

SUMMARY
Cardiac neural crest cells migrate into the pharyngeal arches where they support development of the pharyngeal arch arteries. The pharyngeal endoderm and ectoderm both express high levels of FGF8. We hypothesized that FGF8 is chemotactic for cardiac crest cells. To begin testing this hypothesis, cardiac crest was explanted for migration assays under various conditions. Cardiac neural crest cells migrated more in response to FGF8. Single cell tracing indicated that this was not due to proliferation and subsequent transwell assays showed that the cells migrate toward an FGF8 source. The migratory response was mediated by FGF receptors (FGFR) 1 and 3 and MAPK/ERK intracellular signaling. To test whether FGF8 is chemokinetic and/or chemotactic in vivo, dominant negative FGFR1 was electroporated into the premigratory cardiac neural crest. Cells expressing the dominant negative receptor migrated slower than normal cardiac neural crest cells and were prone to remain in the vicinity of the neural tube and die. Treating with the FGFR1 inhibitor, SU5402 or an FGFR3 function-blocking antibody also slowed neural crest migration. FGF8 over-signaling enhanced neural crest migration. Neural crest cells migrated to an FGF8-sosked bead placed dorsal to the pharynx. Finally, an FGF8 producing plasmid was electroporated into an ectopic site in the ventral pharyngeal endoderm. The FGF8 producing cells attracted a thick layer of mesenchymal cells. DiI labeling of the neural crest as well as quail-to-chick neural crest chimeras showed that neural crest cells migrated to and around the ectopic site of FGF8 expression. These results showing that FGF8 is chemotactic and chemokinetic for cardiac neural crest adds another dimension to understanding the relationship of FGF8 and cardiac neural crest in cardiovascular defects.
Keywords: cardiac neural crest, FGF8, heart development, chemokinesis, chemotaxis, migration
Introduction
FGF8 is produced by the lateral pharyngeal endoderm and ectoderm during development of the pharyngeal arches and is critical for their formation. However, the varied roles of FGF8 in pharyngeal development have been difficult to elucidate because targeted disruption of the fgf8 gene in mice causes the embryos to die at mid-gastrulation (Sun et al., 1999) making it impossible to assess their role in later development. Some information has been available from fgf8 hypomorphic mice that produce enough FGF8 for the embryos to survive through organogenesis and show that FGF8 signaling is critical for normal development of the pharynx and heart as well as neural crest cells migrating to these structures (Abu-Issa et al., 2002; Frank et al., 2002). The neural crest cells required for cardiac development originate from postotic rhombomeres 6, 7 and 8 and migrate to the caudal pharynx (arches 3–6). They are important for normal conversion of the aortic arch arteries to the great arteries (Le Lievre and Le Douarin, 1975; Waldo et al., 1996). A subset of these cells migrates to the arterial pole where they form the aorticopulmonary septation complex which divides the arterial pole into systemic and pulmonary channels (Kirby et al., 1983). Formation of the pharyngeal pouches/grooves which is dependent on FGF8 signaling, is important to restrict the cranial neural crest streams to particular pharyngeal arches (Trumpp et al., 1999).
FGF8 signaling also has important functions during early stages of neural crest development. During early neural crest specification, FGF signaling is needed for expression of slug, a transcription factor required for neural crest delamination and migration. In Xenopus, overexpression of a dominant negative FGF receptor 1 leads to a loss of neural crest formation in Xenopus embryos (Mayor et al., 1997). However, these embryos experience loss of FGFR1 signaling from the single cell stage, making it likely that the neural plate/tube is not normally patterned. Wnt and FGF8 signals act in parallel at the neural border converging on Pax3 activity, which in turn activates slug (Monsoro-Burq et al., 2005).
In an environment of reduced FGF8 signaling, as in fgf8 hypomorphic mice, neural crest cells are specified and seem to begin migration in normal numbers but not enough FGF8 is available to support viability of the cells. Some of the cells die as they leave the neural tube, and those reaching the pharynx undergo massive cell death (Abu-Issa et al., 2002; Frank et al., 2002) leading to various defects in pharyngeal arch artery patterning and in the cardiac outflow tract that represent a combination of outflow malalignment and outflow septation defects.
Our understanding of the role of pharyngeal FGF8 has been further refined by conditional deletion of FGF8 in the pharyngeal ectoderm and endoderm. Conditional deletion in the pharyngeal ectoderm led to failure of arch artery 4 to develop while conditional deletion in the endoderm led to failure in outflow tract septation (Park et al., 2006). Failure in arch artery 4 development was not attributable to neural crest while outflow tract septation was attributed to the reduced number of cardiac neural crest cells. Thus there are several points during neural crest specification and migration into the pharynx that require FGF8 signaling.
It has recently been demonstrated that neural crest cells migrating to pharyngeal arch 2 are attracted there by VEGF signaling (McLennan et al., 2010); however, VEGF is not chemoattractant for cardiac neural crest cells and it is not known if cardiac crest cells are chemoattracted to the caudal pharynx. Some evidence suggests that neural crest cells are attracted to FGF8. After ablation of preotic neural crest which results in a dramatic decrease in FGF8 expression in the frontonasal process and pharyngeal ectoderm, postotic crest can be induced to migrate into the frontonasal process and pharyngeal arch 1 by placing a bead of FGF8 near pharyngeal arch 1 (Creuzet et al., 2002). While it was speculated that the postotic crest was attracted to the region by FGF8, this hypothesis was never rigorously tested.
Two studies have shown FGF8 to be a chemoattractant or chemorepellent for migrating cells depending on context and cell population. Newly gastrulated mesoderm cells migrating from the primitive streak are repelled by FGF8 and attracted to the notochord by FGF4 (Yang et al., 2002). By contrast mesencephalic neural crest cells are attracted to a source of FGF8 in vitro (Kubota and Ito, 2000). In vivo, FGF8 was found to be important for the localization of FGF2, which acted as the chemotactic agent. The chemotaxis was mediated by high affinity FGF receptors (FGFR) 1 and 3.
This paper reports a series of experiments designed to evaluate the role of FGF8 signaling in cardiac neural crest cell migration to the caudal pharyngeal arches. Explanted premigratory cardiac neural crest cells showed a dose-dependent increase in migration index and enhanced single cell migration in the presence of FGF8. Transwell assay showed the explanted cardiac neural crest cells migrate toward an FGF8 source. The migration effect was mediated by FGFR1 and R3 and the Mek/Erk, PI3K and p38 intracellular pathways. In vivo, treatment of embryos with SU5402 to block FGF signaling or expression of dominant negative Fgfr1 delayed cardiac neural crest migration and enhanced cardiac neural crest cell death. Finally, the cardiac neural crest cells are attracted to ectopic sites of FGF8 overexpression. These results indicate that FGF8 is chemotactic and chemokinetic for cardiac neural crest cells and gives another basis for understanding the role of FGF8 in cardiac neural crest-related cardiovascular defects.
Materials and methods
Migration assays
The premigratory neural folds were dissected cleanly from the neural tube between the mid-otic placode and somite 3 as previously described (Kirby et al., 1997). The neural folds were plated on fibronectin (BD Sciences)-coated 8-well glass slides and cultured on a heated stage (LiveCell, Westminster, MD) overnight in 5% FBS in Liebovitz’s L-15 (Gibco) media with high glucose in the absence of CO2. Timelapse images were captured every 10 min for a 16 hr period using a Nikon Eclipse TE2000-U microscope and MetaMorph image analysis software. Cell migration indices were quantified as reported previously (Huang et al., 1998). Differences in migration area resulting from random variation in the size/shape of the explant were controlled by dividing the outgrowth area (mm2) by the perimeter (mm) of the explant. This normalized number is referred to as the migration index and provides an estimate of the migration rate of the neural crest cells. A minimum of nine explants per treatment was recorded.
For single cell analysis total distances that each cell moved were traced and calculated and normalized for the amount of time that the cell was visible in the plane of the recorded images as previously reported by the Lo lab (Xu et al., 2006). At least five cells per experimental condition were quantified. Because directionality of neural crest movement in culture is more dependent on repulsive forces of the neural fold, directionality was not noted so only the total distance the cell moved is reported. Statistical significance was determined using a one-way ANOVA; post-hoc analysis, if necessary (ANOVA p<0.05), was done using an unpaired Student’s t-test with equal variances.
Chemotaxis assays were performed using ChomoTx Chemotaxis System (NeuroProbe, Inc). The assays were performed in 96 well format with an 8 um pore size. For each experiment 24 neural folds were removed from mid-otic placode to somite 3 from HH9 embryos, rinsed in 2%FBS/L-15 medium, and further dissected into small pieces to facilitate cell dissociation. The pieces were dissociated for 5 min at 37°C in 0.25% Trypsin-EDTA (Sigma). Media containing 5% FBS was added and the cells were pelleted by a 5 min centrifugation at 1000rpm. The dissociated cells were then resuspended in 750 ul of 5% FBS/DMEM. The lower well was prepared with the following treatments in replicates of 6: 2% FBS DMEM medium (Control), 2% FBS DMEM with 10ng/ml FGF8 (human recombinant, PeproTech, Inc.); 2% FBS DMEM with 30ng/ml FGF8; 2% FBS DMEM with 10mM SU5402 (Tocris Biosciences). The dissociated cells were pipetted onto the chemotaxis membrane and the plate incubated overnight 37°C, 5% CO2. The membrane was fixed in ice-cold methanol (10 min 4°C), stained with Toluidine blue (0.1% in distilled water) for 3 min at room temperature, rinsed gently in distilled H2O. The cells on the topside of the Chemotaxis membrane were removed with a cotton swab. All of the cells on the underside of the membrane were counted. The experiment was repeated 6 times.
Inhibitors
All inhibitors were used at a concentration of 10µM. The FGFR blocking compound, SU5402 (Calbiochem or Tocris), and the Mek inhibitor, U0126 (Promega), were both resuspended in DMSO. The PLCγ inhibitor, U73122 (ENZO Life Sciences), was resuspended in ethanol. For the in ovo SU5402 treatment, embryos were treated with 20µL of 10µM SU5402 in 1× PBS pH 7.4 as previously described (Hutson et al., 2006). FGFR1 and R3 neutralizing antibodies were made by New England Peptide (Gardner, MA) in rabbit using peptides designed to the ligand binding domain. In the in vitro experiments the function blocking antibodies were used at 5µM concentrations. For in vivo studies, embryos were treated with 20µL of a 5µM FGFR3 antibody in PBS alone or 20µL of a solution containing of 10µM SU5402 and 5µM FGFR3 antibody.
Semi-quantitative PCR and quantitative PCR
RNA was extracted as described previously (Hutson et al., 2006). cDNA was generated using SuperScript® II Reverse Transcriptase according to the manufacturer’s protocol. Primer sequences are shown in Suppl. Table 1. BLAST analysis showed that the sequences only aligned with the targeted genes. Only a partial sequence is available for chick FGFR4; therefore, the exon-exon boundaries for that gene remain unknown.
Real-time quantitative PCR for FGF8 target genes in the caudal pharynx or secondary heart field was performed as previously described (Hutson et al., 2006). The expression levels were normalized to HPRT.
In situ hybridization
Whole mount in situ hybridization was carried out using a standard protocol (Wilkinson, 1992). Previously published probes were used for in situ hybridizations (Dessimoz et al., 2006; Harduf et al., 2005). To confirm that the primers were specific for the FGF receptors, the PCR products were sequenced by the Duke Cancer Biology Sequencing Facility.
Immunohistochemistry, cell proliferation and TUNEL assay
Immunohistochemistry was performed on paraffin or frozen sections as detailed previously (Waldo et al., 1996). Antibodies used were anti-phospho-Histone H3 (pHH3; Upstate), anti-GFP (Molecular probes, Invitrogen), HNK1 (ATCC), anti-phospho-ERK (Cell Signaling Technology) and antiFGFR1 and R3 (see above). Apoptosis was detected using ApopTag Red In Situ Apoptosis Detection Kit (Chemicon) based on the TUNEL assay method. In all cases where immunohistochemistry was performed controls were prepared without addition of primary antibody.
FGF8 and Dominant-negative FGFR1 (dnFGFR1) Plasmids
Two plasmid vectors were used in the experiments to inhibit FGFR1. A green-fluorescent protein (GFP) control plasmid (pCCALL2 EGFP) and a dnFGFR1 plasmid with GFP reporter (pCCALL2 dominant negative FGFRI IRES EGFP). Both plasmids were a gift from Dr. Eric Meyers (currently at St. Luke’s Boise Medical Center).
Two plasmids were used in experiments to overexpress FGF8b. EGFP or FGF8b were cloned into the pMiwSV plasmid vector, which has a chick β-actin promoter and RSV enhancer (Sato et al., 2001; Suemori et al., 1990; Wakamatsu et al., 1997). Both plasmids were gifts from Dr. H. Nakamura (Department of Molecular Neurobiology, Institute of Development, Aging and Cancer, Seiryo-machi 4-1, Aoba-ku, Sendai 980–8575, Japan)
Electroporation
For the foregut electroporation studies eggs were incubated to HH10–11. The eggs were windowed and the vitelline membrane was torn. One or two drops of Ringers solution was applied so that the embryo fell away from the vitelline membrane. Platinum electrodes were placed at the cranial end on either side of the embryo. GFP or FGF8 + GFP expressing plasmid at 0.5 – 3.0 ug/ul was injected into the foregut using a picospritzer. Three 50 msec square wave pulses were immediately applied at 20 V each. If the embryo was electroporated bilaterally, a drop of Ringers was again applied to the embryo, and injection and electroporation was repeated for the other side of the foregut. To label neural crest in some embryos, DiI (Invitrogen) was injected into the neural tubes after electroporation.
For the morpholino studies carboxyfluorescein-tagged start site morpholinos were designed by Gene Tools for FGFR1–3 and a control scrambled morpholino (Suppl. Table 2). The morpholinos (2mM) were injected into the neural tube of HH9–11 chick embryos, and electroporated bilaterally as described above. The electroporated neural folds were removed from the embryo and cultured overnight at 37°C in the presence or absence of 30ng/mL FGF8b in 5% FBS in L-15 with high glucose. Migration analysis of single fluorescently-labeled cells was performed as described above.
For the dnFGFR1 studies, the dnFGFR1 was electroporated into the neural tube as described above. Embryos were harvested at 24–48 after electroporations.
FGF8 Bead Experiments
Heparin beads (Sigma) were soaked overnight in 30ng/ml of FGF8b or PBS. FGF8 or PBS beads were implanted caudal to the otic placode in HH 10 embryos. Embryos were collected at HH14–15 and immunostained for HNK1 in whole mount.
Statistical analysis
Migration data was analyzed by ANOVA followed by posthoc t-tests. Significance was set as p≤0.05. For qPCR the data was tested by Kruskal-Wallis one-way analysis of variance by ranks. Significance was set as p≤0.05.
Results
FGF8 enhances cardiac neural crest migration
To test how FGF8b influences cardiac neural crest cell migration, explanted premigratory cardiac neural crest cells were exposed overnight to FGF8b protein. Cardiac neural crest cells migrated more in the presence of increasing concentrations of FGF8b (2.5–30ng/ml)(Fig. 1A,B). The explants treated with 30ng/ml FGF8b migrated significantly farther than control explants, and treatment with 10µM of the FGFR1 blocking drug SU5402, attenuated this response (Fig. 1A,B) suggesting that FGF8b is chemokinetic for cardiac neural crest cells.
Figure 1.
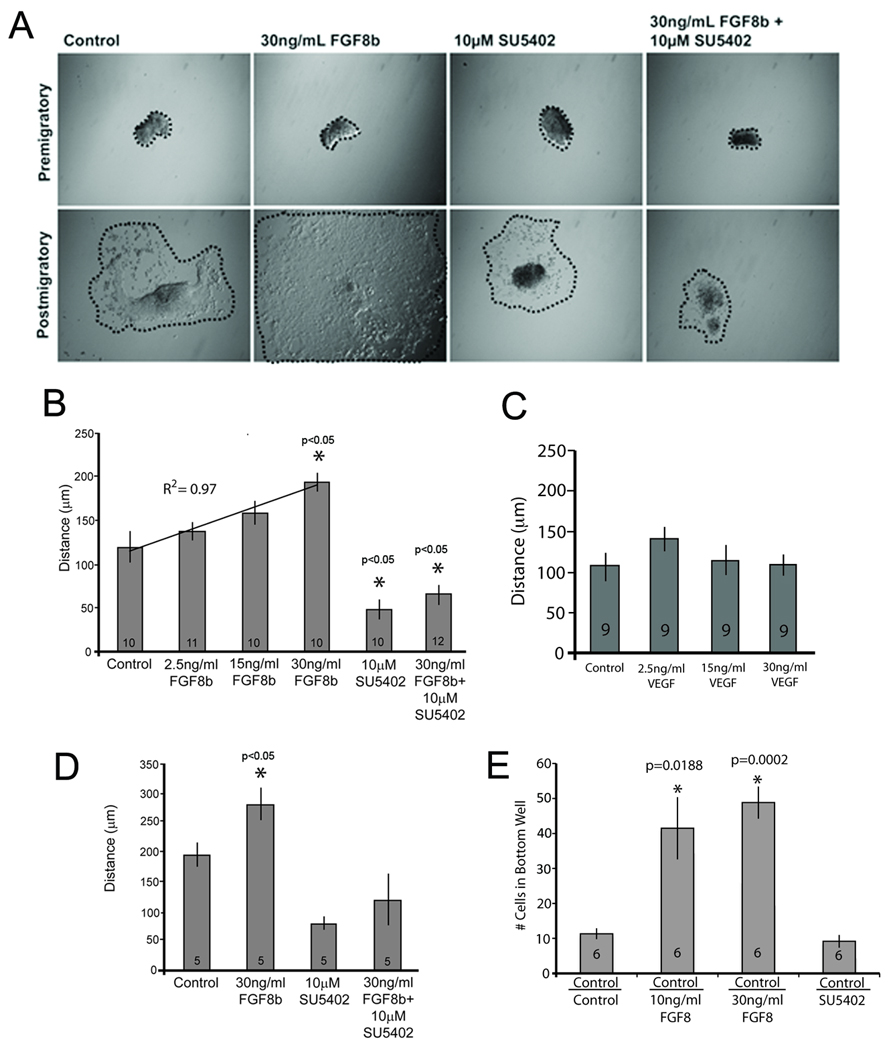
Cardiac neural crest cells migrate farther in response to FGF8. (A) Representative images of premigratory and postmigratory cardiac neural crest explants (10x). Explant circumference is highlighted by a dotted line. Explants were grown overnight with FGF8b, SU5402, or both. (B) Dose-response of cardiac neural crest migration in the presence of FGF8 is linear and can be blocked by the FGFR receptor blocker, SU5402. Asterisks indicate statistical significance when compared to control. (C) Distance migrated by cardiac neural crest cells is not altered by VEGF an established chemotaxic cytokine for neural crest migrating into pharyngeal arch 2. (D) Average length of the migration path of 5 individual neural crest cells from real time imaging shows that individual cell movement is enhanced in the presence of FGF8b and reduced in the presence of SU5402. (E) Transwell assay showing enhanced movement of cardiac neural crest cells toward 10 or 30 ng/ml FGF8. Designation under each bar indicates contents of top and bottom wells. Control in the top well included DMEM with 5% serum. The bottom wells all contained DMEM with 2% serum (Control). Experimental bottom wells included either FGF8 or SU5402. There is no difference in Control/Control versus Control/SU5402 indicating little to no FGF stimulation from the serum in the bottom well. Each bar represents the average of the total number of cells that migrated across the membrane in 6 separate experiments. Within each experiment there were 6 replicates of each treatment condition.
VEGF has been shown to be chemotactic for the preotic cranial crest migrating into pharyngeal arch 2 (McLennan et al., 2010). Since VEGF is also expressed in the caudal pharynx it was tested for its ability to enhance migration of the postotic-derived cardiac neural crest. Cardiac neural crest explants were treated with several concentrations of VEGF. The VEGF-treated cells showed no difference in migration distances compared to controls (Fig. 1C). Because FGF3 is expressed in rhombomere 6, one of the rhombomeres of origin of cardiac neural crest, it was also tested for the ability to promote migration. FGF3 at 10 or 30 ng/ml did not affect cardiac neural crest migration when compared to control (data not shown).
To rule out an effect of FGF8 on cardiac neural crest proliferation that would appear as enhanced migration, individual cells were tracked in time-lapse movies over a 16-hour period. Individual cells treated with 30ng/mL FGF8b migrated significantly farther than control cells (Fig. 1D) and inhibiting FGF signaling with SU5402 blocked this effect. Although less than 1% of the cell behaviors were analyzed in each time-lapse movie all the cells in the FGF treatment group migrated greater distances when compared to control or the SU5402-treated group.
Taken together these results suggest that FGF8 is specifically chemokinetic for the cardiac neural crest cells because it increases the migratory behavior of the cells as a group and individually.
FGF8 is chemotactic for cardiac neural crest in vitro
The results above show that FGF8 may be a chemokinetic factor but the experiments did not test for its capacity to influence the directionality of the migrating cardiac neural cells or chemotaxis. To determine whether FGF8 is chemotactic for cardiac neural crest cells, a standard chemotaxis transwell assay was performed using a filter with 8um pores between the chambers. Dissociated cardiac neural crest cells were equally distributed in the upper chamber with FGF8b containing medium in the bottom well. A significantly greater number of cells migrated across the filter into the bottom chambers containing 10 or 30 ng/ml FGF8b when compared to wells with medium alone or with FGF3 in the bottom well. When SU5402 was added to the chambers, transwell migration was dramatically inhibited in the presence of FGF8 (Fig. 1E). These results show that FGF8 is chemotactic for the cardiac neural crest in vitro.
FGFR1 and R3 mediate FGF8-induced cardiac neural crest cell migration
As mentioned above there are several different FGF ligands that are present in the pharynx, thus specificity of FGF signaling must be achieved through different combinations of ligands and FGF receptors. Using immunohistochemistry, Sarkar et al. (Sarkar et al., 2001) reported expression of FGFR1–3 in mesencephalic chick neural crest cells both in section and in culture. To determine which FGFRs are expressed in premigratory cardiac neural crest and explants cultured for 24 and 48 hrs RT-PCR was performed. All four FGFRs are expressed in the premigratory cardiac neural crest and expression was maintained in culture at all of the times examined (Suppl. Fig. 1).
To determine which FGFRs are required for FGF8 signaling in cardiac neural crest cell migration, we electroporated 3’-carboxyfluorescein-tagged start site morpholinos designed against Fgfrs1–3 into neural tubes at HH9 and explanted the premigratory cardiac neural crest for in vitro migration analysis. The distances that individual fluorescent cells moved over a 16-hour time period were recorded by time-lapse videomicroscopy. Cells electroporated with Fgfr3 morpholino at HH9 migrated significantly less than control (Fig. 2A). Fgfr1-morpholino electroporated cells tended to migrate less than control but there was no change in migration with Fgfr2 knockdown (Fig. 2A).
Figure 2.
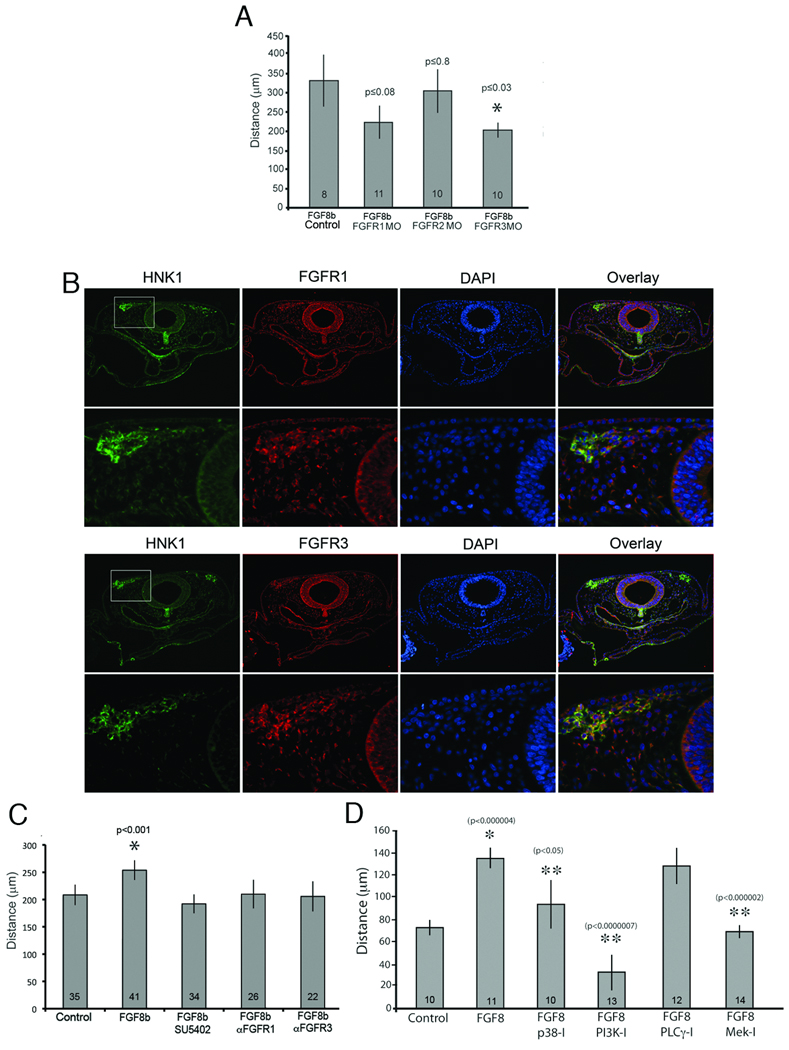
Migratory response of cardiac neural crest cells to FGF8 depends on FGFR1 and R3 and the MAPK/ERK signaling cascade. (A) Cardiac neural crest cells were electroporated at HH9 with scrambled (Control), FGFR1, R2 or R3 morpholinos. The cardiac neural crest region was explanted and individual cell migration followed for 16 hrs in the presence of 30 ug/ml FGF8. FGFR3 morpholino caused a significant reduction in single cell migration while FGFR1 morpholino caused a tendency toward less migration. By contrast FGFR2 morpholino had no effect on migration. (B) Antibodies (red) to FGFR1 and R3 show that these receptors are expressed in the migrating cardiac neural crest cells identified by HNK1 co-immunoreactivity (green) at HH12. (C) The FGFR1 and R3 antibodies are function-blocking due to the significant reduction in migration in response to FGF8b. There was no difference between explants grown with SU5402 or with the antibodies. (D) Migratory response of cardiac neural crest depends on Ras and PI3K intracellular signaling pathways. Cardiac neural crest explants were grown overnight with indicated intracellular inhibitors. Inhibition of p38, PI3K and Mek significantly blocked FGF8b enhanced cardiac neural crest migration. PLCγ inhibition did not block FGF8-induced cardiac neural crest migration. * denotes significant difference from Control; ** denotes significant difference from FGF8 only.
To achieve a greater knockdown effect of FGFR1 and R3 protein expression, blocking antibodies to FGFR1 and FGFR3 were made. Immunofluorescence showed that both antibodies recognized populations of migrating cardiac neural crest in vivo with FGFR3 showing more distinct colocalization with HNK1-positive neural crest cells than FGFR1 (Fig. 2B). FGFR1 was broadly expressed in the pharyngeal mesenchyme and neural tube but was more strongly associated with the “migratory front” of the cardiac neural crest while FGFR3 was more localized to the cardiac neural crest. To determine the ability of these antibodies to block FGF8-induced migration, premigratory cardiac neural crest cells were explanted and treated with FGF8b plus the neutralizing antibodies alone or in combination. Cardiac neural crest cells exposed to either of the function-blocking antibodies migrated significantly less than cells exposed to FGF8 without antibody (Fig. 2C) and were comparable to cultures treated with SU5402. There was no additive effect of FGFR1 and R3 when both antibodies were used together (data not shown).
These results suggest that FGFR1 and R3 are the receptors important for FGF8 enhanced cardiac neural crest cell migration. Because knockdown of either receptor has the same effect while knockdown of both receptors together does not have an additive effect these results are consistent with FGF receptor heterodimerization. Disruption of FGFR3 seems to be slightly more effective at disrupting FGF8-induced migration, which may be a reflection of the increased colocalization with the neural crest observed in the immunostain (Fig 2B).
pERK, PI3K and p38 intracellular signaling is required for cardiac neural crest response to FGF8
FGF signaling activates both ras-dependent and ras-independent intracellular signaling pathways. To ascertain which intracellular signaling pathways are activated by FGFR1 and R3 to modulate cardiac neural crest cell migration, explants were treated with chemical inhibitors of Mek/Erk, p38, phosphotide inositol 3 kinase (PI3K) and the phospholipase C (PLC)γ signaling cascades alone or in combination with 30ng/mL FGF8b. Cultured cardiac neural crest explants treated with Mek inhibitor (U0126) and PI3K inhibitor showed dramatic inhibition of FGF8 induced migration. Inhibition of p38 signaling showed moderate but significant inhibition of migration while inhibition of PLCγ (U73122) had no effect on migration (Fig 2D).
These results indicate that migrating cardiac neural crest cells utilize the Ras (p38 and Mek/Erk) and calcium dependent PI3K intracellular signaling cascades as they migrate towards the caudal pharynx.
FGF8 is chemokinetic and chemotactic for CNC in vivo
Our in vitro results indicated that inhibiting the function of either FGFR1 or R3 blocked the migratory effect of FGF8 on the cardiac neural crest. To determine whether the in vivo chemokinetic effect of FGF8 on cardiac neural crest migration could be altered by knocking down FGFRs, a dominant negative (dn)FGFR1-GFP expressing plasmid was electroporated into the premigratory cardiac neural crest. The embryos were harvested at HH15–18 for assay of migration, proliferation, and apoptosis. GFP-positive cardiac neural crest cells migrated robustly from the neural tube into the caudal pharynx. In contrast, cardiac neural crest cells expressing dnFGFR1-GFP migrated poorly from the neural tube and few to none entered the pharynx (Fig. 3A–D). To measure the distance traveled by individual electroporated cells, the embryos were sectioned and co-immunostained for GFP and HNK1. The dnFGFR1-GFP-positive cardiac neural crest cells lagged behind unelectroporated HNK1-positive migrating cardiac neural crest cells while cells electroporated with GFP were evenly distributed throughout the migrating HNK1-positive cells (Fig. 3E–K). This is somewhat surprising because it suggests that single cardiac neural crest cells do not migrate obligatorily with the group as has been shown for Sdf1 another chemotactic factor for neural crest cells (Kasemeier-Kulesa et al., 2010; Theveneau et al., 2010) although there seems to be a collective effect when the embryos are treated with SU5402 and the FGFR3 blocking antibody (see below). The mitotic index calculated by BrdU incorporation was no different for dnFGFR1, control, and unmarked cardiac neural crest cells during migration (data not shown). However, TUNEL staining for apoptosis indicated that the dnFGFR1-GFP-positive cells showed an increase in cell death near the neural tube (Fig. 3L–T) confirming that FGF not only enhances migration but also is a survival factor for cardiac neural crest as has been shown previously in FGF8 hypomorphic mice (Abu-Issa et al., 2002; Frank et al., 2002).
Figure 3.

DnFGFR1-GFP cardiac neural crest cells show delayed migration and elevated cell death. (A) HH17 chick embryo control expressing GFP (green) shows cardiac neural crest cells migrating into the pharyngeal arches. (B) Magnification of the grey box shown in A showing GFP-positive cells migrating midway into pharyngeal arches. (C) HH18–19 embryo expressing dnFGFR1-GFP in the cardiac neural crest showed delayed cardiac neural crest migration with no cells in the pharynx. (D) Magnification of the grey box shown in C. (E) Transverse section through arch 3 of HH16 chick embryo electroporated with GFP-expressing plasmid. HNK-1 (red) shows migrating cardiac neural crest cells. GFP (green) is expressed by cells throughout the migrating stream. (F) Magnification of right arch 3 showing a wide distribution of GFP electroporated cells throughout the migrating cardiac neural crest stream. (G) Migrating cardiac neural crest stream in right arch 3 with GFP expressing cells mostly located distally. (H) Transverse sections through arch 3 of an HH16 chick embryo electroporated with dnFGFR1-GFP. The migrating cardiac neural crest streams can be seen just dorsal and lateral to the foregut. (I) Magnified view of the cardiac neural crest stream showing that the dnFGFR1-GFP expressing cells are always located dorsally at the tail end of the stream. (J) Magnified view of the cardiac neural crest stream from another section of the same embryo showing a single dnFGFR1-GFP expressing cell also located at the dorsal end of the stream. (K) Graphic representation of the number of control-GFP-positive cells or dnFGFR1-GFP-expressing cells and distance from the neural tube migrated by each population. Distance from the neural tube is indicated as 100% when the cells are in the ventral pharynx on the x-axis. The y-axis indicates the number of cells per section. The majority of dnFGFR1 expressing cells could successfully migrate only about 20% of the distance from the neural tube to the ventral pharynx with a few cells migrating 50% of the distance. By contrast the majority of GFP-positive age matched control cells migrated 40–100% of the distance. TUNEL analysis (L–T) showed increased cell death in the dnFGFR1-GFP expressing cells. Cell death was highest near the neural tube leaving few cells to continue migrating. (L) Transverse section of pharyngeal arch 2–3 level showing the position of panels M, N and O. Apoptosis (red) within cardiac neural crest expressing GFP was rare in the cells nearest the neural tube (M), moderate midway to the arches (N), and more frequent once the cells reach the arch (O). (P) Transverse section of arch 3 level in an embryo electroporated with dnFGFR1-GFPshowing the position of panels Q, R, and S. Apoptosis of cardiac neural crest cells was very high near the neural tube (Q), still moderately high midway to the arches (R) and very low within the arch (S) because almost no dnFGFR1-electroporated cardiac neural crest cells reach the arches. Yellow arrows indicate cells double positive for GFP and TUNEL. (T) Bar graph indicating the number of TUNEL-positive cardiac neural crest cells and their position in the cardiac neural crest migratory pathway.
To determine if blocking FGF signaling globally would alter the migration of cardiac neural crest cells, HH9 embryos were exposed to the FGFR1 receptor blocking drug SU5402 or to our previously tested FGFR3 neutralizing antibody, or both SU5402 and FGFR3 antibody. The embryos were harvested at HH14 when the cells should be migrating into pharyngeal arch 3. In SU5402-treated embryos (7 of 9) and FGFR3 antibody-treated embryos (3 of 6), fewer cardiac neural crest cells migrated and those that did exhibited delayed and poor migration as compared with stage-matched controls (Fig. 3A,B,D and E). Delayed migration by the cardiac neural crest cells was confirmed in whole mount HNK1 stained embryos and in transverse sections (Fig. 4). Treatment with both SU5402 and FGFR3 antibody resulted in severely disrupted migration of the cardiac neural crest cells in 6 out of 6 embryos analyzed (Fig. 4C). This result suggests that migration of the cardiac neural crest in vivo is enhanced by an FGF signal. We have previously shown that treating embryos at these stages results in overriding aorta in more than 50% of embryos treated with SU5402 (Hutson et al., 2006). Because this is not an outflow septation defect but a malalignment defect, it suggests that there are sufficient numbers of cardiac neural crest cells to orchestrate outflow septation but not enough to reduce FGF signaling in the pharynx resulting in FGF oversignaling to the secondary heart field and subsequent arterial pole malalignment defects.
Figure 4.

Blocking FGF8 signaling with SU5402 delays cardiac neural crest migration in vivo. (A–C) Embryos treated with vehicle (control), FGFR3 antibody or SU5402 plus FGFR3 antibody at HH9–10 and viewed in whole mount after HNK1 (green) immunohistochemistry. (A) Control treated embryos show normal cardiac neural crest cell migration into the circumpharyngeal ridge and pharyngeal arch 3 (arrows). (B) Embryos treated with FGFR3 antibody show reduced migration into the circumpharyngeal ridge and pharyngeal arch 3. (C) Embryos treated with SU5402 plus FGFR3 show poor migration of cardiac neural crest cells into the circumpharyngeal ridge and pharyngeal arch 3. (D) Transverse section of HH16 control embryo stained with HNK1 (green) showing cardiac neural crest migration into the 3rd pharyngeal arch approximately 60um caudal to the otic vesicle. Arrows indicate most distal migrating cardiac neural crest cells. (E) Comparable section in an HH16 embryo treated with SU5402 showing delayed migration of the cardiac neural crest cells (green).
These results confirm that enhanced migration in vivo by cardiac neural crest in response to FGF8 is receptor-mediated and further that failure of the cells to leave the proximity of the neural tube is associated with enhanced mortality.
FGF8 is chemotactic for cardiac neural crest cells in vivo
As a first attempt to determine whether FGF8 is chemotactic for cardiac neural crest cells in vivo, GFP-FGF8b plasmid was electroporated into the neural tube at HH10 to establish an ectopic source of FGF8 as the cells migrate. Embryos were harvested at HH13, which is a timepoint after most cardiac neural crest cell migration from the neural tube. The migrated cells at this stage should be in the circumpharyngeal ridge and beginning to enter pharyngeal arch 3 dorsally (Kuratani and Kirby, 1992; Waldo et al., 1996). We found that cardiac neural crest cells ectopically expressing FGF8 delaminated from the neural tube but then formed a clump of cells that did not migrate toward the pharyngeal arches (Suppl. Fig. 3). While this shows that cardiac neural crest cells ectopically expressing FGF8 are not able to migrate distally from the dorsal neural tube, it does not show that FGF8 is a chemotactic signal for the cells.
A definitive test of whether FGF8b serves as a chemotactic signal for cardiac neural crest in vivo would be to show that the cells migrate toward an ectopic source of FGF8. This can be accomplished by placement of beads coated with FGF8 or by electroporation of an FGF8 expressing plasmid into a site that does not normally express FGF8. Individual beads soaked in 30ug/ml recombinant FGF8b or PBS were placed lateral to the postotic neural tube at HH9. At HH14–15 in embryos implanted with control PBS beads, the postotic cardiac neural crest had migrated to the dorsal caudal pharynx as indicated by HNK1 staining (Fig. 5A and C). In embryos with FGF8 beads, the cardiac crest could not be seen in the pharyngeal region but HNK1-positive cells had collected dorsal to the bead (Fig. 5B, D, and E).
Figure 5.
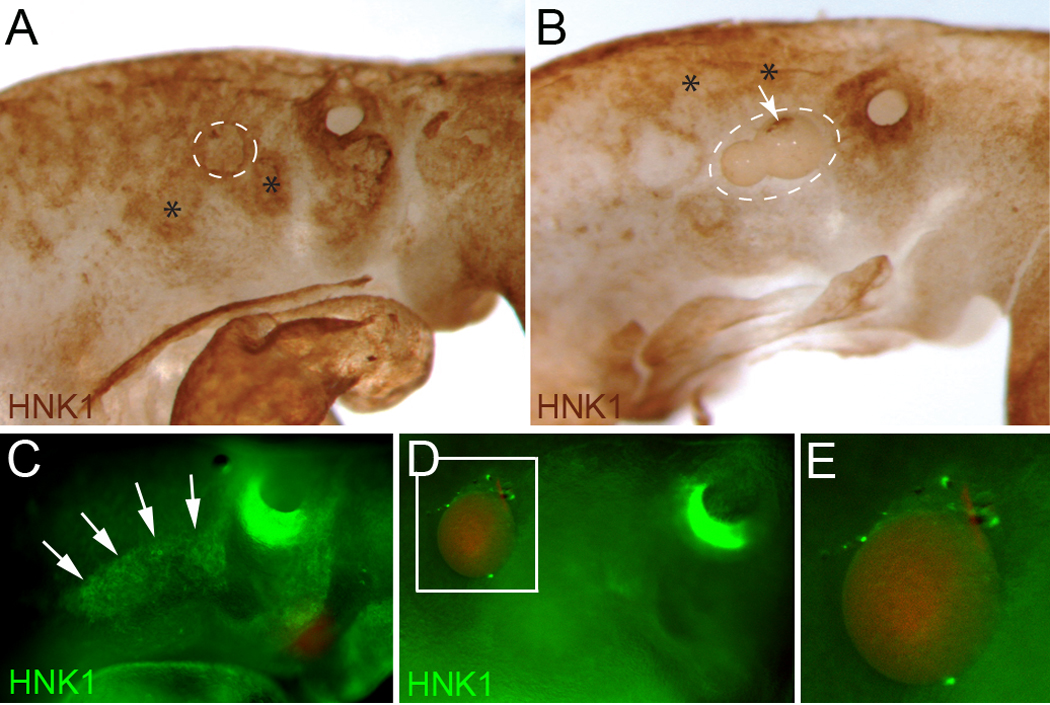
Cardiac neural crest cells migrate toward ectopic sources of FGF8. (A,B,C,D) Whole mount embryos immunostained with HNK1 at HH14. Beads were implanted at HH9 just cranial to somite 1. (A) PBS bead (white circle) with normal cardiac neural crest migration into pharyngeal arch 3 (asterisk) and the circumpharyngeal ridge (asterisk). (B) FGF8 beads (white circle) showing no distal migration of the cardiac neural crest (asterisks) and some cells clustered on the proximal side of the beads (arrow). (C) PBS bead with normal cardiac neural crest cell migration into pharyngeal arch 3 and the circumpharyngeal ridge. (D) FGF8 bead showing no migration of the cardiac neural crest distal to the bead with some HNK1 positive cells collected on the proximal side of the bead. (E) Enlargement of the bead shown in D.
Because bead implantation disturbs the architecture of the pharynx and could disrupt neural crest migration pathways, an FGF8-expressing plasmid was electroporated into an ectopic site in the ventral pharyngeal endoderm. A further advantage of this method is that it provides a constant source of FGF8 and the opportunity to determine whether arterial pole defects result from FGF8 oversignaling in the presence of cardiac neural crest. FGF8 is normally expressed only in the lateral pharyngeal endoderm and ectoderm (Karabagli et al., 2002). Ectopic fgf8 expression was markedly higher than endogenous expression as confirmed by simultaneous assessment of GFP and fgf8 message expression by in situ hybridization (Fig. 6A–D).
Figure 6.

Molecular effects of FGF8 overexpression in the ventral pharynx. (A) Whole mount HH14 embryo after electroporation with FGF8b–GFP (green) showing expression in the pharynx. (B) Transverse section of arch 3 showing that FGF8b–GFP expression is limited to the ventral pharyngeal endoderm. (C) In situ hybridization for fgf8 showing robust expression by the electroporated endodermal cells Arrowhead indicates ectopic FGF8. (D) In situ hybridization showing the normal expression pattern of fgf8 expression in the pharyngeal endoderm and ectoderm (arrowheads) of an unelectroporated embryo. This pattern is not seen in C because the ectopic site of fgf8 expression was too strong for development of the endogenous signal. (E) Quantitative PCR of the ventral pharyngeal mesoderm shows upregulation of all the FGF8 downstream negative regulators but no increase in the pea3 family of positive regulators in embryos electroporated with FGF8b. * indicates statistical significance compared to GFP electroporated controls. (F) Transverse sections of arch 3 in HH16 embryos electroporated with GFP alone (Sham/GFP) or FGF8b–GFP either unilaterally or bilaterally followed by in situ hybridization. Cardiac neural crest cells (arrows) express mkp3 and spry1 at high levels normally and this is not altered by ectopic FGF8 overexpression. However, there is a dramatic increase in spry1 and spry2 expression by the ventral pharyngeal mesoderm also known as secondary heart field (arrowheads). Primer sequences are listed in Supplemental Table 2.
We and others have shown that too much or too little FGF signaling in the pharynx is detrimental to neural crest and secondary heart field development (Abu-Issa et al., 2002; Frank et al., 2002; Hutson et al., 2006). pErk immunohistochemistry was used to assess signaling intensity of the ectopic foci of FGF8 in the pharynx at the protein level. Interestingly, pErk intensity in the cardiac neural crest cells was the same in control and FGF8-overexpressing embryos and there was little to no pErk immunostaining in the secondary heart field (Suppl. Fig. 2). Because strong FGF signaling has the potential to induce expression of negative regulators of pERK, downstream FGF target expression was assessed by in situ hybridization and qPCR. The caudal ventral pharynx in the region of FGF8 overexpression was collected for qPCR of known FGF8 downstream target genes. Genes assessed included positively regulated FGF targets: erm, pea3, and er81; and negative feedback regulators of FGF signaling: mkp3, spry1, spry2, and sef. None of the positive targets of FGF signaling was elevated in the caudal pharynx; however, all of the negative regulators with the exception of sef were upregulated (Fig. 6E).
To resolve how the specific regions of the caudal pharynx were responding to FGF8 overexpression, in situ hybridization was performed for several FGF8 target genes. Expression of the negative feedback genes, mkp3, spry1, spry2, were very high in the cardiac neural crest normally and were not further upregulated by ectopic FGF8 expression (Fig. 6F). However, the negative feedback genes were upregulated in the secondary heart field (ventral pharyngeal splanchnic mesoderm) (Fig 6E). Significantly both spry1 and 2 were significantly up-regulated in the secondary heart field splanchnic mesoderm confirming previous studies showing that this cell population is particularly sensitive FGF8 levels (Hutson et al., 2006).
Interestingly, ectopic FGF8 overexpression in the ventral pharynx of HH16–18 embryos caused a dramatic increase in thickness of the mesenchyme between the endoderm and ventral pharyngeal mesoderm at the site of FGF8 overexpression (Fig. 7A,B). To determine whether the increased mesenchyme was due to increased proliferation, phosphoHistone H3 (pHH3) expression was assayed in the area. No difference in proliferation was found (Fig. 7C,D). Cell death as assayed by TUNEL showed a slight increase in the thickened mesenchyme (Fig. 7E,F).
Figure 7.
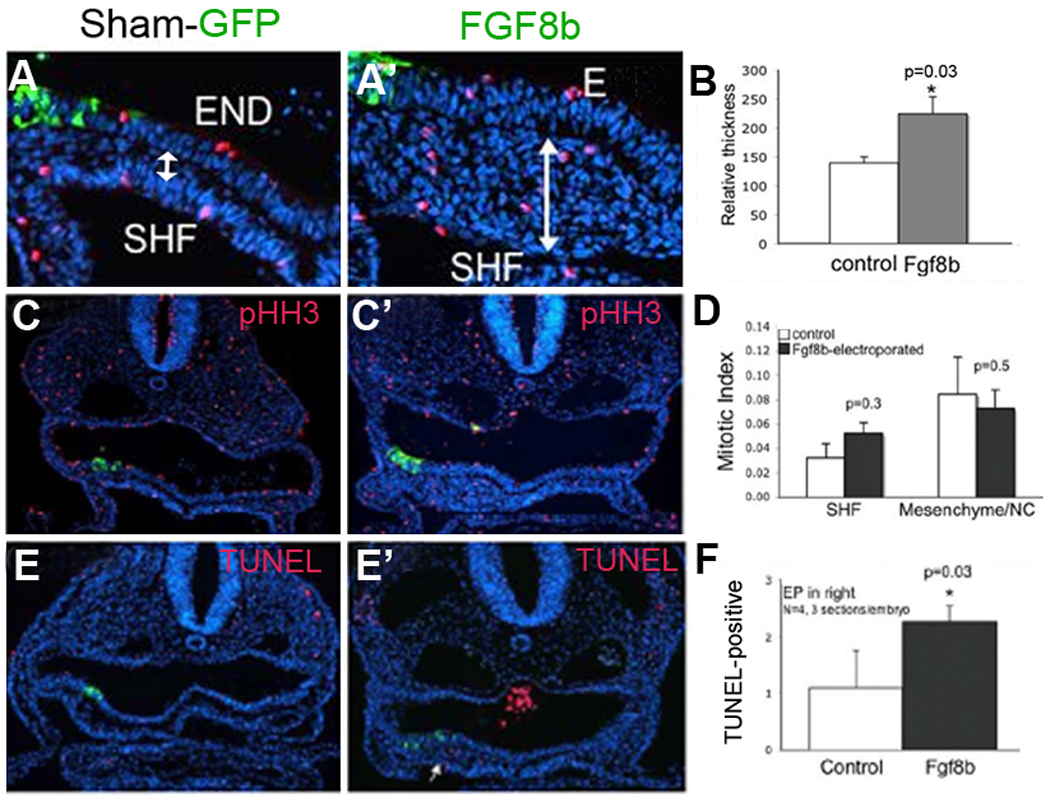
Cells accumulate around an ectopic focus of FGF8 in the ventral pharynx. (A, A’) Transverse sections at HH18 in Sham-GFP and FGF8b–GFP electroporated embryos showing increased thickness (double-headed arrow) of the mesenchyme separating the pharyngeal endoderm and the ventral pharyngeal mesoderm (secondary heart field) in the FGF8b–GFP electroporated embryo when compared to Sham-GFP electroporated embryo. (B) Bar graph illustrates that the thickness of the region around the FGF8 overexpression site is increased by about 30% (n=4 embryos). (C, C’) Transverse sections of arch 3 region of GFP and FGF8b -expressing HH16 embryos stained with phosphohistone (pHH3-red) to indicate dividing cells. (D) Bar graph showing the mitotic index of cells in the secondary heart field (SHF) or pharyngeal mesenchyme is not different. (E, E’) Transverse sections of arch 3 region of GFP (control) and FGF8b expressing HH16 embryos that have been labeled for TUNEL to indicate dying cells (red). (F) Bar graph showing numerical data indicating that apoptosis is slightly increased in secondary heart field ventral to pharyngeal pouch 3 at stage 16.
Another potential explanation of the increased mesenchyme given the chemotactic and chemokinetic effect of FGF8 on cardiac neural crest is that more cardiac neural crest cells are attracted to the ectopic site of FGF8 expression. To establish if this was the case, FGF8b–expressing plasmid was electroporated into the foregut endoderm and the neural tube was injected with DiI to label the premigratory neural crest cells in HH9 embryos. At HH16 DiI-labeled cells had migrated to the ectopic site of FGF8 expression in greater numbers than when compared to the unelectroporated side (Fig. 8A,B) To confirm that these cells were cardiac neural crest cells, cardiac neural crest quail-chick chimeras were constructed in embryos with the FGF8b–expressing plasmid electroporated into the foregut endoderm. The thickened mesenchyme near the site of FGF8b overexpression was populated by a mixture of chick cells and quail cardiac neural crest cells (Fig. 8C,D). The cardiac neural crest cells gathered around the ectopic site of FGF8 expression had moved farther than normal into the ventral pharynx for this time in development. These results confirm the in vitro finding that FGF8b is both chemotactic and chemokinetic for cardiac neural crest cells.
Figure 8.
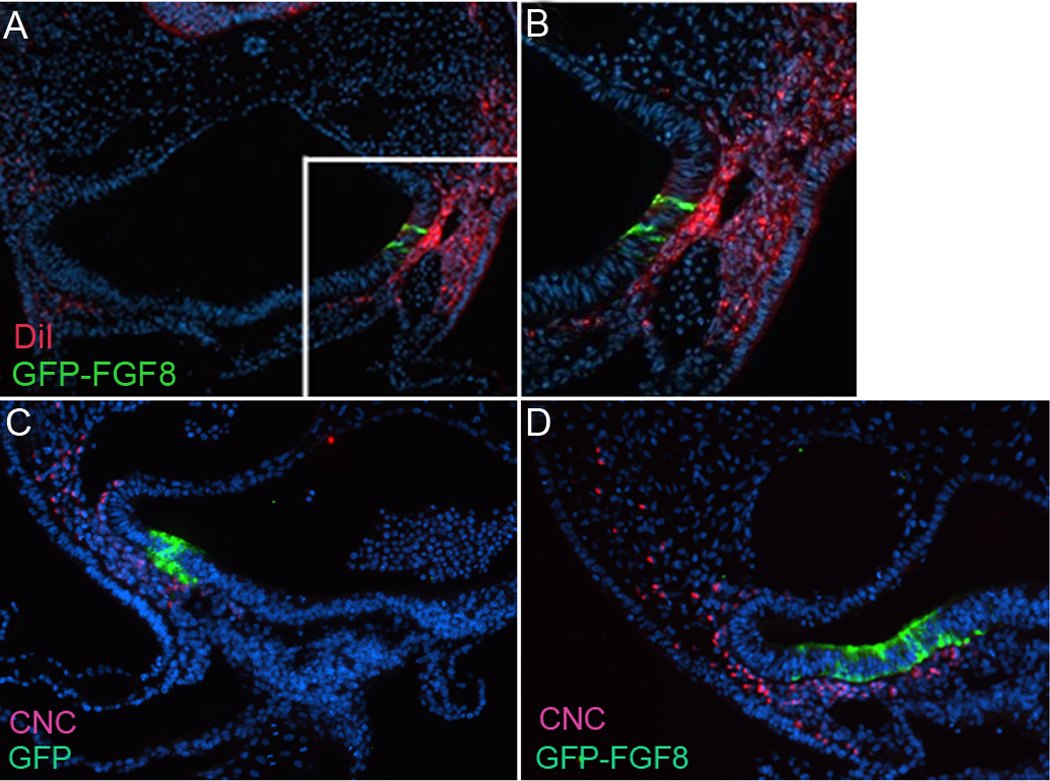
Cardiac neural crest cells are attracted to a site of FGF8 overexpression. (A) Premigratory cardiac neural crest cells labeled with DiI in the neural tube migrate to an ectopic site of FGF8 expression. (B) Enlarged box in A. (C, D) Transverse sections through the arch 3 region of HH16 quail-chick chimeras expressing GFP alone or GFP-FGF8 in the ventral pharynx. Cardiac neural crest cells are recognized by their reactivity with the quail antibody QCPN (red) and can be seen in the vicinity of the FGF8 overexpression (green).
Ectopic overexpression of FGF8 causes subtle changes in outflow tract development
Elevated FGF8 signaling in the caudal pharynx after cardiac neural crest ablation has been shown previously to cause abnormal arterial pole development. Abnormal development is first apparent at HH18 in the form of abnormal cardiac looping and subsequently at day 9 (HH34) as malalignment defects such as overriding aorta and pulmonary stenosis/atresia (Hutson et al., 2006). We hypothesized that ectopic overexpression of FGF8 would result in these cardiac abnormalities even in the presence of cardiac neural crest. However, at HH18 cardiac looping was normal in embryos electroporated with FGF8b–expressing plasmid (not shown). We allowed the electroporated embryos to develop to day 9 of incubation when the development of the 4-chambered heart is essentially complete. While none of the embryos had outflow alignment defects, we found that 30–40% of the overexpressing embryos had slight pulmonary stenosis (Fig. 9, Suppl. Table 3). The pulmonary stenosis was at the level of the pulmonary semilunar valve (Fig. 9G,H). Since the right side of secondary heart field gives rise to the subpulmonary myocardium, we compared whether electroporation of FGF8b bilaterally or unilaterally on the right yielded the same phenotype. A similar rate of pulmonary stenosis was found in either condition (Fig. 9I), possibly because the negative regulators of FGF signaling are maximally activated bilaterally even with unilateral overexpression (Fig. 6E).
Figure 9.

Ectopic overexpression of FGF8 in the ventral pharynx results in mild pulmonary stenosis. (A) HH18 embryo electroporated with GFP-expressing plasmid. (B) Heart from the embryo shown in A at day 9. Development of the arterial pole is normal. (C) HH18 embryo electroporated with FGF8b–GFP-expressing plasmid. (D) Heart from the embryo shown in C at incubation day 9. The pulmonary trunk looks relatively normal (compare with outlined area in B). (E,F) Transverse sections of the arterial pole of the heart in B showing normal position and size of the aorta and pulmonary trunk. (G,H) Transverse sections of the heart shown in D indicate that the position of the aorta (Ao) and pulmonary trunk (PT) is normal but the pulmonary trunk at the level of the semilunar valves is slightly stenotic (compare length of double-headed arrows). (I) Quantitative assessment of hearts with arterial pole defects from embryos with bilateral or unilateral sites of FGF8 overexpression.
Discussion
Our results show that FGF8 is both chemotactic and chemokinetic for cardiac neural crest cells in culture and as they originate from the postotic rhombomeres 6,7, and 8 and migrate into pharyngeal arches 3, 4, and 6. Further we show that the migratory response is mediated by FGFR1 and R3 via Ras and Ca2+–dependent intracellular signaling. More cranial mesencephalic neural crest cells have also been shown to be attracted to FGF8 in vitro but not in vivo where FGF2 was chemotactic (Kubota and Ito, 2000). Interestingly neural crest cells originating from rhombomere 4 (preotic) are attracted into pharyngeal arch 2 by VEGF (McLennan et al., 2010). Cardiac neural crest cells destined for the caudal arches are not attracted by VEGF.
Several FGF ligands are expressed in the appropriate time frame near the caudal rhombencephalon that could act as chemokines. A detailed analysis of the hindbrain distribution of various components of the FGF pathway has been documented by Weisinger and colleagues (2010). The two closest sources of FGF8 to the caudal rhombencephalon are the isthmus (Crossley et al., 1996) and the pharyngeal endoderm/ectoderm (Farrell et al., 2001; Wendling et al., 2000). Both of these sites are at least one hundred microns away from the cardiac neural crest prior to its migration. FGF3 and 19 are also expressed in rhombomere 6 at about the time the cardiac neural crest cells migrate (Weisinger et al., 2010) and FGF19 is expressed in the placodal ectoderm (Kurose et al., 2004) but not in the pharyngeal arches. FGF signaling is likely because pErk1/2 is strongly expressed at HH8 and a target and negative regulator of FGF signaling, MKP3 is strongly expressed in rhombomere 6 at HH9. The FGF target genes, Pea3 and Erm are expressed throughout the hindbrain at the time of cardiac neural crest migration and their expression is downregulated by SU5402 treatment suggesting that their expression is the result of FGF signaling. We have shown that FGF3 does not promote either chemokinesis or chemotaxis by cardiac neural crest. FGF19 could be chemokinetic for cardiac neural crest; however, FGF19 is not present in the caudal pharynx, signals through FGFR4 and requires the cofactor betaKlotho for signaling through this receptor (Ito et al., 2000; Wu et al., 2009). Expression of betaKlotho has not been reported in the caudal pharynx. Thus, we favor FGF8 in the pharyngeal endoderm/ectoderm as being the early chemotactic signal as it seems somewhat unlikely that a chemotactic signal would originate from the source rather than the target of migrating cells (Weisinger et al., 2010) and the caudal pharynx is the target of cardiac crest cells. This is supported by our in vitro studies showing that FGF3 was not chemokinetic for the cardiac neural crest explants while under the same conditions FGF8 was. We have not addressed whether these FGFs might be chemorepellent. While this could certainly be the case, the results from our transwell studies do not favor FGF8 acting as such.
This leaves open the question of how a signal emanating from the pharynx might influence cells in the dorsal neural tube. Graded levels of FGF protein have been shown to span relatively long distances by binding heparan sulfate proteoglycans which localize FGF distribution and enhance the avidity with which FGF binds its receptor (Chen et al., 2009). Using a reporter construct, Chen et al (2009) found a gradient of FGF signaling in the caudal hindbrain. Heparin sulfate proteoglycans are composed of a protein core surrounded by covalently linked heparan sulfate chains composed of disaccharide repeats (Bernfield et al., 1999; Prydz and Dalen, 2000). Several heparin sulfate-modifying enzymes that generate distinct sulfation patterns have been discovered. One of these, Sulf1, is an extracellular endosulfatase that eliminates the sulfate group in position 6-O of glucosamine in highly sulfated regions of HS (Ai et al., 2003; Ai et al., 2007; Morimoto-Tomita et al., 2002). At HH12 Sulf1 is expressed in the developing caudal pharyngeal region (Garcia-Lopez et al., 2009) suggesting that an FGF8 gradient could exist in the caudal pharynx. The fact that sprouty was upregulated bilaterally in the secondary heart field after unilateral overexpression supports the idea that the FGF8 signal is widely distributed.
Neural crest cells have been reported previously to chemotax toward several diffusible signals, including but not limited to FGF2, Sdf1, VEGF and semaphorins (Kasemeier-Kulesa et al., 2010; Kubota and Ito, 2000; McLennan et al., 2010; Olesnicky Killian et al., 2009; Theveneau et al., 2010; Toyofuku et al., 2008). FGF2 is chemotactic for mesencephalic neural crest cells (Kubota and Ito, 2000). These investigators speculated that the chemotaxis was mediated by FGFR1 and R3, which were the two receptors that were highly expressed by mesencephalic neural crest cells. Interestingly, our data also suggest that FGFR1 and R3 are the most highly expressed of the FGFRs in the cardiac neural crest. A double knockdown of FGFR1 and R2 using Wnt1cre or AP2cre did not cause any neural crest-related defects (Park et al., 2008). Thus it is possible that FGFR3 is able to mediate the signaling needed in the cardiac neural crest cells. An alternate explanation is that the receptor knockdown by Wnt1cre or AP2cre could be too late to block the FGF signal.
Park et al also induced conditional overexpression of sprouty in the neural crest (Park et al., 2008). This would be expected to inhibit Mek/Erk signaling which according to our data should disrupt cardiac neural crest migration. However, sprouty overexpression did not affect cardiac neural crest behavior (Park et al., 2008). In mice, the caudal arches are pErk positive at E9.0 but negative by E9.5–10 when cardiac neural crest has populated these arches (Corson et al., 2003). Because our data show that cardiac neural crest cells express high levels of sprouty in the pharyngeal arches, it is possible again that sprouty overexpression would not disrupt neural crest function. By the time the cardiac neural crest cells reach the pharynx, they are most likely refractory to FGF8 signaling because of the high level of expression of negative regulators of FGF8 intracellular signaling.
Further, of great interest to us was the finding that FGF8 overexpression in the presence of cardiac neural crest does not lead to the same type of abnormal development of the secondary heart field and arterial pole that occur when cardiac neural crest does not migrate to the caudal pharynx. This suggests that cardiac neural crest cells not only migrate to a pharyngeal site of FGF8 production but also can modulate the availability of signaling during the window when intracellular inhibitors of FGF8, like sprouty can be upregulated in the secondary heart field. We, and others, have recently shown that signaling through the Mek/Erk pathway is important for inhibiting BMP-induced myocardial differentiation (Hutson et al., 2010; Tirosh-Finkel et al., 2010). Because of the intense sprouty expression in the secondary heart field, signaling through the Mek/Erk pathway may be totally suppressed which would allow premature differentiation of the progenitors. The phenotype seen after FGF8 overexpression is a mild pulmonary stenosis, which would be predicted to result from premature myocardial differentiation at the expense of proliferative expansion of the cardiogenic stem cell population in the secondary heart field. It remains to be determined how cardiac neural crest cells might modulate FGF8 levels.
Finally, Trumpp et al (1999) found that tissue specific inactivation of FGF8 in the ectoderm of pharyngeal arch 1 caused cell death and impaired development of arch 1 (Trumpp et al., 1999). Also hypomorphic expression of FGF8 resulted in increased mesenchymal cell death (Abu-Issa et al., 2002; Frank et al., 2002). Our experiments also indicate that cardiac neural crest cells unable to transduce the FGF8 signal because of the expression of dominant negative FGFR1 results in cell death. However, in our experiments overexpression of FGF8 did not result in an increase in proliferation although cell death was slightly elevated in the ventral pharynx. Thus the increased mesenchymal thickness at a site of ectopic FGF8 expression appears not to be due to either an increase in proliferation or a decrease in cell death.
Supplementary Material
Supplemental Figure 1. Premigratory cardiac neural crest cells express all four FGFRs. Semiquantitative PCR of premigratory cardiac neural crest cells (HH10) showing that all four FGFRs are expressed by these cells. FGFR1 and R3 show the highest expression. HPRT was amplified as a control.
Supplemental Figure 2. pERK is highly expressed by cardiac neural crest cells in the pharyngeal arches but not in the secondary heart field. Immunohistochemistry of pERK on transverse sections through arch 3 region. Embryos electroporated with GFP-expressing plasmid (A) and FGF8b-GFP-expressing plasmid (B) do not show a difference in pERK expression by the cardiac neural crest cells (arrows). The secondary heart field (arrowheads) is negative for pERK.
Supplemental Figure 3. Ectopic expression of FGF8 in the cardiac neural crest blocks movement of the cells away from the neural tube. (A) Embryo electroporated in the right sided of the cardiac region of the neural tube with GFP-FGF8 expressing plasmid and harvested at HH13. (B) Transverse section of the same embryo shown in A. The cardiac crest can be seen on the right as a cap of cells (arrows) on the right side of the neural tube. Cardiac neural crest cells on the contralateral side appear to have migrated normally.
Acknowledgments
This work was funded by a postdoctoral fellowship from the American Heart Association to AS, The Cincinnati Children’s Heart Foundation (MLK), the George and Jean Brumley, Jr. Neonatal-Perinatal Research Institute of Duke University, AHA0830464N (MRH) and NIH grants HL083240 and HL070140 (MLK).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References Cited
- Abu-Issa R, Smyth G, Smoak I, Yamamura K-I, Meyers E. Fgf8 is required for pharyngeal arch and cardiovascular development in the mouse. Development. 2002;129:4613–4625. doi: 10.1242/dev.129.19.4613. [DOI] [PubMed] [Google Scholar]
- Ai X, Do AT, Lozynska O, Kusche-Gullberg M, Lindahl U, Emerson CP., Jr QSulf1 remodels the 6-O sulfation states of cell surface heparan sulfate proteoglycans to promote Wnt signaling. J Cell Biol. 2003;162:341–351. doi: 10.1083/jcb.200212083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ai X, Kitazawa T, Do AT, Kusche-Gullberg M, Labosky PA, Emerson CP., Jr SULF1 and SULF2 regulate heparan sulfate-mediated GDNF signaling for esophageal innervation. Development. 2007;134:3327–3338. doi: 10.1242/dev.007674. [DOI] [PubMed] [Google Scholar]
- Bernfield M, Gotte M, Park PW, Reizes O, Fitzgerald ML, Lincecum J, Zako M. Functions of cell surface heparan sulfate proteoglycans. Annu Rev Biochem. 1999;68:729–777. doi: 10.1146/annurev.biochem.68.1.729. [DOI] [PubMed] [Google Scholar]
- Chen Y, Mohammadi M, Flanagan JG. Graded levels of FGF protein span the midbrain and can instruct graded induction and repression of neural mapping labels. Neuron. 2009;62:773–780. doi: 10.1016/j.neuron.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corson LB, Yamanaka Y, Lai KM, Rossant J. Spatial and temporal patterns of ERK signaling during mouse embryogenesis. Development. 2003;130:4527–4537. doi: 10.1242/dev.00669. [DOI] [PubMed] [Google Scholar]
- Creuzet S, Couly G, Vincent C, Le Douarin NM. Negative effect of Hox gene expression on the development of the neural crest-derived facial skeleton. 2002;129:4301–4313. doi: 10.1242/dev.129.18.4301. [DOI] [PubMed] [Google Scholar]
- Crossley PH, Martinez S, Martin GR. Midbrain development induced by FGF8 in the chick embryo. Nature. 1996;380:66–68. doi: 10.1038/380066a0. [DOI] [PubMed] [Google Scholar]
- Dessimoz J, Opoka R, Kordich JJ, Grapin-Botton A, Wells JM. FGF signaling is necessary for establishing gut tube domains along the anterior-posterior axis in vivo. Mech Dev. 2006;123:42–55. doi: 10.1016/j.mod.2005.10.001. [DOI] [PubMed] [Google Scholar]
- Farrell MJ, Burch JL, Wallis K, Rowley L, Kumiski D, Stadt H, Godt RE, Creazzo TL, Kirby ML. FGF-8 in the ventral pharynx alters development of myocardial calcium transients after neural crest ablation. J. Clin. Invest. 2001;107:1509–1517. doi: 10.1172/JCI9317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank D, Fotheringham L, Brewer J, Muglia L, Tristani-Firouzi, Capecchi M, Moon A. An Fgf8 mouse mutant phenocopies human 22q11 deletion syndrome. Development. 2002;129:4591–4603. doi: 10.1242/dev.129.19.4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Lopez R, Soula C, Martinez S. Expression analysis of Sulf1 in the chick forebrain at early and late stages of development. Dev Dyn. 2009;238:2418–2429. doi: 10.1002/dvdy.22039. [DOI] [PubMed] [Google Scholar]
- Harduf H, Halperin E, Reshef R, Ron D. Sef is synexpressed with FGFs during chick embryogenesis and its expression is differentially regulated by FGFs in the developing limb. Dev Dyn. 2005;233:301–312. doi: 10.1002/dvdy.20364. [DOI] [PubMed] [Google Scholar]
- Huang GY, Cooper ES, Waldo K, Kirby ML, Gilula NB, Lo CW. Gap junction-mediated cell-cell communication modulates mouse neural crest migration. J. Cell. Biol. 1998;143:1725–1734. doi: 10.1083/jcb.143.6.1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutson MR, Zeng XL, Kim AJ, Antoon E, Harward S, Kirby ML. Arterial pole progenitors interpret opposing FGF/BMP signals to proliferate or differentiate. Development. 2010;137:3001–3011. doi: 10.1242/dev.051565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutson MR, Zhang P, Stadt HA, Sato A, Li Y-X, Burch J, Creazzo TL, Kirby ML. Cardiac arterial pole alignment is sensitive to FGF8 signaling in the pharynx. Dev. Biol. 2006;295:486–497. doi: 10.1016/j.ydbio.2006.02.052. [DOI] [PubMed] [Google Scholar]
- Ito S, Kinoshita S, Shiraishi N, Nakagawa S, Sekine S, Fujimori T, Nabeshima YI. Molecular cloning and expression analyses of mouse betaklotho, which encodes a novel Klotho family protein. Mech Dev. 2000;98:115–119. doi: 10.1016/s0925-4773(00)00439-1. [DOI] [PubMed] [Google Scholar]
- Karabagli H, Karabagli P, Ladher RK, Schoenwolf GC. Survey of fibroblast growth factor expression during chick organogenesis. Anat. Rec. 2002;268:1–6. doi: 10.1002/ar.10129. [DOI] [PubMed] [Google Scholar]
- Kasemeier-Kulesa JC, McLennan R, Romine MH, Kulesa PM, Lefcort F. CXCR4 controls ventral migration of sympathetic precursor cells. J Neurosci. 2010;30:13078–13088. doi: 10.1523/JNEUROSCI.0892-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby ML, Gale TF, Stewart DE. Neural crest cells contribute to aorticopulmonary septation. Science. 1983;220:1059–1061. doi: 10.1126/science.6844926. [DOI] [PubMed] [Google Scholar]
- Kirby ML, Hunt P, Wallis KT, Thorogood P. Normal development of the cardiac outflow tract is not dependent on normal patterning of the aortic arch arteries. Dev. Dyn. 1997;208:34–47. doi: 10.1002/(SICI)1097-0177(199701)208:1<34::AID-AJA4>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Kubota Y, Ito K. Chemotactic migration of mesencephalic neural crest cells in the mouse. Developmental Dynamics. 2000;217:170–179. doi: 10.1002/(SICI)1097-0177(200002)217:2<170::AID-DVDY4>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Kuratani SC, Kirby ML. Migration and distribution of circumpharyngeal crest cells in the chick embryo. Formation of the circumpharyngeal ridge and E/C8+crest cells in the vertebrate head region. Anat. Rec. 1992;234:263–280. doi: 10.1002/ar.1092340213. [DOI] [PubMed] [Google Scholar]
- Kurose H, Bito T, Adachi T, Shimizu M, Noji S, Ohuchi H. Expression of Fibroblast growth factor 19 (Fgf19) during chicken embryogenesis and eye development, compared with Fgf15 expression in the mouse. Gene Expr Patterns. 2004;4:687–693. doi: 10.1016/j.modgep.2004.04.005. [DOI] [PubMed] [Google Scholar]
- Le Lievre CS, Le Douarin NM. Mesenchymal derivatives of the neural crest. Analysis of chimaeric quail and chick embryos. J.Embryol.Exp.Morphol. 1975;34:125–154. [PubMed] [Google Scholar]
- Mayor R, Guerrero N, Martinez C. Role of FGF and noggin in neural crest induction. Developmental Biology. 1997;189:1–12. doi: 10.1006/dbio.1997.8634. [DOI] [PubMed] [Google Scholar]
- McLennan R, Teddy JM, Kasemeier-Kulesa JC, Romine MH, Kulesa PM. Vascular endothelial growth factor (VEGF) regulates cranial neural crest migration in vivo. Dev Biol. 2010;339:114–125. doi: 10.1016/j.ydbio.2009.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monsoro-Burq AH, Wang E, Harland R. Msx1 and Pax3 cooperate to mediate FGF8 and WNT signals during Xenopus neural crest induction. Dev Cell. 2005;8:167–178. doi: 10.1016/j.devcel.2004.12.017. [DOI] [PubMed] [Google Scholar]
- Morimoto-Tomita M, Uchimura K, Werb Z, Hemmerich S, Rosen SD. Cloning and characterization of two extracellular heparin-degrading endosulfatases in mice and humans. J Biol Chem. 2002;277:49175–49185. doi: 10.1074/jbc.M205131200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olesnicky Killian EC, Birkholz DA, Artinger KB. A role for chemokine signaling in neural crest cell migration and craniofacial development. Dev Biol. 2009;333:161–172. doi: 10.1016/j.ydbio.2009.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park EJ, Ogden LA, Talbot A, Evans S, Cai CL, Black BL, Frank DU, Moon AM. Required, tissue-specific roles for Fgf8 in outflow tract formation and remodeling. Development. 2006;133:2419–2433. doi: 10.1242/dev.02367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park EJ, Watanabe Y, Smyth G, Miyagawa-Tomita S, Meyers E, Klingensmith J, Camenisch T, Buckingham M, Moon AM. An FGF autocrine loop initiated in second heart field mesoderm regulates morphogenesis at the arterial pole of the heart. Development. 2008;135:3599–3610. doi: 10.1242/dev.025437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prydz K, Dalen KT. Synthesis and sorting of proteoglycans. J Cell Sci. 2000;113(Pt 2):193–205. doi: 10.1242/jcs.113.2.193. [DOI] [PubMed] [Google Scholar]
- Sarkar S, Petiot A, Copp A, Ferretti P, Thorogood P. FGF2 promotes skeletogenic differentiation of cranial neural crest cells. 2001;128:2143–2152. doi: 10.1242/dev.128.11.2143. [DOI] [PubMed] [Google Scholar]
- Sato T, Araki I, Nakamura H. Inductive signal and tissue responsiveness defining the tectum and the cerebellum. Development. 2001;128:2461–2469. doi: 10.1242/dev.128.13.2461. [DOI] [PubMed] [Google Scholar]
- Suemori H, Kadodawa Y, Goto K, Araki I, Kondoh H, Nakatsuji N. A mouse embryonic stem cell line showing pluripotency of differentiation in early embryos and ubiquitous beta-galactosidase expression. Cell Differ Dev. 1990;29:181–186. doi: 10.1016/0922-3371(90)90120-l. [DOI] [PubMed] [Google Scholar]
- Sun X, Meyers EN, Lewandoski M, Martin GR. Targeted disruption of Fgf8 causes failure of cell migration in the gastrulating mouse embryo. Genes Dev. 1999;13:1834–1846. doi: 10.1101/gad.13.14.1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theveneau E, Marchant L, Kuriyama S, Gull M, Moepps B, Parsons M, Mayor R. Collective chemotaxis requires contact-dependent cell polarity. Dev Cell. 2010;19:39–53. doi: 10.1016/j.devcel.2010.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh-Finkel L, Zeisel A, Brodt-Ivenshitz M, Shamai A, Yao Z, Seger R, Domany E, Tzahor E. BMP-mediated inhibition of FGF signaling promotes cardiomyocyte differentiation of anterior heart field progenitors. Development. 2010;137:2989–3000. doi: 10.1242/dev.051649. [DOI] [PubMed] [Google Scholar]
- Toyofuku T, Yoshida J, Sugimoto T, Yamamoto M, Makino N, Takamatsu H, Takegahara N, Suto F, Hori M, Fujisawa H, Kumanogoh A, Kikutani H. Repulsive and attractive semaphorins cooperate to direct the navigation of cardiac neural crest cells. Dev Biol. 2008;321:251–262. doi: 10.1016/j.ydbio.2008.06.028. [DOI] [PubMed] [Google Scholar]
- Trumpp A, Depew MJ, Rubenstein JL, Bishop JM, Martin GR. Cre-mediated gene inactivation demonstrates that FGF8 is required for cell survival and patterning of the first branchial arch. Genes Dev. 1999;13:3136–3148. doi: 10.1101/gad.13.23.3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakamatsu Y, Watanabe Y, Nakamura H, Kondoh H. Regulation of the neural crest cell fate by N-myc: Promotion of ventral migration and neuronal differentiation. 1997;124:1953–1962. doi: 10.1242/dev.124.10.1953. [DOI] [PubMed] [Google Scholar]
- Waldo KL, Kumiski D, Kirby ML. Cardiac neural crest is essential for the persistence rather than the formation of an arch artery. Dev. Dyn. 1996;205:281–292. doi: 10.1002/(SICI)1097-0177(199603)205:3<281::AID-AJA8>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Weisinger K, Kayam G, Missulawin-Drillman T, Sela-Donenfeld D. Analysis of expression and function of FGF-MAPK signaling components in the hindbrain reveals a central role for FGF3 in the regulation of Krox20, mediated by Pea3. Dev Biol. 2010 doi: 10.1016/j.ydbio.2010.06.001. [DOI] [PubMed] [Google Scholar]
- Wendling O, Dennefeld C, Chambon P, Mark M. Retinoid signaling is essential for patterning the endoderm of the third and fourth pharyngeal arches. Development. 2000;127:1553–1562. doi: 10.1242/dev.127.8.1553. [DOI] [PubMed] [Google Scholar]
- Wilkinson DG. In situ hybridization. In: Rickwood D, Hames BD, editors. In Situ Hybridization: A Practical Approach. Oxford: IRS Press; 1992. pp. 75–83. [Google Scholar]
- Wu X, Ge H, Lemon B, Weiszmann J, Gupte J, Hawkins N, Li X, Tang J, Lindberg R, Li Y. Selective activation of FGFR4 by an FGF19 variant does not improve glucose metabolism in ob/ob mice. Proc Natl Acad Sci U S A. 2009;106:14379–14384. doi: 10.1073/pnas.0907812106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Francis R, Wei CJ, Linask KL, Lo CW. Connexin 43-mediated modulation of polarized cell movement and the directional migration of cardiac neural crest cells. Development. 2006;133:3629–3639. doi: 10.1242/dev.02543. [DOI] [PubMed] [Google Scholar]
- Yang X, Dormann D, Munsterberg A, Weijer CJ. Cell movement patterns during gastrulation in the chick are controlled by positive and negative chemotaxis mediated by FGF4 and FGF8. Dev. Cell. 2002;3:425–437. doi: 10.1016/s1534-5807(02)00256-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Premigratory cardiac neural crest cells express all four FGFRs. Semiquantitative PCR of premigratory cardiac neural crest cells (HH10) showing that all four FGFRs are expressed by these cells. FGFR1 and R3 show the highest expression. HPRT was amplified as a control.
Supplemental Figure 2. pERK is highly expressed by cardiac neural crest cells in the pharyngeal arches but not in the secondary heart field. Immunohistochemistry of pERK on transverse sections through arch 3 region. Embryos electroporated with GFP-expressing plasmid (A) and FGF8b-GFP-expressing plasmid (B) do not show a difference in pERK expression by the cardiac neural crest cells (arrows). The secondary heart field (arrowheads) is negative for pERK.
Supplemental Figure 3. Ectopic expression of FGF8 in the cardiac neural crest blocks movement of the cells away from the neural tube. (A) Embryo electroporated in the right sided of the cardiac region of the neural tube with GFP-FGF8 expressing plasmid and harvested at HH13. (B) Transverse section of the same embryo shown in A. The cardiac crest can be seen on the right as a cap of cells (arrows) on the right side of the neural tube. Cardiac neural crest cells on the contralateral side appear to have migrated normally.