time point (~10 min) after NT-3 treatment. We confirmed that VAMP2 and TrkB/C (the antibody cannot distinguish between TrkB and TrkC in these experiments) co-localize in the neurons (Figure 8). We also showed that synaptotagmin,18 a protein that forms a protein complex with VAMP2 as part of the exocytosis machinery, co-localizes with TrkB/C in recycling vesicles (Figure 9). Interestingly, in the SILAC analysis two other proteins that are involved in exocytosis and trafficking, secretory carrier-associated membrane proteins 3 and 1 (Scamp3 and Scamp1)19 were also found to be more abundant in the pY IPs after NT-3 treatment. Taken together, our result suggests that NT-3 signaling through TrkB and/or TrkC activates components of the exocytic machinery and thus may lead to enhanced exocytosis of synaptic vesicles.
Figure 8. Co-localization of VAMP2 and TrkB/C after NT-3 stimulation.

Cortical neurons were stimulated with 25 ng/ml NT-3 for 15 min, fixed and stained for VAMP2 and TrkB/C. Scale bar, 5 μm.
Figure 9. TrkB/C localizes to recycling synaptic vesicles in hippocampal neurons.

Hippocampal neurons after 9 days in vitro were incubated with anti-synaptotagmin and anti-TrkB/C antibodies on ice, warmed to 37°C for 5 min in absence (control) or presence of 50 mM KCl, washed, fixed and counterstained for internalized antibodies. The images show that TrkB/C localizes to areas of synaptic vesicle recycling. Scale bar, 5μm.
Conclusions
The multiplex SILAC labeling strategy does not require complete labeling of the cells, making it easy to perform SILAC using non-dividing cells. We demonstrated that this approach is readily applicable to primary cultured neurons. The labeling and data analysis for partially isotope-labeled samples are greatly simplified and improved by this method. It is however worth noting that the multiplex labeling strategy leads to more complex MS spectra than conventional heavy/light SILAC labeling and label-free approaches. Thus the use of a high resolution MS instrument is desirable. We propose that this labeling strategy will greatly facilitate application of SILAC to a variety of cell models that are difficult to be labeled in a conventional SILAC workflow.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grants P30 NS050276 from NINDS and Shared Instrumentation Grant S10 RR 017990-01 to T. A. N., and EMBO and HFSP funding to K.D. We thank Daniel Spellman for advice and Jhon Sutachan-Rubio for help in animal dissection.
Abbreviations
- SILAC
stable isotope labeling with amino acids in cell culture
- IP
immunoprecipitation
- pY
phosphotyrosine
- LC
liquid chromatography
- MS
mass spectrometry
- MS/MS
tandem mass spectrometry
- BDNF
brain-derived neurotrophic factor
- NT-3
neurotrophin 3
- VAMP2
vesicle-associated membrane protein 2
Footnotes
Supporting Information Available: Supporting Information Table 1 lists all the proteins identified in the NT-3 experiment with their SILAC ratios and extent of label incorporation. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1(5):376–86. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 2.Ong SE, Mann M. A practical recipe for stable isotope labeling by amino acids in cell culture (SILAC) Nat Protoc. 2006;1(6):2650–60. doi: 10.1038/nprot.2006.427. [DOI] [PubMed] [Google Scholar]
- 3.Prokhorova TA, Rigbolt KT, Johansen PT, Henningsen J, Kratchmarova I, Kassem M, Blagoev B. SILAC-labeling and quantitative comparison of the membrane proteomes of self-renewing and differentiating human embryonic stem cells. Mol Cell Proteomics. 2009 doi: 10.1074/mcp.M800287-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spellman DS, Deinhardt K, Darie CC, Chao MV, Neubert TA. Stable isotopic labeling by amino acids in cultured primary neurons: application to brain-derived neurotrophic factor-dependent phosphotyrosine-associated signaling. Mol Cell Proteomics. 2008;7(6):1067–76. doi: 10.1074/mcp.M700387-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liao L, Park SK, Xu T, Vanderklish P, Yates JR. 3rd, Quantitative proteomic analysis of primary neurons reveals diverse changes in synaptic protein content in fmr1 knockout mice. Proc Natl Acad Sci U S A. 2008;105(40):15281–6. doi: 10.1073/pnas.0804678105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Elias JE, Haas W, Faherty BK, Gygi SP. Comparative evaluation of mass spectrometry platforms used in large-scale proteomics investigations. Nat Methods. 2005;2(9):667–75. doi: 10.1038/nmeth785. [DOI] [PubMed] [Google Scholar]
- 7.Selbach M, Schwanhausser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008;455(7209):58–63. doi: 10.1038/nature07228. [DOI] [PubMed] [Google Scholar]
- 8.Imami K, Sugiyama N, Tomita M, Ishihama Y. Quantitative proteome and phosphoproteome analyses of cultured cells based on SILAC labeling without requirement of serum dialysis. Mol Biosyst. 6(3):594–602. doi: 10.1039/b921379a. [DOI] [PubMed] [Google Scholar]
- 9.Maisonpierre PC, Belluscio L, Squinto S, Ip NY, Furth ME, Lindsay RM, Yancopoulos GD. Neurotrophin-3: a neurotrophic factor related to NGF and BDNF. Science. 1990;247(4949 Pt 1):1446–51. doi: 10.1126/science.247.4949.1446. [DOI] [PubMed] [Google Scholar]
- 10.Schecterson LC, Bothwell M. Novel roles for neurotrophins are suggested by BDNF and NT-3 mRNA expression in developing neurons. Neuron. 1992;9(3):449–63. doi: 10.1016/0896-6273(92)90183-e. [DOI] [PubMed] [Google Scholar]
- 11.Katoh-Semba R, Kaisho Y, Shintani A, Nagahama M, Kato K. Tissue distribution and immunocytochemical localization of neurotrophin-3 in the brain and peripheral tissues of rats. J Neurochem. 1996;66(1):330–7. doi: 10.1046/j.1471-4159.1996.66010330.x. [DOI] [PubMed] [Google Scholar]
- 12.Minichiello L, Klein R. TrkB and TrkC neurotrophin receptors cooperate in promoting survival of hippocampal and cerebellar granule neurons. Genes Dev. 1996;10(22):2849–58. doi: 10.1101/gad.10.22.2849. [DOI] [PubMed] [Google Scholar]
- 13.Ma L, Harada T, Harada C, Romero M, Hebert JM, McConnell SK, Parada LF. Neurotrophin-3 is required for appropriate establishment of thalamocortical connections. Neuron. 2002;36(4):623–34. doi: 10.1016/s0896-6273(02)01021-8. [DOI] [PubMed] [Google Scholar]
- 14.Reichardt LF. Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond B Biol Sci. 2006;361(1473):1545–64. doi: 10.1098/rstb.2006.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blanquet PR, Lamour Y. Brain-derived neurotrophic factor increases Ca2+/calmodulin-dependent protein kinase 2 activity in hippocampus. J Biol Chem. 1997;272(39):24133–6. doi: 10.1074/jbc.272.39.24133. [DOI] [PubMed] [Google Scholar]
- 16.Barbacid M. Structural and functional properties of the TRK family of neurotrophin receptors. Ann N Y Acad Sci. 1995;766:442–58. doi: 10.1111/j.1749-6632.1995.tb26693.x. [DOI] [PubMed] [Google Scholar]
- 17.Rossi V, Picco R, Vacca M, D’Esposito M, D’Urso M, Galli T, Filippini F. VAMP subfamilies identified by specific R-SNARE motifs. Biol Cell. 2004;96(4):251–6. doi: 10.1016/j.biolcel.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 18.Fernandez-Alfonso T, Kwan R, Ryan TA. Synaptic vesicles interchange their membrane proteins with a large surface reservoir during recycling. Neuron. 2006;51(2):179–86. doi: 10.1016/j.neuron.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 19.Castle A, Castle D. Ubiquitously expressed secretory carrier membrane proteins (SCAMPs) 1–4 mark different pathways and exhibit limited constitutive trafficking to and from the cell surface. J Cell Sci. 2005;118(Pt 16):3769–80. doi: 10.1242/jcs.02503. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.