

Abstract
Background
Evidence suggests that the noradrenergic and corticotrophin-releasing factor (CRF) systems play critical roles in relapse and stress related behaviors. In particular, behavioral studies point to a serial signaling process initiated by β-adrenergic receptors that requires CRF receptor (CRFR)-dependent signaling in the bed nucleus of the stria terminalis (BNST) to produce stress-induced relapse to cocaine seeking.
Methods
We used whole cell patch clamp recordings from acutely prepared mouse brain slices to examine the actions of β-adrenergic receptors and CRFR1 on excitatory transmission in BNST. We examined the effects of agonists of these receptors in slices prepared from naïve, sham, and cocaine conditioned mice.
Results
β1-adrenergic receptor activation within the BNST produces an enhancement of excitatory synaptic transmission that requires CRFR1-dependent signaling. We show that chronic cocaine administration transiently disrupts β1-adrenergic- and CRFR1-dependent enhancement of glutamatergic transmission, that this disruption wanes with time, and that it can be reintroduced with a cocaine challenge.
Conclusions
In total, these studies identify a circuit mechanism within the BNST that may play an important role in CRF and NE regulated behaviors.
Keywords: Addiction, cocaine, glutamate, CRF, beta adrenergic, norepinephrine
Introduction
Drug addiction is a chronically relapsing disorder, presenting a major clinical challenge for effective treatment. Addiction research has traditionally focused on dopamine (DA) and positive reinforcement-based behaviors, but in an effort to explain persistent behavioral effects there has been an increase in the discovery of targets and actions of addictive drugs on transmitters beyond the DA system. In particular, increased focus has been placed on negative reinforcement as a key driver in the addiction process. Noradrenergic and corticotrophin releasing factor (CRF) signaling systems have been heavily implicated in negative reinforcement, particularly within the extended amygdala (1–3). Both norepinephrine (NE) and CRF play crucial roles in integrating the body’s overall response to stress (4, 5). NE and CRF are critical in behavioral aspects of addiction, including the reinforcing properties of drugs (6, 7), anxiogenic effects of drug withdrawal (8–12), and reinstatement of drug seeking (13–18).
Evidence specifically suggests a serial NE-CRF receptor (CRFR)-dependent process within the bed nucleus of the stria terminalis (BNST), a key component of the extended amygdala that mediates stress-induced reinstatement of drug seeking behavior. For example, intracerebroventricular (icv) injections of NE induces drug seeking behavior that is blocked by pretreatment with a CRF antagonist (19). There is an increase in the release of NE in the BNST following chronic morphine or during morphine withdrawal (20, 21) and infusion of β-adrenergic receptor (β-AR) antagonists into the BNST attenuates morphine withdrawal-induced conditioned place aversion (20). The effects of both CRF and NE on stress-induced reinstatement to cocaine seeking have been localized to the BNST as antagonists of β-ARs and CRFRs administered into the BNST attenuate this behavior in rats (13, 18, 22). Further, these receptors likely act via increasing activity of BNST neurons, as intra-BNST injection of a GABAA receptor agonist blocks yohimbine-induced reinstatement of drug seeking behavior (23). Activation of β-ARs and CRFR1 can alter excitatory transmission within the BNST (24–28), however at present they have not been shown to interact in a functional way.
In this study we demonstrate an enhancement of excitatory transmission by β-AR activation in the BNST that is dependent on intact CRFR1 signaling. Given the involvement of CRFR and β-AR signaling within the BNST in addiction behaviors we predicted that this signaling would be altered by cocaine exposure and withdrawal. Indeed, we find that this enhancement of excitatory transmission is disrupted by repeated cocaine but not during withdrawal from repeated cocaine administration. Further, prior cocaine experience influences how CRFR signaling will respond to subsequent cocaine challenges.
Materials and Methods
Animals, Brain Slice Preparation & Electrophysiology
All procedures were performed according to Vanderbilt University Institutional Animal Care and Use Committee approved procedures. Brain slices of 300 μm thickness were prepared from 6 – 9 week old male C57BL/6J mice and recordings were performed as described previously in (25, 26, 28). Whole-cell voltage clamp recordings of AMPA receptor-mediated spontaneous excitatory postsynaptic currents (EPSCs) and synaptically stimulated evoked EPSCs were made at −70 mV and pharmacologically isolated by the addition of 25 μM picrotoxin to the ACSF [ACSF: (in mM) 124 NaCl, 4.4 KCl, 2 CaCl2, 1.2 MgSO4, 1 NaH2PO4, 10.0 glucose, and 26.0 NaHCO3]. sEPSC recordings were acquired and analyzed in 2 minute gap-free blocks. Cells in which the frequency was below 0.2Hz (n=2) were not included in the data analysis. Access resistance was monitored continuously throughout the duration of evoked experiments and between blocks during sEPSC recordings. Those experiments in which the access resistance changed by greater than 20% were not included in the data analyses. Recording electrodes for whole-cell experiments were filled with (in mM) Cs+-gluconate (135), NaCl (5), HEPES (10), EGTA (0.6), ATP (4), GTP (0.4), pH 7.2, 290–295 mOsmol. In experiments where applications of 300 nM urocortin 1 and 3 μM isoproterenol were performed the application lasted for 10 minutes in order to ensure sufficient slice wash-in. Applications of 1μM DA lasted for 5 minutes as sufficient wash-in is achieved in that time (25). For experiments in which the effects of antagonists were determined, the antagonist was applied for at least 15 minutes prior to application of the agonist and then remained on for the duration of the experiment. Mice were killed either 30 minutes or 10 days following last injection of cocaine or saline, and the brains rapidly removed for slice preparation. Following dissection, slices were transferred to a holding chamber where they were heated (29–30°C). Slices were allowed to equilibrate for at least 1 h before being transferred to a submerged perfusion chamber for subsequent whole-cell patch clamp recording. All electrophysiological recordings were made using either Clampex 8.2 or 9.2 and analyzed using Clampfit 9.2 (Molecular Devices, Sunnyvale, CA).
Pharmacology
The drugs cocaine (Sigma, NIDA), Isoproterenol (Tocris), NBI-27914 (Sigma), Betaxolol (Tocris), ICI 118,511(Tocris), Urocortin 1 (Tocris), picrotoxin (Tocris) and DA (Tocris, Ellisville, MO) were bath-applied at final concentrations which are noted in the experimental design. Dimethylsulfoxide (DMSO) is the solvent used for stock solutions of NBI-27914 where the maximum final concentration of DMSO was 0.02% by volume.
Statistical Analyses
Statistical analyses were performed using Microsoft Excel, Graphpad Prism, and Microcal Origin. When determining if a drug had a significant effect, a Student's paired t-test was used to compare the baseline value to the drug effect value. When comparing drug effects across experimental conditions (saline versus cocaine, for example) a Student’s unpaired t-test was used. Paired comparisons were made between baseline (the average of the first two recording blocks in the timecourse) and the two recording blocks immediately following removal of drug after 10 minute application unless otherwise noted. For unpaired comparisons, a percent of baseline was computed from the baseline and drug epochs defined above, and this mean was compared across experimental conditions. When comparing antagonist effects on the isoproterenol-induced increase in sEPSCs, a one-way ANOVA was used followed by a Dunnett post-test to determine the significance of specific comparisons. Statistical tests used and means compared are described in Results.
Results
β-AR activation increases spontaneous glutamatergic transmission through β1 adrenergic receptors in a CRFR dependent manner
To examine potential crosstalk between β-AR and CRFR1 signaling, we focused on neurons within the oval and undifferentiated anterolateral portions of the dorsal BNST. We focused on these regions because we previously reported that β-AR (29) and CRFR (25) activation in this region can increase glutamatergic synaptic transmission, and previous studies have indicated that dorsal BNST β-ARs are important in withdrawal responses (30). We examined sEPSCs recorded using whole-cell patch clamp from neurons located in the dlBNST (basal freq of naïve cells=2.28 ± 0.54Hz, average basal amplitude=30 ± 1.0pA, n=24) (Figure 1A, representative trace) from acutely prepared coronal slices from male C57BL/6J mice (as diagrammed in (25). We bath-applied the β-AR agonist isoproterenol (3 μM) for 10 minutes while spontaneous glutamatergic transmission was monitored and found that this short application resulted in an increase in the frequency of sEPSCs (159.9 ± 13.9% of basal frequency, p<0.01, basal versus isoproterenol, Student’s paired t-test, n=6) (Figure 1A,B) with no change in the amplitude (95 ± 3.0% of basal, n=6) (Figure 1C). We assessed the ability of two β-AR antagonists, betaxolol and ICI118,551, to disrupt the effects of isoproterenol on sEPSC frequency. Our results (Figure 1D,E) showed an effect of antagonist treatment [F(2, 13)=4.7, p<0.05] with the β1-AR antagonist 10 μM betaxolol (n=5) preventing the ability of isoproterenol to increase sEPSC frequency (p<0.05, Dunnett's post-test), while the β2-AR antagonist 10 μM ICI118,551 (n=5) was ineffective (p=0.4). The effect of isoproterenol on sEPSC frequency but not amplitude is consistent with a presynaptic site of action in enhancing glutamatergic synaptic function. To further assess this possibility, we examined the actions of isoproterenol on evoked EPSCs so that paired-pulse ratios (PPR) of evoked responses could be monitored. Isoproterenol (3 μM) elicited an enhancement of evoked EPSC amplitudes (average of minutes 20–25 = 116.3 ± 1.7% of baseline p<0.01, Student’s paired t-test, basal versus isoproterenol n=5) (Figure 1F) that was associated with a decrease in PPR, further suggesting an increase in presynaptic glutamate release (PPR of paired responses with a 50 ms interstimulus interval before isoproterenol application 1.26 ±0.03, after application 1.14 ± 0.04, p<0.05, Student’s paired t-test, basal versus isoproterenol n=5) (Figure 1F, inset).
Figure 1. Beta adrenergic receptor activation increases spontaneous glutamatergic transmission in a CRFR1-dependent manner.

A) Representative sEPSC recordings in the dlBNST demonstrating the ability of isoproterenol to enhance glutamatergic transmission. Calibration: 20 pA, 50 ms.
B) Application (10 minutes) of 3 μM isoproterenol increases sEPSC frequency in the dlBNST
C) Application (10 minutes) of 3 μM isoproterenol has no effect on sEPSC amplitude in the dlBNST
D) In the presence of the β1 adrenergic receptor antagonist betaxolol (10 μM) but not the β2 antagonist ICI118,551 (10 μM) application (10 minutes) of 3 μM isoproterenol does not alter sEPSC frequency
E) Bar graph representing the lack of effect of isoproterenol on sEPSC frequency in the presence of the β1 adrenergic receptor antagonist betaxolol but not the β2 antagonist ICI118,511 (*p<0.05).
F) Application of 3 μM isoproterenol produces a modest increase in evoked EPSCs. Inset representative traces before (black) and after (red) isoproterenol application, calibration: 100 pA, 1ms.
We previously reported that another catecholamine, DA, also enhances spontaneous excitatory transmission in this region and that this enhancement occurs through activation of endogenous CRFR1 signaling (25). A subpopulation of neurons in the BNST are CRF positive (31, 32) and the BNST also receives exogenous CRF from the central amygdala (33). Since 1) behavioral evidence suggests an interaction between NE and CRF (19) in mediating stress-induced reinstatement, and 2) an anatomical interaction exists within the BNST between NE and CRF (34), it is intriguing to consider that β-AR mediated increases in sEPSCs may also be mediated through CRFR signaling. To investigate this hypothesis we pre-applied the CRFR1 antagonist NBI27914 (1 μM) and found that this inhibited the isoproterenol-induced increase in sEPSC frequency (117 ± 13% of basal frequency, n=7) (Figure 2A). While the CRFR1 antagonist disrupted β-AR mediated increases in sEPSCs, the β1-AR antagonist betaxolol, which blocked the actions of isoproterenol, did not prevent 300 nM urocortin from enhancing sEPSC frequency (204±15% of basal frequency, n=5, p<0.01, Student’s paired t-test, basal versus urocortin plus betaxolol, data not shown).
Figure 2. Isoproterenol and DA regulate distinct excitatory synapses in the dlBNST.
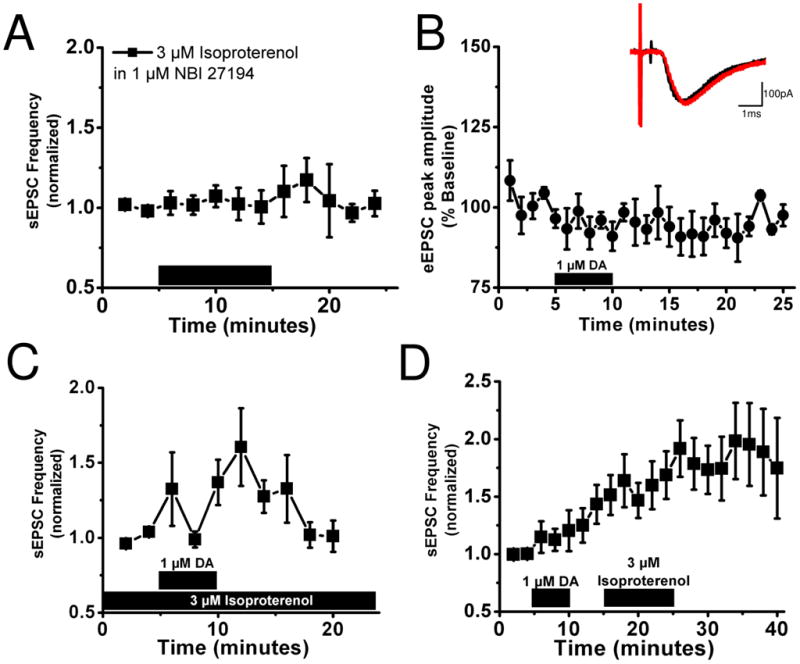
A) In the presence of 1 μM of the CRF-R1 antagonist NBI27914 application of 3 μM isoproterenol does not alter sEPSC frequency
B) Application of 1 μM DA has no effect on evoked EPSCs. Inset, representative traces before (black) and after (red) DA application, calibration: 100 pA, 1 ms.
C) Bath application of 1 μM DA increases sEPSC frequency in the presence of 3 μM isoproterenol.
D) Application of 1 μM DA increases sEPSC frequency but does not occlude the increase in sEPSC frequency produced by application of 3 μM isoproterenol.
Isoproterenol facilitates dlBNST glutamatergic synapses not facilitated by DA
Since both DA through DA receptors (25) and isoproterenol through β-ARs increase spontaneous excitatory transmission in BNST through a CRFR1 signaling dependent manner, we next sought to determine if these effects are through modulation of the same glutamate synapse population. Curiously, we found that in contrast to its actions on sEPSCs, bath application of DA (1 μM) had no effect on evoked EPSCs (average minutes 20–25 = 95.8 ± 2.3% baseline, n=5) (Figure 2B). This evidence suggests that DA and isoproterenol may be enhancing excitatory transmission at different synapses. This hypothesis was further tested by pre-applying isoproterenol (3 μM 22 minutes before DA application) to the slice followed by an application of DA (1 μM) to determine if DA could further increase sEPSC frequency. We determined that 3 μM was a saturating concentration, in that 10 μM isoproterenol did not produce a statistically significant difference in response amplitude (data not shown). Pre-application of isoproterenol failed to occlude an increase in sEPSCs frequency with subsequent DA application (160.5 ± 25.9% peak percent of frequency following DA application, p<0.05, Student’s paired t-test, isoproterenol versus isoproterenol plus dopamine, n=7) (Figure 2C). In addition, DA alone increased the sEPSC frequency to 149±18% of basal frequency but addition of isoproterenol increased the sEPSC frequency further to 163 ± 22% of DA alone (236±29% increase over initial baseline n=6, p<0.05, Student's paired t-test for Iso versus DA plus Iso; Figure 2D). Altogether, these data suggest that isoproterenol facilitates glutamatergic synapses that are not facilitated by DA.
Repeated in vivo cocaine blocks excitatory actions of CRFR1 and β1AR activation
Because of evidence implicating the BNST in behavioral actions of cocaine, we wanted to assess the impact of repeated cocaine administration on β-AR and CRFR-dependent signaling within the BNST. Therefore, mice were first habituated to handling then given intraperitoneal (ip) injections of cocaine (20 mg/kg) or saline for 10 days in a blinded design. Thirty minutes following the tenth injection of cocaine or saline, brain slices were prepared for electrophysiological experiments (Figure 3A, 4A) (See Figure 7 for summary of all conditions). We measured sEPSCs in dBNST neurons from cocaine and saline treated animals and found no gross changes in basal frequency or amplitude of sEPSCs between treatments (2.0 ± 0.6Hz and 25.3 ± 2.0 pA for cocaine, n=11 from 8 animals, 1.9 ± 0.5Hz and 24.3 ± 1.2 pA for saline, n=12 from 7 animals) (Figure S1 in the Supplement) nor any significant differences in the AMPA/NMDA ratios of evoked EPSCs (Figure S1 in the Supplement). Urocortin 1, an endogenous agonist of CRFRs (35), produced an enhancement of sEPSC frequency in slices prepared from saline-treated mice in a manner similar to that previously observed in naïve animals (Figure 3B)(25). In contrast, urocortin 1 failed to enhance sEPSC frequency in animals receiving repeated cocaine injections (cocaine 98.6 ± 5.4% of basal frequency n=6 from 4 animals, saline 146.2 ± 9.6% of basal frequency n=7 from 4 animals, p<0.01, Student’s unpaired t-test, cocaine versus saline, Figure 3B).
Figure 3. Repeated in vivo cocaine blocks excitatory actions of CRFR1 activation.
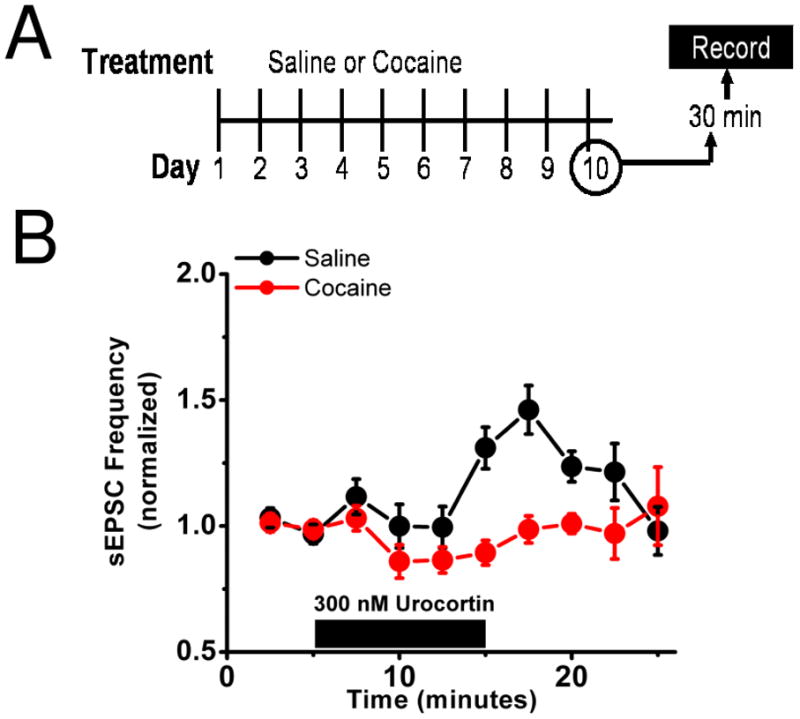
A) Diagrammatic representation of the experimental setup. Mice were given either 10 days of ip cocaine (20mg/kg) or saline. On day 10 brain slices were prepared 30 minutes after the tenth injection.
B) In mice receiving 10 days of cocaine (red) bath application of 300 nM urocortin had no effect on sEPSC frequency but increases it in those mice receiving saline (black).
Figure 4. Repeated in vivo cocaine disrupts excitatory effects of isoproterenol.
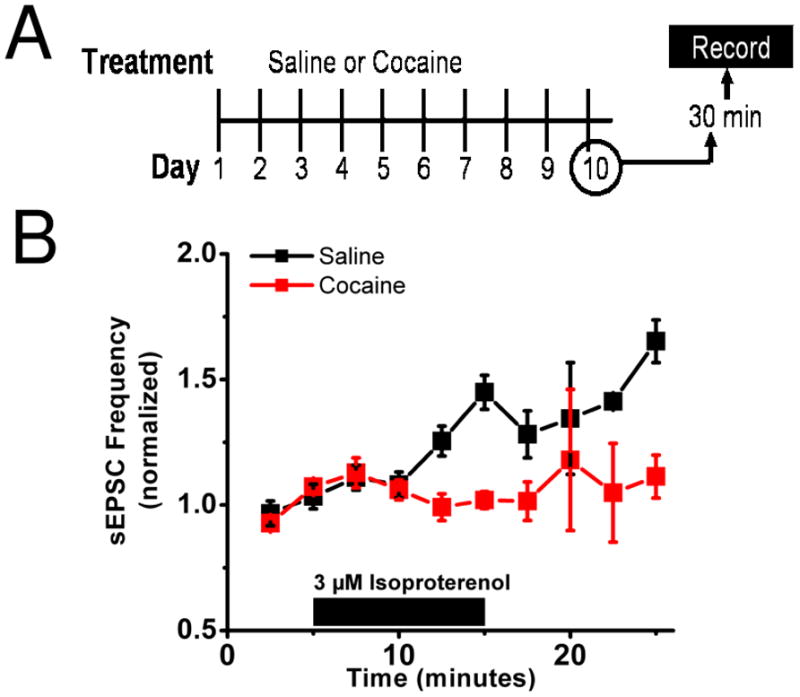
A) Diagrammatic representation of the experimental setup. Mice were given either 10 days of ip cocaine (20mg/kg) or saline. On day 10 brain slices were prepared 30 minutes after the tenth injection.
B) In mice receiving 10 days of cocaine (red) bath application of 3 μM isoproterenol had no effect on sEPSC frequency but increases it in those mice receiving saline (black).
Figure 7. Summary of effects of cocaine and withdrawal on urocortin regulation of sEPSC frequency.

Bar graph summarizing the effects of repeated cocaine or saline on the effect of 300nm urocortin on sEPSC frequency (*p<0.05, Student’s unpaired t-test,)
Since β-AR activation increased sEPSCs in a CRFR1-dependent manner (Figure 2A), we next examined the effects of chronic cocaine administration on isoproterenol actions on sEPSCs. Application of isoproterenol produced an increase in sEPSC frequency in mice that received repeated saline injections, but did not alter sEPSC frequency in slices prepared from mice that received repeated cocaine injections (cocaine 101.5 ± 7.7% of basal frequency n=5 from 4 animals, saline 128.2 ± 9.4% of basal frequency n=5 from 3 animals, p<0.05 Student’s unpaired t-test, saline versus cocaine) (Figure 4B).
CRFR1-dependent enhancement of excitatory transmission recovers during withdrawal from repeated cocaine but is disrupted following a cocaine challenge
Previous studies have identified cocaine-induced changes at glutamatergic synapses within the mesocorticolimbic reward circuit that differ between acute and extended withdrawal (36–38), or only emerge after extended withdrawal (36, 39). Protracted withdrawal from alcohol, cocaine, and heroin has been recently found to impair excitability in the juxtacapsular nucleus of the BNST (40). To determine if cocaine withdrawal time is also a critical parameter for the excitatory functions of BNST CRFRs, we examined the effect of urocortin on sEPSCs in mice 10 days following 10 injections of cocaine or saline (Figure 5A). Unlike 30 minutes post cocaine, where we observed ablation of urocortin actions, urocortin was able to increase sEPSCs 10 days after cocaine administration similar to saline injection controls (cocaine 121.5 ± 9.1% of basal frequency n=9 from 6 animals, saline 134.2 ± 13.5% of basal frequency n=7 from 5 animals, p=0.4 cocaine versus saline, Student’s unpaired t-test,) (Figure 5B).
Figure 5. Actions of repeated in vivo cocaine on excitatory effects of urocortin are absent during withdrawal.
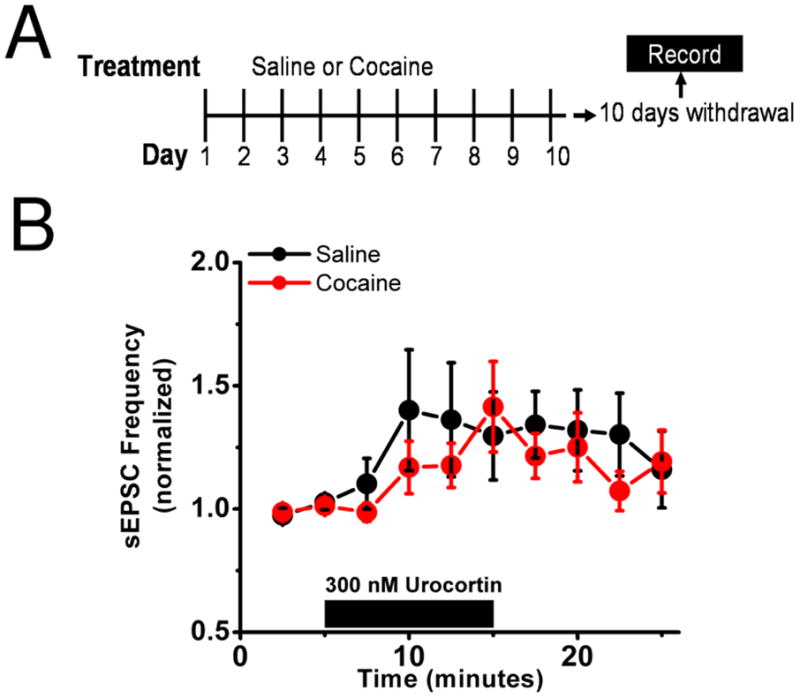
A) Diagrammatic representation of the experimental setup. Mice were given either 10 days of ip cocaine (20mg/kg) or saline followed by 10 days of withdrawal. On the tenth day after there last injection brain slices were prepared for electrophysiological recordings.
B) In mice receiving 10 days of cocaine (red) bath application of 300 nM urocortin produces a modest increase in sEPSC frequency and similarly increases it in those mice receiving saline (black).
A single drug exposure can reinstate drug-seeking behavior in humans and animal models (41). To test the hypothesis that cocaine history may alter the effects of subsequent cocaine administration on BNST function, we again treated mice with 10 days of ip cocaine or saline injections followed by a 10 day withdrawal period. All mice were then subsequently challenged with an ip injection of cocaine on the tenth day of cocaine/saline withdrawal (Figure 6A). Brain slices were prepared for electrophysiological experiments from mice 30 minutes after the challenge injection. Urocortin application enhanced sEPSC frequency after a single cocaine challenge in mice with a history of saline injections, but not those with a chronic cocaine injection history (cocaine 99.5 ± 9.5% of basal frequency n=5 from 4 animals, saline 152.0 ± 15.5% of basal frequency n=4 from 3 animals, p<0.05, Student’s unpaired t-test, saline versus cocaine) (Figure 6B). Taken together, these results demonstrate the ability of repeated cocaine exposure to differentially modify CRFR1 signaling in the BNST (Figure 7).
Figure 6. Urocortin enhancement of excitatory transmission is disrupted by a cocaine challenge during withdrawal.
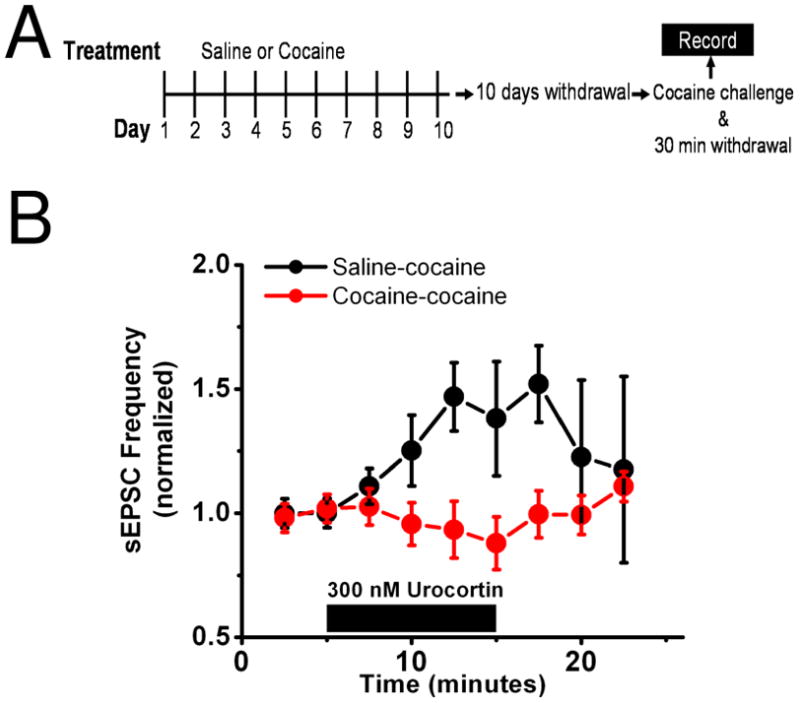
A) Diagrammatic representation of the experimental setup. Mice were given either 10 days of ip cocaine (20mg/kg) or saline followed by 10 days of withdrawal. On the tenth day of withdrawal all animals received a challenge injection of cocaine (20mg/kg) and brain slices were prepared 30 minutes following this injection.
B) In mice who had received 10 days of cocaine before the challenge (red) bath application of 300 nM urocortin had no effect on sEPSC frequency but increases it in those mice who had received saline (black).
Discussion
In the current study we report that β-AR activation increased sEPSCs in the dlBNST, a component of the extended amygdala that receives a dense noradrenergic projection and is activated by both stressors and drugs of abuse (42–47). These effects were mediated through β1-ARs and required CRFR1 signaling. Our earlier studies indicated that NE-induced increases in evoked field potentials are dependent upon β2-AR activation (29), suggesting that multiple β-AR subtypes participate in regulation of excitatory transmission in the dlBNST. In parallel, differing roles of BNST β-AR subtypes have been proposed at the behavioral level. In animals highly reactive to morphine withdrawal, a β1-AR antagonist injected into the BNST blocks withdrawal induced conditioned place aversion (30). On the other hand, a β2-AR antagonist injected into the BNST dose-dependently attenuated intraplantar-formalin-induced CPA (48).
We previously found that DA, through multiple DA receptors, also increases sEPSC frequency in a CRFR1-dependent manner in the dlBNST (25). Interestingly, while both the actions of DA receptors and β-ARs on excitatory transmission in BNST required CRFR1 signaling, the actions of DA were specific to sEPSCs, while β-AR activation modulates both sEPSCs and evoked EPSCs. In addition, we found that DA could still produce a further enhancement of sEPSCs under a period of maximal β-AR activation and vice versa. Thus these data suggest the interesting possibility that DA and NE may modulate distinct populations of excitatory synapses in the BNST.
Finally, we found that CRFR1 dependent modulation of excitatory transmission in the dlBNST is disrupted by repeated cocaine administration. The CRFR1 actions return after 10 days of withdrawal, but now appear to be more labile, as they are disrupted by a single cocaine challenge.
β-AR activation enhances BNST glutamate release in a CRFR1-dependent manner
The BNST receives both dopaminergic projections (49, 50) as well as a dense noradrenergic input from the nucleus tractus solitarius via the ventral noradrenergic bundle (51). Our previous and current results indicate that both DA and NE can enhance glutamatergic transmission in the dlBNST. We speculate that in both cases the CRFR1s that mediate this increased glutamate release are localized on presynaptic glutamatergic terminals in the dlBNST. Indeed, immunohistochemical evidence suggests that CRFR1 is expressed on excitatory terminals in BNST(52).
The BNST is an important region for the integration of stress and reward information. There is an acute increase in NE in the BNST after restraint stress (44) as well as an increase in DA during administration of drugs of abuse (42). Therefore, our data indicate that both acute stress and drugs of abuse or reward may enhance excitatory transmission via activation of CRFR1 signaling. It is interesting to speculate that perhaps NE and DA are activating different populations of CRFR1, with DA enhancing CRF release from local dlBNST neurons and NE activating extrinsic CRF afferents to the regions (for example from the central nucleus of the amygdala).
Repeated cocaine disrupts CRFR1 signaling and decreases output from the dlBNST
In naive slices both DA receptor and β-AR signaling enhance excitatory transmission through CRFR1 actions in the dlBNST. There is evidence for downregulation of CRF signaling following repeated administration of drugs of abuse throughout the brain. For example, in the central nucleus of the amygdala (CeA) CRF release (53) and mRNA levels (54) are decreased after repeated administration of cocaine. Further, CRFR1 is internalized 30 minutes following a 14-day escalating dose morphine treatment (52). Thus, the disruption of CRFR1 signaling that we observe after repeated cocaine administration likely reflects a downregulation of CRFR1 function. This raises the interesting possibility that NE signaling may be effectively re-routed through previously described α1 and α2-ARs signaling, which leads to reductions in BNST excitatory transmission (27–29, 55).
Withdrawal from repeated cocaine sensitizes the CRF system in the dlBNST to cocaine challenge
While it appears that there is a downregulation of CRF signaling following repeated administration of drugs of abuse, there is evidence that an opposite phenomenon may occur during extended withdrawal. In rats in withdrawal from ethanol there is a marked increase in CRF release in the BNST as measured by microdialysis (56). There is also an increase in CRF release in the CeA during opiate and cocaine withdrawal (53, 57). Moreover, Roberto and colleagues recently demonstrated that chronic ethanol exposure and withdrawal enhances CRF-dependent signaling in the CeA(58). Our results show that while there may be increased CRF during withdrawal, CRF signaling remains intact as urocortin still increases sEPSC frequency. However, if a challenge of cocaine is given during this period CRFR1 signaling is once again disrupted.
A single injection of cocaine to drug-naïve animals produces no disruption of CRF signaling (see saline-cocaine animals, Figure 6) and in fact has been shown to enhance short term plasticity (25), but animals that have previously received cocaine now have a disruption of CRF signaling with a single challenge injection. One possible explanation is that CRF signaling within the BNST may be highly sensitized during withdrawal. Perhaps in the presence of the higher levels of CRF that are present during withdrawal there is a change in the properties of the CRFRs that are returned to the membrane during the withdrawal state. CRFRs are known for promiscuous signaling (59) and in fact in the lateral septal nucleus CRFR2 receptors shift from signaling through PKA to largely through PKC following chronic cocaine administration (60). Another possibility is a shift in the relative number of CRFR1 and CRFR2 within the BNST following cocaine withdrawal. In the hippocampus, blocking CRFR1 but not CRFR2 attenuates LTP in naïve rats but these effects are reversed in rats in withdrawal from cocaine where now CRFR2 antagonism attenuates LTP (61).
One or a combination of these mechanisms could produce BNST neurons that are highly sensitive to CRF-signaling. In this new state, when a cocaine injection is given which would presumably increase DA within the BNST (42) CRF signaling may be rapidly activated, producing either a quick removal of CRF receptors from the membrane, a change in the type of CRF receptors present, or attenuation in their ability to be activated. Therefore, the ability to further activate this system is occluded and no effect is observed by the application of urocortin in animals 30 minutes after cocaine administration.
Implications for reinstatement models
The BNST, and specifically β-AR and CRF signaling within the BNST, are critically involved in stress-induced reinstatement models of drug seeking (18, 62). In the model proposed here, a stressor during the withdrawal period, which is known to increase NE release in the BNST, would be predicted to activate the system in a similar manner as a cocaine challenge. The disrupted state of CRF signaling by repeated administration and the subsequent highly-sensitized system that develops during withdrawal may be what is setting the molecular switch that is triggered for stressed-induced relapse. A drug challenge is likely activating other systems in addition to what is described by this model which are critical for drug priming induced reinstatement, such as CRF signaling within the accumbens (63). This may in part explain why CRFR antagonism in the BNST has no effect on drug priming models of reinstatement (41), yet an important effect on stress-induced reinstatement. Furthermore, this model projects a cellular mechanism for the finding that icv norepinephrine failed to induce reinstatement when it was preceded by a pretreatment of CRF antagonist (19).
In conclusion, our results indicate a novel interaction between NE and CRF to enhance excitatory transmission within a region that plays a key role in mediating aspects of anxiety, reward, and relapse-related behaviors. Further, we describe how this system is disrupted and subsequently altered by repeated cocaine and withdrawal.
Supplementary Material
Acknowledgments
Research supported by the NIAAA and NIDA.
Footnotes
DISCLOSURE OF BIOMEDICAL FINANCIAL INTERESTS AND POTENTIAL CONFLICTS OF INTEREST: The authors reported no biomedical financial interests or potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Koob GF. A Role for Brain Stress Systems in Addiction. Neuron. 2008;59:11–34. doi: 10.1016/j.neuron.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Piazza PV, Le Moal M. The role of stress in drug self-administration. Trends in Pharmacological Sciences. 1998;19:67–74. doi: 10.1016/s0165-6147(97)01115-2. [DOI] [PubMed] [Google Scholar]
- 3.Sinha R. Chronic stress, drug use, and vulnerability to addiction. Ann N Y Acad Sci. 2008;1141:105–130. doi: 10.1196/annals.1441.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsigos C, Chrousos GP. Hypothalamic-pituitary-adrenal axis, neuroendocrine factors and stress. J Psychosom Res. 2002;53:865–871. doi: 10.1016/s0022-3999(02)00429-4. [DOI] [PubMed] [Google Scholar]
- 5.Koob GF. Corticotropin-releasing factor, norepinephrine, and stress. Biol Psychiatry. 1999;46:1167–1180. doi: 10.1016/s0006-3223(99)00164-x. [DOI] [PubMed] [Google Scholar]
- 6.Goeders NE, Guerin GF. Effects of the CRH receptor antagonist CP-154,526 on intravenous cocaine self-administration in rats. Neuropsychopharmacology. 2000;23:577–586. doi: 10.1016/S0893-133X(00)00148-2. [DOI] [PubMed] [Google Scholar]
- 7.Piazza PV, Le Moal ML. Pathophysiological basis of vulnerability to drug abuse: role of an interaction between stress, glucocorticoids, and dopaminergic neurons. Annu Rev Pharmacol Toxicol. 1996;36:359–378. doi: 10.1146/annurev.pa.36.040196.002043. [DOI] [PubMed] [Google Scholar]
- 8.Delfs JM, Zhu Y, Druhan JP, Aston-Jones G. Noradrenaline in the ventral forebrain is critical for opiate withdrawal-induced aversion. Nature. 2000;403:430–434. doi: 10.1038/35000212. [DOI] [PubMed] [Google Scholar]
- 9.Heinrichs SC, Menzaghi F, Schulteis G, Koob GF, Stinus L. Suppression of corticotropin-releasing factor in the amygdala attenuates aversive consequences of morphine withdrawal. Behav Pharmacol. 1995;6:74–80. [PubMed] [Google Scholar]
- 10.Menzaghi F, Rassnick S, Heinrichs S, Baldwin H, Pich EM, Weiss F, et al. The role of corticotropin-releasing factor in the anxiogenic effects of ethanol withdrawal. Ann N Y Acad Sci. 1994;739:176–184. doi: 10.1111/j.1749-6632.1994.tb19819.x. [DOI] [PubMed] [Google Scholar]
- 11.Rodriguez de Fonseca F, Carrera MR, Navarro M, Koob GF, Weiss F. Activation of corticotropin-releasing factor in the limbic system during cannabinoid withdrawal. Science. 1997;276:2050–2054. doi: 10.1126/science.276.5321.2050. [DOI] [PubMed] [Google Scholar]
- 12.Sarnyai Z, Biro E, Gardi J, Vecsernyes M, Julesz J, Telegdy G. Brain corticotropin-releasing factor mediates 'anxiety-like' behavior induced by cocaine withdrawal in rats. Brain Res. 1995;675:89–97. doi: 10.1016/0006-8993(95)00043-p. [DOI] [PubMed] [Google Scholar]
- 13.Erb S, Hitchcott PK, Rajabi H, Mueller D, Shaham Y, Stewart J. Alpha-2 adrenergic receptor agonists block stress-induced reinstatement of cocaine seeking. Neuropsychopharmacology. 2000;23:138–150. doi: 10.1016/S0893-133X(99)00158-X. [DOI] [PubMed] [Google Scholar]
- 14.Erb S, Shaham Y, Stewart J. The role of corticotropin-releasing factor and corticosterone in stress- and cocaine-induced relapse to cocaine seeking in rats. J Neurosci. 1998;18:5529–5536. doi: 10.1523/JNEUROSCI.18-14-05529.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Le AD, Harding S, Juzytsch W, Watchus J, Shalev U, Shaham Y. The role of corticotrophin-releasing factor in stress-induced relapse to alcohol-seeking behavior in rats. Psychopharmacology (Berl) 2000;150:317–324. doi: 10.1007/s002130000411. [DOI] [PubMed] [Google Scholar]
- 16.Shaham Y, Funk D, Erb S, Brown TJ, Walker CD, Stewart J. Corticotropin-releasing factor, but not corticosterone, is involved in stress-induced relapse to heroin-seeking in rats. J Neurosci. 1997;17:2605–2614. doi: 10.1523/JNEUROSCI.17-07-02605.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shaham Y, Highfield D, Delfs J, Leung S, Stewart J. Clonidine blocks stress-induced reinstatement of heroin seeking in rats: an effect independent of locus coeruleus noradrenergic neurons. Eur J Neurosci. 2000;12:292–302. doi: 10.1046/j.1460-9568.2000.00899.x. [DOI] [PubMed] [Google Scholar]
- 18.Leri F, Flores J, Rodaros D, Stewart J. Blockade of stress-induced but not cocaine-induced reinstatement by infusion of noradrenergic antagonists into the bed nucleus of the stria terminalis or the central nucleus of the amygdala. J Neurosci. 2002;22:5713–5718. doi: 10.1523/JNEUROSCI.22-13-05713.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brown Z, Tribe E, D’souza N, Erb S. Interaction between noradrenaline and corticotrophin-releasing factor in the reinstatement of cocaine seeking in the rat. Psychopharmacology. 2009;203:121–130. doi: 10.1007/s00213-008-1376-4. [DOI] [PubMed] [Google Scholar]
- 20.Aston-Jones G, Delfs JM, Druhan J, Zhu Y. The bed nucleus of the stria terminalis. A target site for noradrenergic actions in opiate withdrawal. Annals of the New York Academy of Sciences. 1999;877:486–498. doi: 10.1111/j.1749-6632.1999.tb09284.x. [DOI] [PubMed] [Google Scholar]
- 21.Harris GC, Aston-Jones G. Activation in extended amygdala corresponds to altered hedonic processing during protracted morphine withdrawal. Behavioural Brain Research. 2007;176:251–258. doi: 10.1016/j.bbr.2006.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Erb S, Stewart J. A Role for the Bed Nucleus of the Stria Terminalis, But Not the Amygdala, in the Effects of Corticotropin-Releasing Factor on Stress-Induced Reinstatement of Cocaine Seeking. J Neurosci. 1999;19:35RC–35RC. doi: 10.1523/JNEUROSCI.19-20-j0006.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Buffalari DM, See RE. Inactivation of the bed nucleus of the stria terminalis in an animal model of relapse: effects on conditioned cue-induced reinstatement and its enhancement by yohimbine. Psychopharmacology (Berl) 2010 doi: 10.1007/s00213-010-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Egli RE, Kash TL, Choo K, Savchenko V, Matthews RT, Blakely RD, et al. Norepinephrine Modulates Glutamatergic Transmission in the Bed Nucleus of the Stria Terminalis. Neuropsychopharmacology. 2004;30:657–668. doi: 10.1038/sj.npp.1300639. [DOI] [PubMed] [Google Scholar]
- 25.Kash TL, Nobis WP, Matthews RT, Winder DG. Dopamine enhances fast excitatory synaptic transmission in the extended amygdala by a CRF-R1-dependent process. J Neurosci. 2008;28:13856–13865. doi: 10.1523/JNEUROSCI.4715-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kash TL, Winder DG. Neuropeptide Y and corticotropin-releasing factor bi-directionally modulate inhibitory synaptic transmission in the bed nucleus of the stria terminalis. Neuropharmacology. 2006;51:1013–1022. doi: 10.1016/j.neuropharm.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 27.McElligott ZA, Winder DG. Alpha1-adrenergic receptor-induced heterosynaptic long-term depression in the bed nucleus of the stria terminalis is disrupted in mouse models of affective disorders. Neuropsychopharmacology. 2008;33:2313–2323. doi: 10.1038/sj.npp.1301635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McElligott ZA, Klug JR, Nobis WP, Patel S, Grueter BA, Kash TL, et al. Distinct forms of Gq-receptor-dependent plasticity of excitatory transmission in the BNST are differentially affected by stress. Proc Natl Acad Sci U S A. 2010;107:2271–2276. doi: 10.1073/pnas.0905568107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Egli RE, Kash TL, Choo K, Savchenko V, Matthews RT, Blakely RD, et al. Norepinephrine modulates glutamatergic transmission in the bed nucleus of the stria terminalis. Neuropsychopharmacology. 2005;30:657–668. doi: 10.1038/sj.npp.1300639. [DOI] [PubMed] [Google Scholar]
- 30.Cecchi M, Capriles N, Watson SJ, Akil H. Beta1 adrenergic receptors in the bed nucleus of stria terminalis mediate differential responses to opiate withdrawal. Neuropsychopharmacology. 2007;32:589–599. doi: 10.1038/sj.npp.1301140. [DOI] [PubMed] [Google Scholar]
- 31.Day HE, Curran EJ, Watson SJ, Jr, Akil H. Distinct neurochemical populations in the rat central nucleus of the amygdala and bed nucleus of the stria terminalis: evidence for their selective activation by interleukin-1beta. J Comp Neurol. 1999;413:113–128. [PubMed] [Google Scholar]
- 32.Rodaros D, Caruana DA, Amir S, Stewart J. Corticotropin-releasing factor projections from limbic forebrain and paraventricular nucleus of the hypothalamus to the region of the ventral tegmental area. Neuroscience. 2007;150:8–13. doi: 10.1016/j.neuroscience.2007.09.043. [DOI] [PubMed] [Google Scholar]
- 33.Erb, Salmaso, Rodaros, Stewart A role for the CRF-containing pathway from central nucleus of the amygdala to bed nucleus of the stria terminalis in the stress-induced reinstatement of cocaine seeking in rats. Psychopharmacology. 2001;158:360–365. doi: 10.1007/s002130000642. [DOI] [PubMed] [Google Scholar]
- 34.Phelix CF, Liposits Z, Paull WK. Catecholamine-CRF synaptic interaction in a septal bed nucleus: afferents of neurons in the bed nucleus of the stria terminalis. Brain Res Bull. 1994;33:109–119. doi: 10.1016/0361-9230(94)90056-6. [DOI] [PubMed] [Google Scholar]
- 35.Vaughan J, Donaldson C, Bittencourt J, Perrin MH, Lewis K, Sutton S, et al. Urocortin, a mammalian neuropeptide related to fish urotensin I and to corticotropin-releasing factor. Nature. 1995;378:287–292. doi: 10.1038/378287a0. [DOI] [PubMed] [Google Scholar]
- 36.Kourrich S, Rothwell PE, Klug JR, Thomas MJ. Cocaine Experience Controls Bidirectional Synaptic Plasticity in the Nucleus Accumbens. J Neurosci. 2007;27:7921–7928. doi: 10.1523/JNEUROSCI.1859-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saal D, Dong Y, Bonci A, Malenka RC. Drugs of Abuse and Stress Trigger a Common Synaptic Adaptation in Dopamine Neurons. Neuron. 2003;37:577–582. doi: 10.1016/s0896-6273(03)00021-7. [DOI] [PubMed] [Google Scholar]
- 38.Ungless MA, Whistler JL, Malenka RC, Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature. 2001;411:583–587. doi: 10.1038/35079077. [DOI] [PubMed] [Google Scholar]
- 39.Fu Y, Pollandt S, Liu J, Krishnan B, Genzer K, Orozco-Cabal L, et al. Long-term potentiation (LTP) in the central amygdala (CeA) is enhanced after prolonged withdrawal from chronic cocaine and requires CRF1 receptors. J Neurophysiol. 2007;97:937–941. doi: 10.1152/jn.00349.2006. [DOI] [PubMed] [Google Scholar]
- 40.Francesconi W, Berton F, Repunte-Canonigo V, Hagihara K, Thurbon D, Lekic D, et al. Protracted withdrawal from alcohol and drugs of abuse impairs long-term potentiation of intrinsic excitability in the juxtacapsular bed nucleus of the stria terminalis. J Neurosci. 2009;29:5389–5401. doi: 10.1523/JNEUROSCI.5129-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shaham Y, Shalev U, Lu L, De Wit H, Stewart J. The reinstatement model of drug relapse: history, methodology and major findings. Psychopharmacology (Berl) 2003;168:3–20. doi: 10.1007/s00213-002-1224-x. [DOI] [PubMed] [Google Scholar]
- 42.Carboni E, Silvagni A, Rolando MT, Di Chiara G. Stimulation of in vivo dopamine transmission in the bed nucleus of stria terminalis by reinforcing drugs. J Neurosci. 2000;20:RC102. doi: 10.1523/JNEUROSCI.20-20-j0002.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Funk D, Li Z, Lê AD. Effects of environmental and pharmacological stressors on c-fos and corticotropin-releasing factor mRNA in rat brain: Relationship to the reinstatement of alcohol seeking. Neuroscience. 2006;138:235–243. doi: 10.1016/j.neuroscience.2005.10.062. [DOI] [PubMed] [Google Scholar]
- 44.Ma S, Morilak DA. Norepinephrine release in medial amygdala facilitates activation of the hypothalamic-pituitary-adrenal axis in response to acute immobilisation stress. J Neuroendocrinol. 2005;17:22–28. doi: 10.1111/j.1365-2826.2005.01279.x. [DOI] [PubMed] [Google Scholar]
- 45.Morilak DA, Barrera G, Echevarria DJ, Garcia AS, Hernandez A, Ma S, et al. Role of brain norepinephrine in the behavioral response to stress. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29:1214–1224. doi: 10.1016/j.pnpbp.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 46.Valjent E, Pages C, Herve D, Girault JA, Caboche J. Addictive and non-addictive drugs induce distinct and specific patterns of ERK activation in mouse brain. Eur J Neurosci. 2004;19:1826–1836. doi: 10.1111/j.1460-9568.2004.03278.x. [DOI] [PubMed] [Google Scholar]
- 47.Park J, Kile BM, Wightman RM. In vivo voltammetric monitoring of norepinephrine release in the rat ventral bed nucleus of the stria terminalis and anteroventral thalamic nucleus. Eur J Neurosci. 2009;30:2121–2133. doi: 10.1111/j.1460-9568.2009.07005.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deyama S, Katayama T, Ohno A, Nakagawa T, Kaneko S, Yamaguchi T, et al. Activation of the beta-adrenoceptor-protein kinase A signaling pathway within the ventral bed nucleus of the stria terminalis mediates the negative affective component of pain in rats. J Neurosci. 2008;28:7728–7736. doi: 10.1523/JNEUROSCI.1480-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meloni EG, Jackson A, Gerety LP, Cohen BM, Carlezon WA., Jr Role of the bed nucleus of the stria terminalis (BST) in the expression of conditioned fear. Ann N Y Acad Sci. 2006;1071:538–541. doi: 10.1196/annals.1364.059. [DOI] [PubMed] [Google Scholar]
- 50.Phelix CF, Liposits Z, Paull WK. Monoamine innervation of bed nucleus of stria terminalis: an electron microscopic investigation. Brain Res Bull. 1992;28:949–965. doi: 10.1016/0361-9230(92)90218-m. [DOI] [PubMed] [Google Scholar]
- 51.Forray MI, Gysling K. Role of noradrenergic projections to the bed nucleus of the stria terminalis in the regulation of the hypothalamic-pituitary-adrenal axis. Brain Res Brain Res Rev. 2004;47:145–160. doi: 10.1016/j.brainresrev.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 52.Jaferi A, Lane DA, Pickel VM. Subcellular plasticity of the corticotropin-releasing factor receptor in dendrites of the mouse bed nucleus of the stria terminalis following chronic opiate exposure. Neuroscience. 2009;163:143–154. doi: 10.1016/j.neuroscience.2009.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Richter RM, Weiss F. In vivo CRF release in rat amygdala is increased during cocaine withdrawal in self-administering rats. Synapse. 1999;32:254–261. doi: 10.1002/(SICI)1098-2396(19990615)32:4<254::AID-SYN2>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 54.Maj M, Turchan J, Smialowska M, Przewlocka B. Morphine and cocaine influence on CRF biosynthesis in the rat central nucleus of amygdala. Neuropeptides. 2003;37:105–110. doi: 10.1016/s0143-4179(03)00021-0. [DOI] [PubMed] [Google Scholar]
- 55.Davis AR, Shields AD, Brigman JL, Norcross M, McElligott ZA, Holmes A, et al. Yohimbine impairs extinction of cocaine-conditioned place preference in an alpha2-adrenergic receptor independent process. Learn Mem. 2008;15:667–676. doi: 10.1101/lm.1079308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Olive MF, Koenig HN, Nannini MA, Hodge CW. Elevated extracellular CRF levels in the bed nucleus of the stria terminalis during ethanol withdrawal and reduction by subsequent ethanol intake. Pharmacol Biochem Behav. 2002;72:213–220. doi: 10.1016/s0091-3057(01)00748-1. [DOI] [PubMed] [Google Scholar]
- 57.Iredale PA, Alvaro JD, Lee Y, Terwilliger R, Chen YL, Duman RS. Role of corticotropin-releasing factor receptor-1 in opiate withdrawal. J Neurochem. 2000;74:199–208. doi: 10.1046/j.1471-4159.2000.0740199.x. [DOI] [PubMed] [Google Scholar]
- 58.Roberto M, Cruz MT, Gilpin NW, Sabino V, Schweitzer P, Bajo M, et al. Corticotropin releasing factor-induced amygdala gamma-aminobutyric Acid release plays a key role in alcohol dependence. Biol Psychiatry. 2010;67:831–839. doi: 10.1016/j.biopsych.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Blank T, Nijholt I, Grammatopoulos DK, Randeva HS, Hillhouse EW, Spiess J. Corticotropin-releasing factor receptors couple to multiple G-proteins to activate diverse intracellular signaling pathways in mouse hippocampus: role in neuronal excitability and associative learning. J Neurosci. 2003;23:700–707. doi: 10.1523/JNEUROSCI.23-02-00700.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu J, Yu B, Orozco-Cabal L, Grigoriadis DE, Rivier J, Vale WW, et al. Chronic cocaine administration switches corticotropin-releasing factor2 receptor-mediated depression to facilitation of glutamatergic transmission in the lateral septum. J Neurosci. 2005;25:577–583. doi: 10.1523/JNEUROSCI.4196-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guan X, Zhang R, Xu Y, Li S. Cocaine withdrawal enhances long-term potentiation in rat hippocampus via changing the activity of corticotropin-releasing factor receptor subtype 2. Neuroscience. 2009;161:665–670. doi: 10.1016/j.neuroscience.2009.04.035. [DOI] [PubMed] [Google Scholar]
- 62.Erb S, Stewart J. A role for the bed nucleus of the stria terminalis, but not the amygdala, in the effects of corticotropin-releasing factor on stress-induced reinstatement of cocaine seeking. J Neurosci. 1999;19:RC35. doi: 10.1523/JNEUROSCI.19-20-j0006.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang J, Fang Q, Liu Z, Lu L. Region-specific effects of brain corticotropin-releasing factor receptor type 1 blockade on footshock-stress- or drug-priming-induced reinstatement of morphine conditioned place preference in rats. Psychopharmacology (Berl) 2006;185:19–28. doi: 10.1007/s00213-005-0262-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.