

Non-technical summary
Previous studies show that oestrogen is beneficial for maintaining blood glucose levels and helping the body respond to insulin. Despite these previous findings, the mechanism by which oestrogen acts is unknown. We show that specific activation of oestrogen receptor α (ERα) increases glucose uptake into skeletal muscle when insulin is present. Activation of oestrogen receptor β (ERβ) alone or activation of both ERα and ERβ together did not increase glucose uptake into skeletal muscle. This suggests that oestrogen's beneficial effect occurs by activating ERα. These results have important implications for understanding the mechanisms of glucose homeostasis, particularly in postmenopausal women with low oestrogen levels.
Abstract
Abstract
Previous studies suggest oestrogen receptor α (ERα) is involved in oestrogen-mediated regulation of glucose metabolism and is critical for maintenance of whole body insulin action. Despite this, the effect of direct ERα modulation in insulin-responsive tissues is unknown. The purpose of the current study was to determine the impact of ERα activation, using the ER subtype-selective ligand propylpyrazoletriyl (PPT), on skeletal muscle glucose uptake. Two-month-old female Sprague–Dawley rats, ovariectomized for 1 week, were given subcutaneous injections of PPT (10 mg kg−1), oestradiol benzoate (EB; 20 μg kg−1), the ERβ agonist diarylpropionitrile (DPN, 10 mg kg−1) or vehicle every 24 h for 3 days. On the fourth day, insulin-stimulated skeletal muscle glucose uptake was measured in vitro and insulin signalling intermediates were assessed via Western blotting. Activation of ERα with PPT resulted in increased insulin-stimulated glucose uptake into the slow-twitch soleus and fast-twitch extensor digitorum longus (EDL) muscles, activation of insulin signalling intermediates (as measured by phospho-Akt (pAkt) and pAkt substrate (PAS)) and phosphorylation of AMP-activated protein kinase (AMPK). GLUT4 protein was increased only in the EDL muscle. Rats treated with EB or DPN for 3 days did not show an increase in insulin-stimulated skeletal muscle glucose uptake compared to vehicle-treated animals. These new findings reveal that direct activation of ERα positively mediates glucose uptake and insulin action in skeletal muscle. Evidence that oestrogens and ERα stimulate glucose uptake has important implications for understanding mechanisms of glucose homeostasis, particularly in postmenopausal women.
Introduction
Numerous clinical and basic studies demonstrate that oestrogens contribute to glucose homeostasis (Louet et al. 2004). The beneficial effects of oestrogens on insulin action and glucose homeostasis are supported by studies showing insulin sensitivity is higher in premenopausal women compared with age-matched men (Nuutila et al. 1995; Donahue et al. 1997). Following menopause, a significant decline in insulin sensitivity occurs along with a corresponding increase in fat mass (Lindheim et al. 1994; Carr 2003; Alonso et al. 2006; Moreno et al. 2010). Ovariectomy has also been shown to impair insulin sensitivity and glucose metabolism in animal models (Kumagai et al. 1993; Wagner et al. 1998). In addition, oestrogen replacement can ameliorate the increased risk for type 2 diabetes in postmenopausal women and improve whole body (Lindheim et al. 1994; Margolis et al. 2004; Alonso et al. 2006; Riant et al. 2009; Moreno et al. 2010) and skeletal muscle glucose metabolism (Riant et al. 2009; Moreno et al. 2010).
The physiological actions of oestrogens are mediated by two receptors, oestrogen receptor (ER) α and ERβ. Both ERα and ERβ are expressed in a variety of tissues, with ERα more highly expressed in insulin-sensitive tissue (Ribas et al. 2009). Increased adiposity occurs in humans and mice as a result of decreased ERα activation (Smith et al. 1994; Heine et al. 2000), and mice with global knockout of ERα exhibit impaired glucose tolerance and skeletal muscle insulin resistance (Heine et al. 2000; Bryzgalova et al. 2006; Riant et al. 2009). Based on this evidence, the beneficial effects of oestrogens on glucose metabolism are thought to be mediated by ERα. However, while there is strong clinical evidence demonstrating a relationship between ERα expression levels and the incidence of insulin resistance and increased adiposity, the ability of ERα to positivity mediate insulin action and increase glucose uptake in vivo is unknown.
The purpose of the current study was to determine the impact of in vivo ERα activation on skeletal muscle glucose uptake and insulin action. Skeletal muscle accounts for 75% of glucose regulation in the body (DeFronzo et al. 1985) and, as a result, has a significant impact on whole body glucose homeostasis. In the current study, we utilized oestradiol benzoate (EB) and the compound propylpyrazoletriyl (PPT), a potent ERα agonist. PPT is capable of binding with high affinity and 400-fold preference to ERα, and exhibits almost no binding to ERβ (Stauffer et al. 2000). For comparison, the ERβ agonist diarylpropionitrile (DPN), which binds to ERβ at a 70-fold higher affinity than ERα (Meyers et al. 2001), was also used. Our results demonstrate that activation of ERα with PPT increases insulin-stimulated glucose uptake and insulin signalling in skeletal muscle. These findings provide new insight into the role of oestrogen receptors in mediating glucose uptake, a finding with important implications for postmenopausal women at increased risk for type 2 diabetes.
Methods
Ethical approval
The authors have read ‘Reporting ethical matters in The Journal of Physiology: standards and advice’ (Drummond, 2009), and our experiments comply with the policies and regulations of The Journal of Physiology and the UK regulations on animal experimentation. All protocols were approved by the Animal Care and Use Committee of the University of Kansas Medical Center.
Materials
GLUT4 antibody (ab654) and tubulin (ab7291) were purchased from Abcam (Cambridge, MA, USA). pAkt (S473), total Akt, phospho-(ser-thr) Akt substrate (pAS), pERα (S118), pAMPK (T172), and total AMPK were purchased from Cell Signalling (Beverly, MA, USA). PAS-160 (T642) and total AS160 were purchased from Millipore (Billerica, MA, USA). ERα (MC-20) was purchased from Santa Cruz (Santa Cruz, CA, USA) and ERβ (PA1-310B) was purchased from Thermo Fisher Scientific (Rockford, IL, USA). Donkey anti-rabbit HRP-conjugated secondary antibody was purchased from Jackson (West Grove, PA, USA) and goat anti-mouse HRP-conjugated secondary antibody was purchased from Bio-Rad (Hercules, CA, USA). Enhanced chemiluminescence reagents were purchased from Fisher Scientific (Pittsburg, PA, USA). [14C]mannitol and 2-deoxy-[1,2-3H]glucose were purchased from American Radiolabeled Chemicals (St Louis, MO, USA). Oestradiol benzoate (E9000; EB) was purchased from Sigma. Propylpyrazoletriyl (PPT) and diarylpropionitrile (DPN) were purchased from Tocris Bioscience (Ellisville, MO, USA). All other reagents were obtained from Sigma.
Experimental animals and treatment
Two-month-old female Sprague–Dawley rats were purchased from Charles River Laboratories (Wilmington, MA, USA) and housed in a temperature-controlled (22 ± 2°C) room with 12 h light and dark cycles and given free access to food and water. Animals underwent bilateral ovariectomy (OVX) under ketamine–atropine–xylazine anaesthesia (intraperitoneal injection of 60 mg (kg body wt)−1 ketamine, 0.4 mg (kg body wt)−1 atropine and 8 mg (kg body wt)−1 xylazine). Bilateral flank incisions were made under aseptic conditions. The ovaries were identified and bilaterally removed via cauterization. In a subset of six animals, incisions were made but the ovaries were left intact for evaluation of endogenous oestradiol levels at killing. Wounds were closed using sutures and wound clips. One week following surgery, OVX animals received subcutaneous injections once every 24 h for 3 days (N = 6 animals per group) of EB (20 μg (kg body wt)−1) dissolved in 90% corn oil–10% ethanol, PPT (10 mg (kg body wt)−1) dissolved in DMSO, or DPN (10 mg (kg body wt)−1) dissolved in DMSO. This dose of PPT and DPN has previously been used in in vivo rodent studies (Harris et al. 2002; Lee et al. 2005). The dose of EB was chosen to produce physiological levels of serum oestradiol (Hurn & Macrae, 2000; Haim et al. 2003). EB is commonly used in research studies and is a conjugate-salt form of 17β-oestradiol. Like all conjugate molecules, the benzoate moiety dissociates from the 17β-oestradiol moiety when dissolved in solution. Thus, the solution of EB injected into the animals contains free 17β-oestradiol which binds to the ERs. Vehicle treatments were 90% corn oil–10% ethanol or DMSO, as appropriate. No differences in uterine weight or glucose uptake were observed between the two vehicle treatments, and therefore these measurements were combined in the results. Rats were fasted 10 h prior to muscle incubation and glucose transport experiments. Twenty-four hours following the final injection, the animals were anaesthetized with an intraperitoneal injection of pentobarbital sodium (2.5 mg (100 g body wt)−1) for removal of the soleus and extensor digitorum longus (EDL) muscles. The uterus was also removed and weighed. Rats were killed by cervical dislocation.
Muscle incubation
The soleus and EDL muscles were dissected and each split longitudinally into two strips to allow for adequate diffusion of substrates, as described previously (Henriksen & Holloszy, 1991; Gupte et al. 2008). Two muscle strips per rat were assessed for glucose transport and two strips for Western blot analysis. Muscle strips designated for Western blot analysis recovered from the dissection for 60 min in flasks containing 2 ml of Krebs–Henseleit bicarbonate buffer (KHB) with 8 mm glucose, 32 mm mannitol, and a gas phase of 95% O2–5% CO2 (recovery medium). The flasks were placed in a shaking incubator maintained at 35°C. Following recovery, one muscle strip was transferred to recovery medium containing 2 mU ml−1 insulin, and the other muscle strip was left without insulin (basal) in recovery medium for 30 min and then clamp frozen in liquid nitrogen.
Measurement of glucose transport activity
Glucose transport was measured in soleus and EDL muscle strips. Muscle strips were incubated after dissection in recovery medium for 60 min at 35°C and then rinsed for 30 min at 29°C in 2 ml of oxygenated KHB containing 40 mm mannitol, with or without insulin (2 mU ml−1). After the rinse step, muscles were incubated for 20 min at 29°C in flasks containing 2 ml KHB with 4 mm 2-[1,2-3H]deoxyglucose (2-DG) (1.5 μCi ml−1) and 36 mm[14C]mannitol (0.2 μCi ml−1), with or without insulin (2 mU ml−1), with a gas phase of 95% O2–5% CO2 in a shaking incubator. The muscles were then lightly blotted, clamp frozen in liquid nitrogen, and processed as described previously (Young et al. 1986; Geiger et al. 2006) for determination of intracellular 2-DG accumulation (3H dpm) and extracellular space (14C dpm) on a scintillation counter.
Serum oestradiol measurement
Blood samples were collected at time of killing and allowed to clot at room temperature for 30 min. Samples were spun at 17,500 g for 20 min at 4°C. Serum oestradiol levels were measured by Estradiol E2 Coat-a-Count Assay (Siemens Diagnostics, TKE21).
Western blotting
Muscles incubated with and without insulin and clamp frozen in liquid nitrogen were homogenized in a 12:1 (volume-to-weight) ratio of ice-cold buffer from Biosource (Invitrogen, Camarillo, CA, USA) containing 10 mm Tris-HCl (pH 7.4); 100 mm NaCl; 1 mm each of EDTA, EGTA, NaF and phenylmethylsulfonyl fluoride; 2 mm Na3VO4; 20 mm Na4P2O7; 1% Triton X-100; 10% glycerol; 0.1% SDS; 0.5% deoxycholate; and 250 μl/5 ml protease inhibitor cocktail. Homogenized samples were rotated for 30 min at 4°C and then centrifuged for 20 min at 3000 rpm at 4°C. The protein concentration of the supernatant was determined by the Bradford method (Bio-Rad). Samples were prepared in 5× Laemmli buffer containing 100 mm dithiothreitol and boiled in a water bath for 5 min. Samples analysed for GLUT4 protein were not boiled. Protein (30–100 μg) was separated on a SDS-PAGE (7.5–10%) gel followed by a wet transfer to a nitrocellulose membrane for 60–90 min (200 mA). Total protein was visualized by Ponceau staining, and GLUT4 blots were normalized to the 45 kDa band. Membranes were blocked for 1 h at room temperature in 5% non-fat dried milk in Tris-buffered saline with 0.1% Tween 20 (TBST) and then incubated overnight with the appropriate primary antibodies. Antibodies were diluted in 5% non-fat dried milk in TBST or in 1% bovine serum albumin in TBST. Blots were incubated in a HRP-conjugated secondary antibody in 1% non-fat dried milk in TBST for 1 h at room temperature and visualized by Enhanced chemiluminescence (ECL). Western blots first probed for phosphorylated proteins were stripped and probed for total protein expression for normalization, and non-phosphorylated proteins were stripped and probed for tubulin expression for normalization. Blots were stripped for 20 min at 55°C in buffer containing 62.5 mm Tris-HCl, 2% SDS and 100 mm 2-mercaptoethanol. Blots were then rinsed three times in TBST for 15 min each, blocked in 5% milk in TBST for 1 h, and incubated in the appropriate primary antibody overnight. The GLUT4 antibody only works with non-denatured protein, and the tubulin antibody requires denaturation. Therefore, Western blots that were run with non-denatured samples and probed for GLUT4 were not able to be stripped and re-probed for tubulin for normalization. Therefore, Ponceau staining, which does not require denaturing, was used for normalizing as previously described (Gupte et al. 2008). We identified the 56 kDa ERβ protein by using the rat hypothalamus as a positive control (Fig. 1). This tissue has previously been used as a positive control (Kalbe et al. 2007). Bands were quantified using Image J densitometry.
Figure 1. ERβ protein identified by the hypothalamus.
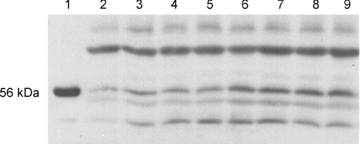
Female Sprague–Dawley rat hypothalamus and soleus muscles were homogenized and total protein was measured as stated in the Methods. ERβ protein was measured by Western blot analysis. Lane 1 represents the hypothalamus (3 μg protein) and lanes 2–9 represent soleus muscle samples (100 μg protein).
Statistical analysis
Results are presented as means ± SE, and statistical significance was set at P < 0.05. Symbols on the uterine weight, pERα, total ERα, pAMPK and GLUT4 graphs represent differences determined by one-way ANOVA. Symbols on the glucose transport, pAkt and PAS160 graphs represent differences determined by a Student–Newman–Keuls post hoc test following a significant interaction as determined by two-way ANOVA.
Results
In vivo effects of oestrogen and PPT treatment
In order to control for endogenous oestrogen levels, all animals underwent OVX for 1 week. At killing, serum oestadiol levels were significantly decreased as a result of OVX (11.9 ± 1.8 pg ml−1vs. 6.8 ± 0.4 pg ml−1 for intact and OVX animals, respectively; P < 0.01). Serum oestradiol levels were significantly greater in EB-treated animals (14.5 ± 2.0 pg ml−1) compared to vehicle-treated animals (6.8 ± 0.4 pg ml−1; P < 0.001). Oestradiol levels for EB-treated animals were within physiological values for cycling rodents (Hurn & Macrae, 2000; Haim et al. 2003).
Administration of oestrogen and PPT has been shown to increase uterine weight by activation of ERα (Harris et al. 2002; Frasor et al. 2003; Stygar et al. 2007). In this manner, uterine weight can serve as a bioassay for the in vivo effects of oestrogen and PPT. In OVX female rats, administration of EB or PPT for 3 days resulted in a significant and similar increase in uterine weight compared to vehicle-treated controls (Fig. 2), which suggests that ERα is being activated to the same extent in the uterus. Consistent with previous findings (Harris et al. 2002; Frasor et al. 2003), a higher dose of PPT than EB was needed to increase uterine weight. In contrast, administration of the ERβ agonist DPN for 3 days had no effect on uterine weight (Fig. 2) as previously shown (Frasor et al. 2003).
Figure 2. Administration of EB and PPT activates ERα as indicated by an increase in uterine weight.
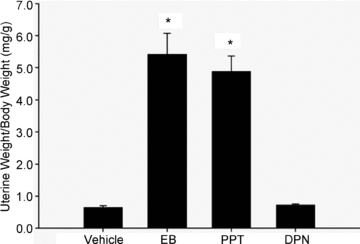
Female Sprague–Dawley rats were given subcutaneous injections of EB (20 μg kg−1), PPT (10 mg kg−1), DPN (10 mg kg−1) or vehicle for 3 days. Uterine weight was measured at time of killing. Symbol represents differences determined by one-way ANOVA. *P < 0.001 vs. vehicle; N = 6 per group.
Prior to treatment with EB, PPT or DPN, body weight was not different among groups. After treatment, body weight did not change in animals treated with EB (vehicle 179.2 ± 3.2 g vs. EB 172.2 ± 2.8 g) or DPN (vehicle 184.8 ± 3.6 g vs. DPN 181.5 ± 2.9 g) but decreased in animals treated with PPT (vehicle 183.3 ± 2.0 g vs. PPT 169.3 ± 1.8 g; P < 0.001).
PPT increases insulin-stimulated glucose transport in soleus and EDL muscle
To determine the impact of ERα stimulation on skeletal muscle glucose uptake, we performed in vitro 2-DG uptake assays on the predominately slow-twitch soleus or the predominately fast-twitch EDL muscles following EB, PPT or DPN administration. Insulin-stimulated glucose uptake was significantly increased above basal in all treatment groups in both the soleus and EDL muscles (Fig. 3A and B, respectively). Three day treatment with EB did not augment insulin-stimulated glucose uptake compared to that observed with vehicle alone. However, specifically activating ERα with PPT resulted in a greater increase in insulin-stimulated glucose transport in both the soleus and EDL muscles compared to vehicle-treated controls. This observed increase in the soleus and EDL muscles was 108% and 55%, respectively, greater than insulin-stimulated glucose transport in rats treated with vehicle. Treatment with the ERβ agonist DPN had no effect on insulin-stimulated skeletal muscle glucose uptake.
Figure 3. Skeletal muscle glucose transport in female rats treated with vehicle, EB, PPT and DPN.
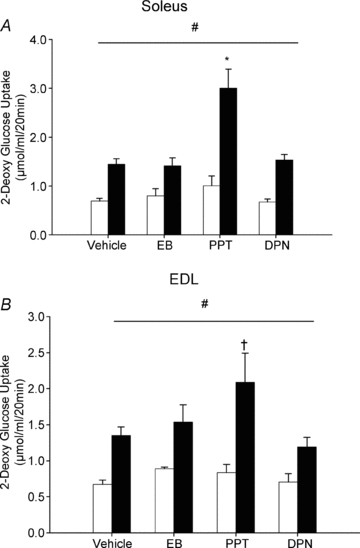
Female Sprague–Dawley rats were give subcutaneous injections of EB (20 μg kg−1), PPT (10 mg kg−1), DPN (10 mg kg−1) or vehicle for 3 days. Insulin-stimulated glucose transport was measured in soleus (A) and EDL (B) muscles as described in the Methods. Briefly, muscles were incubated in the absence of insulin (open bars) or in the presence of insulin (2 mU ml−1, filled bars), along with 2-[1,2-3H]deoxyglucose. *P < 0.001 and †P < 0.05 vs. insulin-stimulated vehicle indicates a significant interaction as determined by two-way ANOVA. The horizontal line indicates a significant main effect of insulin relative to basal across all treatment groups, #P < 0.05; N = 6 per group.
PPT activates ERα in skeletal muscle
Given the significant effect of PPT on skeletal muscle glucose uptake, we next investigated the cell signalling pathways altered by PPT in skeletal muscle. Phosphorylation of ERα, a measure of protein activation (Weigel, 1996; Joel et al. 1998), significantly increased in soleus and EDL muscles (Fig. 4C and D, respectively) following 3 days of PPT administration compared to vehicle-treated controls. Total ERα also decreased following PPT treatment (Fig. 4C and D). No change in ERβ occurred in soleus or EDL muscles as a result of PPT treatment (Fig. 4C and D), highlighting the specificity of PPT for ERα in skeletal muscle.
Figure 4. PPT activates ERα in skeletal muscle.
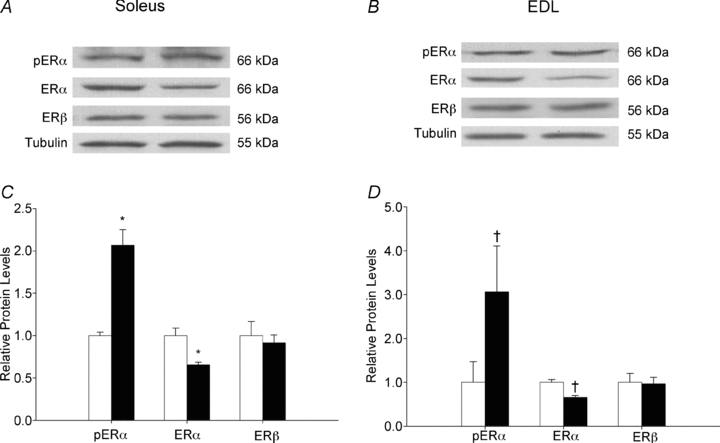
Female Sprague–Dawley rats were given subcutaneous injections of PPT (10 mg kg−1) or vehicle for 3 days. Western blot analysis measured phosphorylation of ERα normalized to total ERα, and total ERα and total ERβ normalized to tubulin in the soleus (C) and EDL (D) for vehicle (open bars) and PPT-treated (filled bars) animals. Representative blots are shown for the soleus (A) and EDL (B). *P < 0.05 and †P < 0.01 vs. vehicle as determined by one-way ANOVA; N = 6 per group.
PPT increases insulin-stimulated phosphorylation of Akt and AMPK in soleus and EDL muscle
Akt is a protein kinase in the insulin–insulin receptor substrate-1 (IRS-1)–phosphoinositide 3-kinase (PI3K) signalling cascade, and activation of this protein is crucial for insulin-stimulated glucose transport. Our results demonstrate that activation of Akt in response to insulin stimulation, as measured by phosphorylation of Akt on serine 473, was increased in the soleus and EDL muscles from rats treated with PPT (Fig. 5A and B) with a significantly greater increase in PPT-treated rats compared to vehicle. An additional pathway for signalling GLUT4 translocation to the membrane and increasing glucose transport, independent of insulin, is by phosphorylation of AMP-activated protein kinase (AMPK). PPT treatment resulted in increased phosphorylation of AMPK in both the soleus and EDL muscles (Fig. 5C and D, respectively).
Figure 5. In vivo activation of ERα via PPT increases insulin-stimulated phosphorylation of Akt and AMPK in the soleus and EDL.
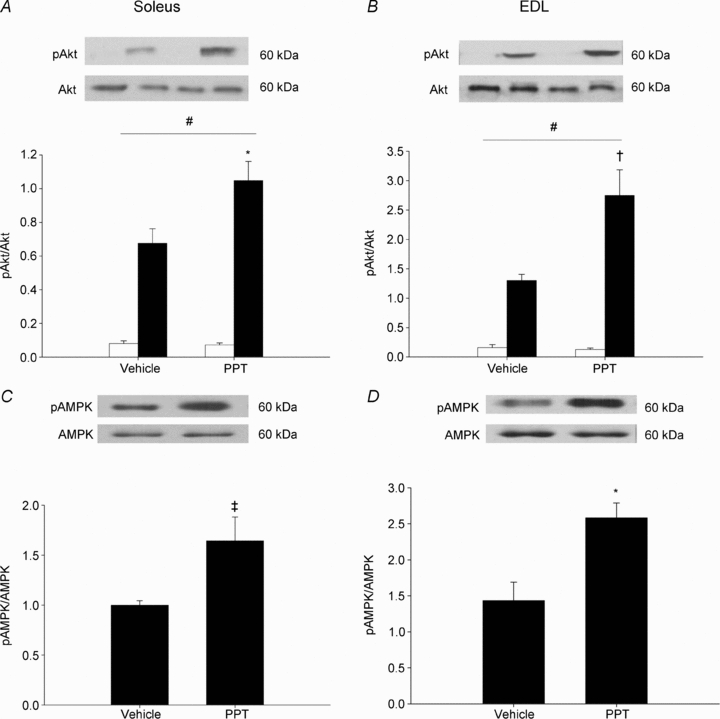
Female Sprague–Dawley rats were treated with PPT as stated in the Methods. Activation of Akt was measured by Western blot analysis of pAkt normalized to Akt in the absence of insulin (open bars) or in the presence of insulin (2 mU ml−1, filled bars) in the soleus (A) and EDL (B). *P < 0.01, †P < 0.001 on A and B vs. insulin-stimulated vehicle indicates a significant interaction as determined by two-way ANOVA. The horizontal lines indicate a significant main effect of insulin relative to basal, #P < 0.05. Activation of AMPK was measured by Western blot analysis of pAMPK normalized to total AMPK in the soleus (C) and EDL (D). Symbols on C and D represent differences determined by one-way ANOVA. *P < 0.01, ‡P < 0.05 vs. vehicle; N = 6 per group.
PPT increases insulin-stimulated phosphorylation of PAS160 in soleus and EDL muscle
Activation of Akt and AMPK results in downstream activation of Akt substrates with a molecular mass of 160 kDa, including AS160. Detection of activated Akt substrates can collectively be identified by a phospho-Akt substrate (pAS) antibody. Activation of Akt substrates with a molecular mass of 160 kDa (PAS-160) in response to insulin stimulation was increased in the soleus and EDL muscles from rats treated with PPT (Fig. 6A and B). In addition, insulin-stimulated activation of PAS-160 was significantly greater with PPT compared to vehicle alone. We measured specific activation of AS160 by phosphorylation of threonine 642 and found no additional increase in insulin-stimulated activation in the soleus and EDL muscles from rats treated with PPT compared to rats treated with vehicle (Fig. 6C and D).
Figure 6. In vivo activation of ERα via PPT increases insulin-stimulated phosphorylation of PAS-160 in the soleus and EDL.
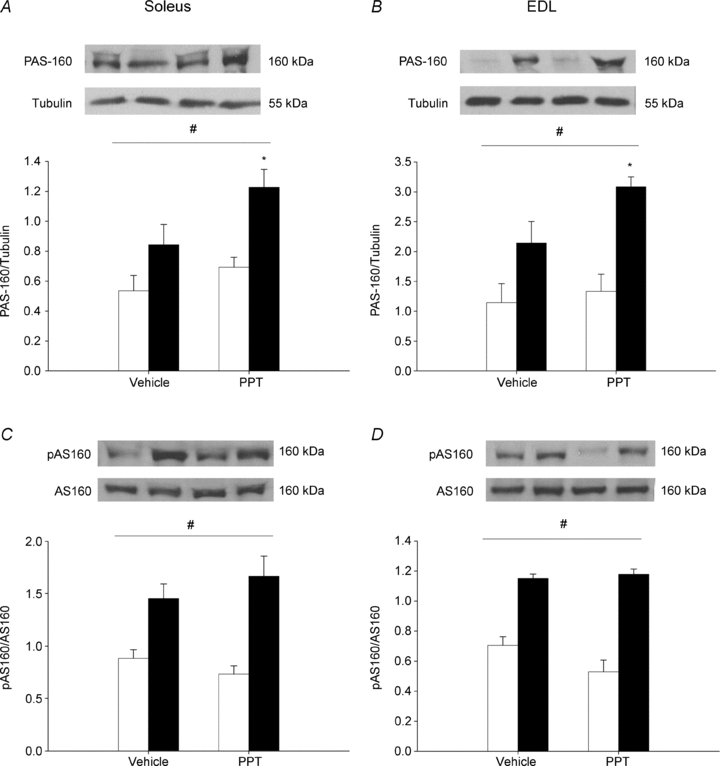
Female Sprague–Dawley rats were treated with PPT as stated in the Methods. Downstream activation of Akt and AMPK was measured by Western blot analysis of PAS-160 normalized to tubulin (A and B) and PAS-160 normalized to total AS160 (C and D) in the absence of insulin (open bars) or in the presence of insulin (2 mU ml−1, filled bars) in the soleus (A and C) and EDL (B and D). *P < 0.05 vs. insulin-stimulated vehicle indicates a significant interaction as determined by two-way ANOVA. The horizontal lines indicate a significant effect of insulin relative to basal, #P < 0.05; N = 6 per group.
PPT increases GLUT4 protein in the EDL muscle
Insulin and AMPK both signal translocation of GLUT4 to the cell membrane where GLUT4 then transports glucose into the cell. Rats treated with PPT demonstrated increased GLUT4 protein levels in EDL muscles (Fig. 7B). In contrast, GLUT4 protein was unaltered in response to PPT in soleus muscles (Fig. 7A). GLUT4 levels were not altered as a result of EB or DPN treatment (Fig. 7C–F).
Figure 7. In vivo activation of ERα via PPT increases GLUT4 protein in the EDL muscle.
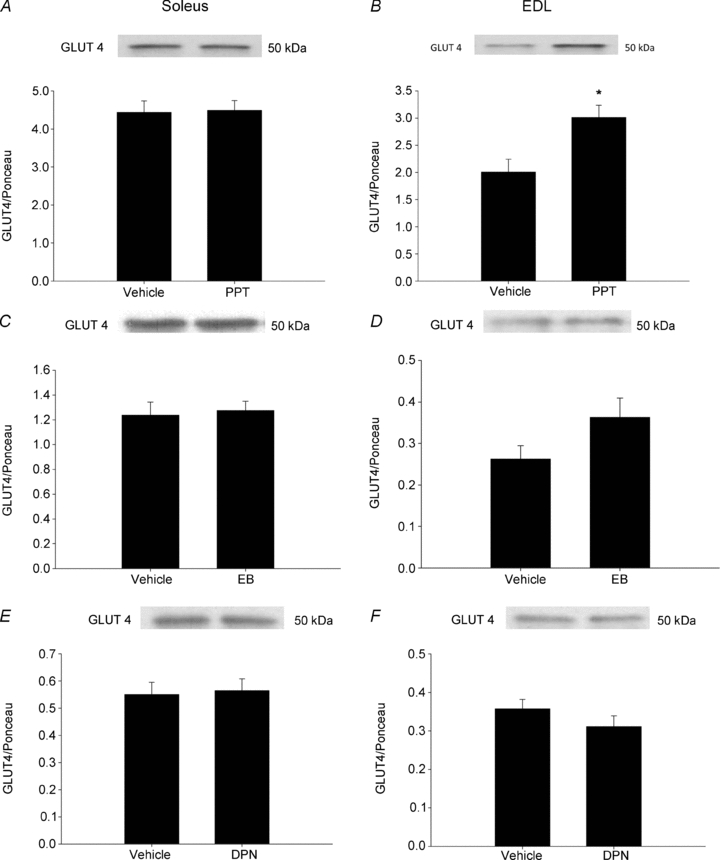
Female Sprague–Dawley rats were treated with PPT, EB or DPN as stated in the Methods. GLUT4 was measured by Western blot analysis and normalized to total protein as measured by Ponceau staining in animals treated with PPT (A and B), EB (C and D) and DPN (E and F). Symbol represents differences determined by one-way ANOVA. *P < 0.05 vs. vehicle; N = 6 per group.
Discussion
The purpose of the current study was to determine the effect of direct ERα modulation on skeletal muscle glucose uptake. While studies utilizing ERα-deficient mice demonstrate impaired glucose tolerance in the absence of ERα (Heine et al. 2000; Bryzgalova et al. 2006; Riant et al. 2009), in vivo activation of ERα in control female rodents had not previously been tested. Three day treatment with PPT, a specific agonist of ERα, resulted in increased glucose uptake and activation of Akt, PAS-160 and AMPK in both soleus and EDL muscles. In addition, activation of ERα resulted in increased GLUT4 protein in the EDL muscle. This new evidence of the ability of short-term modulation of ERα to increase glucose uptake has important implications for understanding the regulation of glucose uptake, particularly in postmenopausal women.
In a previous study by Ribas et al. (2009), ERα knockout mice demonstrated impaired glucose tolerance and reduced insulin sensitivity in liver and skeletal muscle while on a normal chow diet. The decrease in glucose disposal rate in the knockout mice was attributed primarily to impaired insulin action in skeletal muscle. This is in contrast to a study by Bryzgalova et al. (2006) that attributed deceased glucose tolerance in ERα knockout mice primarily to the liver. Our current findings demonstrating a dramatic increase in insulin-stimulated skeletal muscle glucose uptake in control animals treated with the ERα agonist PPT support the idea that ERα plays an important role in skeletal muscle insulin sensitivity. Using ER subtype-specific ligands is an alternative and complementary approach to using knockout animals. It is encouraging that both methods demonstrate an important role for ERα in mediating skeletal muscle insulin sensitivity.
To our knowledge, only one previous study has looked at the effect of PPT on glucose uptake in skeletal muscle. In this study, ob/ob mice were treated with PPT for 7 days prior to measurement of insulin-stimulated glucose uptake (Lundholm et al. 2008). In contrast with the current study, there was no increase in soleus or EDL muscle glucose uptake in ob/ob mice treated with PPT. This difference can most likely be attributed to the lower dose of PPT used in the previous study (1mg (kg body wt)−1vs. 10 mg (kg body wt)−1). Further, the use of hyperglycaemic and hyperinsulinaemic ob/ob mice in the previous study is markedly different from the control OVX rats used in the current study. Additional experiments will be needed to determine the ability of a higher dose of PPT to improve insulin sensitivity in insulin-resistant or type 2 diabetes animal models.
Oestrogen and PPT have been shown to potentiate the insulin signalling pathway and increase glucose transport in adipocytes in culture (Muraki et al. 2006; Nagira et al. 2006), and oestrogen has been shown to increase phosphorylation of Akt (Vasconsuelo et al. 2008) and AMPK (D'Eon et al. 2008) in C2C12 muscle cells. In the first study to test the effects of oestrogen on skeletal muscle in vitro, acute incubations (5 and 10 min) with oestrogen increased phosphorylation of Akt, AMPK and TBC1D1/4 in soleus muscle (Rogers et al. 2009). However, incubation in oestrogen for 10 min did not increase insulin-stimulated glucose transport. Our findings show that 3 days of oestrogen treatment in vivo also had no effect on insulin-stimulated glucose uptake in soleus or EDL muscles. These findings are the first to show that both in isolated rodent skeletal muscle and following in vivo administration, acute oestrogen treatment did not produce a measurable increase in skeletal muscle glucose uptake. This may be due to the specific expression pattern and activation of ERs in skeletal muscle.
Previous studies indicate that oestrogens and oestrogen receptor modulators will produce a distinct phenotype in cells that express predominately ERα compared to those expressing predominantly ERβ (Kian Tee et al. 2004). A number of studies suggest that ERα is more highly expressed in insulin-sensitive tissues (Deroo & Korach, 2006; Heldring et al. 2007), and this expression pattern was also recently demonstrated in mouse skeletal muscle (Ribas et al. 2009; Baltgalvis et al. (2010). In contrast, Barros et al. (2009) suggest that ERβ expression predominates in skeletal muscle, although these investigators primarily focused on nuclear ER expression and the results were not quantified. ERα and ERβ are known to demonstrate a complex inter-regulatory relationship that varies with the target tissue. For example, activation of ERβ can oppose the action of ERα and act as a negative regulator in glucose metabolism (Matthews & Gustafsson, 2003; Barros et al. 2006a,b;). As oestrogen activates both ERs, the lack of an effect of oestrogen on skeletal muscle glucose uptake in the present study could be due to ERβ activation off-setting any stimulation of ERα via oestrogen. As DPN administration did not decrease insulin-stimulated glucose uptake in the present study, stimulation of both ERα and ERβ by oestrogen could prevent direct activation of ERα due to the unique active conformation formed between oestrogen and these two receptors. In addition to the inter-regulatory actions of the ERs on each other, oestrogen and oestrogen receptor modulators exert distinct tissue-specific effects by recruiting different co-regulatory proteins to ERs (Shang & Brown, 2002; Kian Tee et al. 2004). This difference in co-regulatory protein recruitment can result in a modulator having agonist or antagonist properties. The specific co-regulatory proteins involved in ERα activation by PPT and oestrogen in skeletal muscle have yet to be identified. Previous studies have shown that long-term oestrogen treatment can improve whole body and skeletal muscle glucose metabolism in animals fed a high-fat diet or as a result of ageing (Riant et al. 2009; Moreno et al. 2010). Ageing or metabolic disease could alter the ratio of ERα to ERβ in skeletal muscle and change the tissue response to oestrogen and oestrogen receptor modulators. Future studies are needed to determine the impact of insulin resistance, and subsequent chronic oestrogen treatment, on ER expression and activation patterns in skeletal muscle.
In the current study, PPT increased ERα phosphorylation in skeletal muscle relative to total ERα, while total ERα expression was decreased compared to vehicle-treated controls. Phosphorylation of ERα may occur in the cytoplasm or the nucleus and is important for receptor dimerization and DNA binding (Arnold et al. 1995a,b;). However, the extent to which phosphorylation of ERα contributes to the involvement of ERα in cell signalling cascades is unknown, and little is known regarding the effect of PPT on phosphorylation of ERα. To our knowledge, this is the first report of ERα activation and expression characterized in skeletal muscle as a result of PPT treatment. Studies thus far have mainly focused on oestrogen's phosphorylation of ERα. Phosphorylation on tyrosine residues probably represents basal phosphorylation of ERα in the absence of oestrogen (Migliaccio et al. 1986; Arnold et al. 1995a), and activation of ERα results in phosphorylation of serine residues (Washburn et al. 1991; Weigel, 1996). Specifically, Joel et al. (1998) report that oestrogen treatment in MCF-7 cells results in increased phosphorylation on Ser118. In addition, mutation of Ser118 resulted in a 40% reduction in transactivation activity in response to oestrogen (Le Goff et al. 1994). We chose to assess Ser118 due to a more recent report showing increased phosphorylation of ERα on Ser118 in the soleus muscle due to resveratrol treatment, which resulted in enhanced glucose uptake (Deng et al. 2008).
In contrast to the previously mentioned studies on ERα phosphorylation, the present study used PPT as an ERα agonist. In MCF-7 cells, Joel et al. (1998) demonstrated an increase in oestrogen-stimulated pERα on Ser118 relative to total ERα, with no decrease in total ERα. In contrast, while our study in rat skeletal muscle demonstrates an increase in PPT-stimulated pERα on Ser118 relative to total ERα, we also saw a decrease in total ERα. In fact, more recent reports demonstrate that phosphorylation of ERα on Ser118 leads to protein degradation of ERα (reviewed in Murphy et al. 2011). Ultimately, our study demonstrates that of the total ERα present, more ERα is phosphorylated on Ser118 in the PPT-treated animals than in the vehicle-treated animals. A recent study by Baltgalvis et al. (2010) demonstrated that 1 week of OVX resulted in an increase in ERα gene and protein expression compared to sham-treated animals. Two days of oestradiol treatment in OVX female mice in this same study resulted in a decrease in ERα levels, similar to our results with PPT. In addition, we have recently shown that ER protein levels are altered in the skeletal muscle and adipose tissue in response to OVX and a high-fat diet (Gorres et al. 2011). The specificity of PPT for ERα in skeletal muscle is further demonstrated by the lack of effect of PPT on ERβ expression. In addition to regulation of ERα by phosphorylation, localization of ERα proteins (nuclear, cytosolic, membrane-associated) could also play an important role in activation and regulation in response to PPT and should be pursued in future studies.
PPT also results in direct activation of the insulin signalling pathway as shown by increased phosphorylation of Akt. To our knowledge, this is the first evidence of the effects of PPT on insulin signalling. In support of our findings, stimulation of the insulin signalling pathway with resveretrol in C2C12 myotubes was shown to be dependent on ERα activation (Deng et al. 2008). Phosphorylation of AS160 on threonine 642 is commonly used to assess activation of AS160 and, hence, activation of the insulin signalling pathway downstream of Akt. Our results indicate that rats treated with PPT have increased insulin-stimulated phospho-Akt substrate (pAS), but not as a result of a significant increase in PAS-160 on threonine 642. The PAS immunoreactivity at 160 kDa includes both AS160 and a paralogue of AS160, TBC1D1. As a result, an additional site of phosphorylation on AS160 or the TBC1D1 protein could be activated with PPT and result in increased glucose transport.
Specific activation of ERαin vivo with PPT results in increased pAMPK in soleus and EDL muscles. In support of this finding, ERα knockout (ERα-KO) mice demonstrate decreased pAMPK in skeletal muscle (Ribas et al. 2009). Furthermore, skeletal muscle stimulated with oestrogen in vivo and in vitro can increase AMPK activation (D'Eon et al. 2008; Riant et al. 2009; Rogers et al. 2009), with a recent study showing that oestrogen-induced AMPK activation is mediated by ERα (Rogers et al. 2009). Together, these findings suggest that ERα acts as a positive modulator of AMPK activation. AMPK activation can result in increased basal glucose transport, an effect that was not observed in the present study. The amount of AMPK phosphorylation may have been insufficient to alter basal glucose uptake in the present study. AMPK can phosphorylate both AS160 (Treebak et al. 2006; Chen et al. 2008) and TBC1D1 (Geraghty et al. 2007; Pehmoller et al. 2009), although with phospho-specific sites distinct from those activated by Akt, to stimulate an increase in glucose uptake. This potential for differential regulation has important implications for the regulation of glucose uptake and will need to be further explored in the context of ERα activation.
The ERs have been shown to be involved in modulation of GLUT4 transcription, with ERα acting as a positive modulator and ERβ acting as a negative modulator (Barros et al. 2006a, 2009). Barros et al. (2006a,b); have proposed a mechanism by which ERα could bind to nuclear factor-kappa B (NF-κB), a transcription factor with the potential to repress GLUT4 expression. The binding of ERα to NF-κB could inhibit this transcription factor's repression of GLUT4 and thereby increase GLUT4 expression. Without ERα present, GLUT4 is decreased in the gastrocnemius muscle of male ERα-KO mice (Barros et al. 2006b). However, a more recent report shows that female ERα-KO mice do not have decreased GLUT4 in the quadriceps or soleus muscle (Ribas et al. 2009). In the current study, activation of ERα resulted in increased GLUT4 in the EDL (fast twitch) but not in the soleus (slow twitch), which suggests that the ability of ERα to regulate GLUT4 may be fibre type specific. As we measured total GLUT4 protein levels, we do not know if the increase in GLUT4 in the EDL contributed to the increase in insulin-stimulated glucose uptake. It is possible that the increase in total GLUT4 did not contribute to an increase in GLUT4 at the membrane or in glucose transport. If so, then this would explain the disparity between the soleus and EDL GLUT4 and glucose transport data. However, as GLUT4 is an important protein for insulin-stimulated glucose uptake, it is important to understand factors which modulate GLUT4, and the ability to modulate GLUT4 by direct ERα activation has not previously been shown. While numerous transcriptional pathways regulate GLUT4 (Murgia et al. 2009), acute ERα activation may be an additional mechanism for modulating GLUT4.
A recent study reported that 8 weeks of OVX increased circulating glycerol, non-esterified fatty acids (NEFA) and glucose, and oestrogen treatment reversed this (Wohlers & Spangenburg, 2010). While long-term OVX and oestrogen treatment had significant systemic metabolic effects, it is unlikely that 1 week of OVX or 3 days of PPT treatment resulted in significant changes in plasma glucose, insulin or lipids, although we did not measure these factors in the current study. We think the PPT effects demonstrated in this study are a result of acute changes in signalling and protein expression in the skeletal muscle, although future studies will be needed to confirm a lack of systemic effect on plasma glucose, insulin and lipid levels. In addition, the decrease in body weight with PPT treatment suggests that ERα activation may affect body weight and metabolism. These factors may also play an important role in increasing skeletal muscle glucose uptake and insulin signalling and will be addressed in future studies.
In summary, our study demonstrates that the ERα agonist PPT results in increased insulin-stimulated glucose uptake into the skeletal muscle via potentiating the insulin signalling pathway, activating AMPK, and increasing GLUT4 protein. Specific activation of ERα may provide an additional means by which drug treatments can be developed that have a positive impact on glucose metabolism. Future studies are needed to determine the long-term effects of oestrogens, insulin resistance and type 2 diabetes on ER expression and activation in skeletal muscle.
Acknowledgments
The authors would like to thank Anisha Gupte and Susan Smittkamp for their technical assistance with this manuscript. The project described was supported by NIH grants AG031575 and P20 RR016475 from the National Centre for Research Resources (NCRR), with core support provided by Kansas Intellectual and Developmental Disabilities Research Centre grant HD02528. The authors would also like to thank the University of Kansas Medical Centre Biomedical Research Training Program awarded to B.K.G. for financial support.
Glossary
Abbreviations
- AMPK
AMP-activated protein kinase
- DPN
diarylpropionitrile
- EB
oestradiol benzoate
- EDL
extensor digitorum longus
- ERα
oestrogen receptor α
- ERβ
oestrogen receptor β
- GLUT4
glucose transporter 4
- OVX
ovariectomy
- pAkt
phospho-Akt
- PAS
phospho-Akt substrate
- PPT
propylpyrazoletriyl
Author contributions
B.K.G. and P.C.G. designed the experiments, analysed and interpreted data, and wrote the paper. B.K.G., G.L.B., J.K.M. and P.C.G. collected the data. All authors approved the final version of the manuscript. These experiments were carried out at the University of Kansas Medical Centre.
References
- Alonso A, Fernandez R, Moreno M, Ordonez P, Gonzalez-Pardo H, et al. Positive effects of 17β-estradiol on insulin sensitivity in aged ovariectomized female rats. J Gerontol A Biol Sci Med Sci. 2006;61:419–426. doi: 10.1093/gerona/61.5.419. [DOI] [PubMed] [Google Scholar]
- Arnold SF, Obourn JD, Jaffe H, Notides AC. Phosphorylation of the human estrogen receptor on tyrosine 537 in vivo and by src family tyrosine kinases in vitro. Mol Endocrinol. 1995a;9:24–33. doi: 10.1210/mend.9.1.7539106. [DOI] [PubMed] [Google Scholar]
- Arnold SF, Obourn JD, Yudt MR, Carter TH, Notides AC. In vivo and in vitro phosphorylation of the human estrogen receptor. J Steroid Biochem Mol Biol. 1995b;52:159–171. doi: 10.1016/0960-0760(94)00166-j. [DOI] [PubMed] [Google Scholar]
- Baltgalvis KA, Greising SM, Warren GL, Lowe DA. Estrogen regulates estrogen receptors and antioxidant gene expression in mouse skeletal muscle. PLoS One. 2010;5:e10164. doi: 10.1371/journal.pone.0010164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barros RP, Gabbi C, Morani A, Warner M, Gustafsson JA. Participation of ERα and ERβ in glucose homeostasis in skeletal muscle and white adipose tissue. Am J Physiol Endocrinol Metab. 2009;297:E124–E133. doi: 10.1152/ajpendo.00189.2009. [DOI] [PubMed] [Google Scholar]
- Barros RP, Machado UF, Gustafsson JA. Estrogen receptors: new players in diabetes mellitus. Trends Mol Med. 2006a;12:425–431. doi: 10.1016/j.molmed.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Barros RP, Machado UF, Warner M, Gustafsson JA. Muscle GLUT4 regulation by estrogen receptors ERβ and ERα. Proc Natl Acad Sci U S A. 2006b;103:1605–1608. doi: 10.1073/pnas.0510391103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryzgalova G, Gao H, Ahren B, Zierath JR, Galuska D, Steiler TL, et al. Evidence that oestrogen receptor-α plays an important role in the regulation of glucose homeostasis in mice: insulin sensitivity in the liver. Diabetologia. 2006;49:588–597. doi: 10.1007/s00125-005-0105-3. [DOI] [PubMed] [Google Scholar]
- Carr MC. The emergence of the metabolic syndrome with menopause. J Clin Endocrinol Metab. 2003;88:2404–2411. doi: 10.1210/jc.2003-030242. [DOI] [PubMed] [Google Scholar]
- Chen S, Murphy J, Toth R, Campbell DG, Morrice NA, Mackintosh C. Complementary regulation of TBC1D1 and AS160 by growth factors, insulin and AMPK activators. Biochem J. 2008;409:449–459. doi: 10.1042/BJ20071114. [DOI] [PubMed] [Google Scholar]
- D'Eon TM, Rogers NH, Stancheva ZS, Greenberg AS. Estradiol and the estradiol metabolite, 2-hydroxyestradiol, activate AMP-activated protein kinase in C2C12 myotubes. Obesity (Silver Spring) 2008;16:1284–1288. doi: 10.1038/oby.2008.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFronzo RA, Gunnarsson R, Bjorkman O, Olsson M, Wahren J. Effects of insulin on peripheral and splanchnic glucose metabolism in noninsulin-dependent (type II) diabetes mellitus. J Clin Invest. 1985;76:149–155. doi: 10.1172/JCI111938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng JY, Hsieh PS, Huang JP, Lu LS, Hung LM. Activation of estrogen receptor is crucial for resveratrol-stimulating muscular glucose uptake via both insulin-dependent and -independent pathways. Diabetes. 2008;57:1814–1823. doi: 10.2337/db07-1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deroo BJ, Korach KS. Estrogen receptors and human disease. J Clin Invest. 2006;116:561–570. doi: 10.1172/JCI27987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donahue RP, Bean JA, Donahue RA, Goldberg RB, Prineas RJ. Insulin response in a triethnic population: effects of sex, ethnic origin, and body fat. Miami Community Health Study. Diabetes Care. 1997;20:1670–1676. doi: 10.2337/diacare.20.11.1670. [DOI] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frasor J, Barnett DH, Danes JM, Hess R, Parlow AF, Katzenellenbogen BS. Response-specific and ligand dose-dependent modulation of estrogen receptor (ER) α activity by ERβ in the uterus. Endocrinology. 2003;144:3159–3166. doi: 10.1210/en.2002-0143. [DOI] [PubMed] [Google Scholar]
- Geiger PC, Han DH, Wright DC, Holloszy JO. How muscle insulin sensitivity is regulated: testing of a hypothesis. Am J Physiol Endocrinol Metab. 2006;291:E1258–E1263. doi: 10.1152/ajpendo.00273.2006. [DOI] [PubMed] [Google Scholar]
- Geraghty KM, Chen S, Harthill JE, Ibrahim AF, Toth R, Morrice NA, et al. Regulation of multisite phosphorylation and 14-3-3 binding of AS160 in response to IGF-1, EGF, PMA and AICAR. Biochem J. 2007;407:231–241. doi: 10.1042/BJ20070649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorres BK, Bomhoff GL, Gupte AA, Geiger PC. Altered estrogen receptor expression in skeletal muscle and adipose tissue of female rats fed a high-fat diet. J Appl Physiol. 2011 doi: 10.1152/japplphysiol.00541.2010. Jan 13, E-Pub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupte AA, Bomhoff GL, Geiger PC. Age-related differences in skeletal muscle insulin signaling: the role of stress kinases and heat shock proteins. J Appl Physiol. 2008;105:839–848. doi: 10.1152/japplphysiol.00148.2008. [DOI] [PubMed] [Google Scholar]
- Haim S, Shakhar G, Rossene E, Taylor AN, Ben-Eliyahu S. Serum levels of sex hormones and corticosterone throughout 4- and 5-day estrous cycles in Fischer 344 rats and their simulation in ovariectomized females. J Endocrinol Invest. 2003;26:1013–1022. doi: 10.1007/BF03348201. [DOI] [PubMed] [Google Scholar]
- Harris HA, Katzenellenbogen JA, Katzenellenbogen BS. Characterization of the biological roles of the estrogen receptors, ERα and ERα, in estrogen target tissues in vivo through the use of an ERα-selective ligand. Endocrinology. 2002;143:4172–4177. doi: 10.1210/en.2002-220403. [DOI] [PubMed] [Google Scholar]
- Heine PA, Taylor JA, Iwamoto GA, Lubahn DB, Cooke PS. Increased adipose tissue in male and female estrogen receptor-α knockout mice. Proc Natl Acad Sci U S A. 2000;97:12729–12734. doi: 10.1073/pnas.97.23.12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldring N, Pike A, Andersson S, Matthews J, Cheng G, Hartman J, et al. Estrogen receptors: how do they signal and what are their targets. Physiol Rev. 2007;87:905–931. doi: 10.1152/physrev.00026.2006. [DOI] [PubMed] [Google Scholar]
- Henriksen EJ, Holloszy JO. Effect of diffusion distance on measurement of rat skeletal muscle glucose transport in vitro. Acta Physiol Scand. 1991;143:381–386. doi: 10.1111/j.1748-1716.1991.tb09249.x. [DOI] [PubMed] [Google Scholar]
- Hurn PD, Macrae IM. Estrogen as a neuroprotectant in stroke. J Cereb Blood Flow Metab. 2000;20:631–652. doi: 10.1097/00004647-200004000-00001. [DOI] [PubMed] [Google Scholar]
- Joel PB, Traish AM, Lannigan DA. Estradiol-induced phosphorylation of serine 118 in the estrogen receptor is independent of p42/p44 mitogen-activated protein kinase. J Biol Chem. 1998;273:13317–13323. doi: 10.1074/jbc.273.21.13317. [DOI] [PubMed] [Google Scholar]
- Kalbe C, Mau M, Wollenhaupt K, Rehfeldt C. Evidence for estrogen receptor a and b expression in skeletal muscle of pigs. Histochem Cell Biol. 2007;127:95–107. doi: 10.1007/s00418-006-0224-z. [DOI] [PubMed] [Google Scholar]
- Kian Tee M, Rogatsky I, Tzagarakis-Foster C, Cvoro A, An J, Christy RJ, et al. Estradiol and selective estrogen receptor modulators differentially regulate target genes with estrogen receptors α and β. Mol Biol Cell. 2004;15:1262–1272. doi: 10.1091/mbc.E03-06-0360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai S, Holmang A, Bjorntorp P. The effects of oestrogen and progesterone on insulin sensitivity in female rats. Acta Physiol Scand. 1993;149:91–97. doi: 10.1111/j.1748-1716.1993.tb09596.x. [DOI] [PubMed] [Google Scholar]
- Le Goff P, Montano MM, Schodin DJ, Katzenellenbogen BS. Phosphorylation of the human estrogen receptorIdentification of hormone-regulated sites and examination of their influence on transcriptional activity. J Biol Chem. 1994;269:4458–4466. [PubMed] [Google Scholar]
- Lee GS, Kim HJ, Jung YW, Choi KC, Jeung EB. Estrogen receptor α pathway is involved in the regulation of Calbindin-D9k in the uterus of immature rats. Toxicol Sci. 2005;84:270–277. doi: 10.1093/toxsci/kfi072. [DOI] [PubMed] [Google Scholar]
- Lindheim SR, Buchanan TA, Duffy DM, Vijod MA, Kojima T, Stanczyk FZ, Lobo RA. Comparison of estimates of insulin sensitivity in pre- and postmenopausal women using the insulin tolerance test and the frequently sampled intravenous glucose tolerance test. J Soc Gynecol Investig. 1994;1:150–154. doi: 10.1177/107155769400100210. [DOI] [PubMed] [Google Scholar]
- Louet JF, LeMay C, Mauvais-Jarvis F. Antidiabetic actions of estrogen: insight from human and genetic mouse models. Curr Atheroscler Rep. 2004;6:180–185. doi: 10.1007/s11883-004-0030-9. [DOI] [PubMed] [Google Scholar]
- Lundholm L, Bryzgalova G, Gao H, Portwood N, Falt S, Berndt KD, et al. The estrogen receptor α-selective agonist propyl pyrazole triol improves glucose tolerance in ob/ob mice; potential molecular mechanisms. J Endocrinol. 2008;199:275–286. doi: 10.1530/JOE-08-0192e. [DOI] [PubMed] [Google Scholar]
- Margolis KL, Bonds DE, Rodabough RJ, Tinker L, Phillips LS, Allen C, et al. Effect of oestrogen plus progestin on the incidence of diabetes in postmenopausal women: results from the Women's Health Initiative Hormone Trial. Diabetologia. 2004;47:1175–1187. doi: 10.1007/s00125-004-1448-x. [DOI] [PubMed] [Google Scholar]
- Matthews J, Gustafsson JA. Estrogen signaling: a subtle balance between ERα and ERβ. Mol Interv. 2003;3:281–292. doi: 10.1124/mi.3.5.281. [DOI] [PubMed] [Google Scholar]
- Meyers MJ, Sun J, Carlson KE, Marriner GA, Katzenellenbogen BS, Katzenellenbogen JA. Estrogen receptor-β potency-selective ligands: structure-activity relationship studies of diarylpropionitriles and their acetylene and polar analogues. J Med Chem. 2001;44:4230–4251. doi: 10.1021/jm010254a. [DOI] [PubMed] [Google Scholar]
- Migliaccio A, Rotondi A, Auricchio F. Estradiol receptor: phosphorylation on tyrosine in uterus and interaction with anti-phosphotyrosine antibody. EMBO J. 1986;5:2867–2872. doi: 10.1002/j.1460-2075.1986.tb04581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno M, Ordonez P, Alonso A, Diaz F, Tolivia J, Gonzalez C. Chronic 17β-estradiol treatment improves skeletal muscle insulin signaling pathway components in insulin resistance associated with aging. Age (Dordr) 2010;32:1–13. doi: 10.1007/s11357-009-9095-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muraki K, Okuya S, Tanizawa Y. Estrogen receptor a regulates insulin sensitivity through IRS-1 tyrosine phosphorylation in mature 3T3-L1 adipocytes. Endocr J. 2006;53:841–851. doi: 10.1507/endocrj.k06-005. [DOI] [PubMed] [Google Scholar]
- Murgia M, Jensen TE, Cusinato M, Garcia M, Richter EA, Schiaffino S. Multiple signalling pathways redundantly control glucose transporter GLUT4 gene transcription in skeletal muscle. J Physiol. 2009;587:4319–4327. doi: 10.1113/jphysiol.2009.174888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy LC, Seekallu SV, Watson PH. Clinical significance of estrogen receptor phosphorylation. Endocr Relat Cancer. 2011;18:R1–R14. doi: 10.1677/ERC-10-0070. [DOI] [PubMed] [Google Scholar]
- Nagira K, Sasaoka T, Wada T, Fukui K, Ikubo M, Hori S, et al. Altered subcellular distribution of estrogen receptor α is implicated in estradiol-induced dual regulation of insulin signaling in 3T3-L1 adipocytes. Endocrinology. 2006;147:1020–1028. doi: 10.1210/en.2005-0825. [DOI] [PubMed] [Google Scholar]
- Nuutila P, Knuuti MJ, Maki M, Laine H, Ruotsalainen U, Teras M, et al. Gender and insulin sensitivity in the heart and in skeletal muscles. Studies using positron emission tomography. Diabetes. 1995;44:31–36. doi: 10.2337/diab.44.1.31. [DOI] [PubMed] [Google Scholar]
- Pehmoller C, Treebak JT, Birk JB, Chen S, Mackintosh C, Hardie DG, et al. Genetic disruption of AMPK signaling abolishes both contraction- and insulin-stimulated TBC1D1 phosphorylation and 14-3-3 binding in mouse skeletal muscle. Am J Physiol Endocrinol Metab. 2009;297:E665–E675. doi: 10.1152/ajpendo.00115.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riant E, Waget A, Cogo H, Arnal JF, Burcelin R, Gourdy P. Estrogens protect against high-fat diet-induced insulin resistance and glucose intolerance in mice. Endocrinology. 2009;150:2109–2117. doi: 10.1210/en.2008-0971. [DOI] [PubMed] [Google Scholar]
- Ribas V, Nguyen MT, Henstridge DC, Nguyen AK, Beaven SW, Watt MJ, Hevener AL. Impaired oxidative metabolism and inflammation are associated with insulin resistance in ERα-deficient mice. Am J Physiol Endocrinol Metab. 2009;298:E304–E319. doi: 10.1152/ajpendo.00504.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers NH, Witczak CA, Hirshman MF, Goodyear LJ, Greenberg AS. Estradiol stimulates Akt, AMP-activated protein kinase (AMPK) and TBC1D1/4, but not glucose uptake in rat soleus. Biochem Biophys Res Commun. 2009;382:646–650. doi: 10.1016/j.bbrc.2009.02.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang Y, Brown M. Molecular determinants for the tissue specificity of SERMs. Science. 2002;295:2465–2468. doi: 10.1126/science.1068537. [DOI] [PubMed] [Google Scholar]
- Smith EP, Boyd J, Frank GR, Takahashi H, Cohen RM, Specker B, et al. Estrogen resistance caused by a mutation in the estrogen-receptor gene in a man. N Engl J Med. 1994;331:1056–1061. doi: 10.1056/NEJM199410203311604. [DOI] [PubMed] [Google Scholar]
- Stauffer SR, Coletta CJ, Tedesco R, Nishiguchi G, Carlson K, Sun J, Katzenellenbogen BS, Katzenellenbogen JA. Pyrazole ligands: structure-affinity/activity relationships and estrogen receptor-α-selective agonists. J Med Chem. 2000;43:4934–4947. doi: 10.1021/jm000170m. [DOI] [PubMed] [Google Scholar]
- Stygar D, Masironi B, Eriksson H, Sahlin L. Studies on estrogen receptor (ER) α and β responses on gene regulation in peripheral blood leukocytes in vivo using selective ER agonists. J Endocrinol. 2007;194:101–119. doi: 10.1677/JOE-06-0060. [DOI] [PubMed] [Google Scholar]
- Treebak JT, Glund S, Deshmukh A, Klein DK, Long YC, Jensen TE, et al. AMPK-mediated AS160 phosphorylation in skeletal muscle is dependent on AMPK catalytic and regulatory subunits. Diabetes. 2006;55:2051–2058. doi: 10.2337/db06-0175. [DOI] [PubMed] [Google Scholar]
- Vasconsuelo A, Milanesi L, Boland R. 17β-estradiol abrogates apoptosis in murine skeletal muscle cells through estrogen receptors: role of the phosphatidylinositol 3-kinase/Akt pathway. J Endocrinol. 2008;196:385–397. doi: 10.1677/JOE-07-0250. [DOI] [PubMed] [Google Scholar]
- Wagner JD, Thomas MJ, Williams JK, Zhang L, Greaves KA, Cefalu WT. Insulin sensitivity and cardiovascular risk factors in ovariectomized monkeys with estradiol alone or combined with nomegestrol acetate. J Clin Endocrinol Metab. 1998;83:896–901. doi: 10.1210/jcem.83.3.4628. [DOI] [PubMed] [Google Scholar]
- Washburn T, Hocutt A, Brautigan DL, Korach KS. Uterine estrogen receptor in vivo: phosphorylation of nuclear specific forms on serine residues. Mol Endocrinol. 1991;5:235–242. doi: 10.1210/mend-5-2-235. [DOI] [PubMed] [Google Scholar]
- Weigel NL. Steroid hormone receptors and their regulation by phosphorylation. Biochem J. 1996;319:657–667. doi: 10.1042/bj3190657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlers LM, Spangenburg EE. 17β-estradiol supplementation attenuates ovariectomy-induced increases in ATGL signaling and reduced perilipin expression in visceral adipose tissue. J Cell Biochem. 2010;110:420–427. doi: 10.1002/jcb.22553. [DOI] [PubMed] [Google Scholar]
- Young DA, Uhl JJ, Cartee GD, Holloszy JO. Activation of glucose transport in muscle by prolonged exposure to insulin. Effects of glucose and insulin concentrations. J Biol Chem. 1986;261:16049–16053. [PubMed] [Google Scholar]