

Non-technical summary
A high concentration of cholesterol in the blood, known as hypercholersterolaemia, in the absence of overt atherosclerotic disease induces changes throughout the circulation including an inability to fully respond to vasodilatory stimuli. Here we show that skin blood flow responses are reduced in hypercholersterolaemic men and women partly due to an upregulation of the arginase pathway. Arginase competes for the common substrate l-arginine for the synthesis of the vasoprotective molecule nitric oxide. After 3 months of oral atrovastatin (cholesterol lowering medication) intervention, arginase activity was decreased and skin blood flow responses resembled those of healthy men and women. This suggests that upregulated arginase contributes to decreased vasoreactivity in hyperocholesterolaemic humans and that atrovastatin therapy restores functional vasodilatory properties by decreasing arginase activity.
Abstract
Abstract
Elevated low-density lipoproteins (LDLs) are associated with vascular dysfunction evident in the cutaneous microvasculature. We hypothesized that uncoupled endothelial nitric oxide synthase (NOS3) through upregulated arginase contributes to cutaneous microvascular dysfunction in hyperocholesterolaemic (HC) humans and that a statin intervention would decrease arginase activity. Five microdialysis fibres were placed in the skin of nine normocholesterolaemic (NC: LDL level 95 ± 4 mg dl−1) and nine hypercholesterolaemic (HC: LDL: 177 ± 6 mg dl−1) men and women before and after 3 months of systemic atrovastatin. Sites served as control, NOS inhibited, arginase inhibited, l-arginine supplemented and arginase inhibited plus l-arginine supplemented. Skin blood flow was measured while local skin heating (42°C) induced NO-dependent vasodilatation. l-NAME was infused after the established plateau in all sites to quantify NO-dependent vasodilatation. Data were normalized to maximum cutaneous vascular conductance (CVCmax). Skin samples were obtained to measure total arginase activity and arginase I and arginase II protein. Vasodilatation was reduced in hyperocholesterolaemic subjects (HC: 76 ± 2 vs. NC: 94 ± 3%CVCmax, P < 0.001) as was NO-dependent vasodilatation (HC: 43 ± 5 vs. NC: 62 ± 4%CVCmax, P < 0.001). The plateau and NO-dependent vasodilatation were augmented in HC with arginase inhibition (92 ± 2, 67 ± 2%CVCmax, P < 0.001), l-arginine (93 ± 2, 71 ± 5%CVCmax, P < 0.001) and combined treatments (94 ± 4, 65 ± 5%CVCmax, P < 0.001) but not in NC. After statin intervention (LDL: 98 ± 5 mg dl−1) there was no longer a difference between control sites (88 ± 4, 61 ± 5%CVCmax) and localized microdialysis treatment sites (all P > 0.05). Arginase activity and protein were increased in HC skin (P < 0.05 vs. NC) and activity decreased with atrovastatin treatment (P < 0.05). Reduced NOS3 substrate availability through upregulated arginase contributes to cutaneous microvascular dysfunction in hyperocholesterolaemic humans, which is corrected with atorvastatin therapy.
Introduction
Hypercholesterolaemia with elevated oxidized low density lipoprotein (oxLDL) is a major risk factor for the development of atherosclerosis (Toshima et al. 2000; Inoue et al. 2001; Vasankari et al. 2001). One early indicator of hyperocholesterolaemia-associated vascular disease is a decrease in endothelial derived nitric oxide (NO), which plays a crucial vasoprotective role by promoting vasodilatation and by inhibiting leukocyte adhesion, vascular smooth muscle proliferation, and platelet aggregation. The human cutaneous circulation has emerged as an easily accessible and representative regional circulation for investigating mechanisms of microvascular dysfunction in clinical populations (Abularrage et al. 2005; Cracowski et al. 2006; Holowatz, 2008; Debbabi et al. 2010). Hyperocholesterolaemically induced microvascular dysfunction involving a reduction in NO-dependent vasodilatation is clearly evident in the cutaneous circulation (Binggeli et al. 2003; Rossi et al. 2009). Hypercholesterolaemic humans exhibit a significantly attenuated cutaneous vasodilatory response to local skin warming (Stulc et al. 2003), a stimulus known to induce vasodilatation predominantly through the production of NO via endothelial nitric oxide synthase (NOS3) (Minson et al. 2001; Kellogg et al. 2008). Furthermore, systemic statin interventions have been shown to improve the overall cutaneous vascular responsiveness to vasodilatory stimuli (Binggeli et al. 2003). However, the mechanisms underlying impaired cutaneous microvascular function with hypercholesterolaemia and the effects of statin therapy on these mechanisms are unknown.
One putative mechanism mediating hyperocholesterolaemically induced vascular dysfunction is through the upregulation of arginase (Ryoo et al. 2006; Lim et al. 2007). Two isoforms of arginase are present in the vasculature having different subcellular locations with arginase I being localated in the cytosol and arginase II being constrained to the mitochondria (Ash, 2004). Arginase activity is differentially upregulated in animal models of aging (Berkowitz et al. 2003), hypertension (Demougeot et al. 2006), and atherosclerosis and is known to restrain NOS3 production of functional NO by competing for l-arginine substrate pools and contributing to NOS3 uncoupling. Furthermore, vascular arginase II activity is reduced in atherosclerotic animal models with statin treatment via a pleiotropic effect of stabilizing the cellular microtubular structure via RhoA-dependent mechanisms resulting in delocalizing arginase from NOS3 (Lim et al. 2007; Ryoo et al. 2010).
Therefore, we sought to determine the role of arginase and freely exchangeable l-arginine in reduced cutaneous NO-dependent vasodilatation in hyperocholesterolaemic humans before and after statin therapy. We hypothesized that uncoupled NOS3 through upregulated arginase contributes to cutaneous microvascular dysfunction in hyperocholesterolaemic humans and would be reduced with a 3 month atorvastatin systemic intervention. Because isoform specific arginase inhibitors are not available, we tested the hypothesis that acute arginase I and II inhibition or l-arginine supplementation would functionally augment NO-dependent cutaneous vasodilatation in hyperocholesterolaemic humans. We further hypothesized that a short term oral atrovastatin intervention would augment NO-dependent vasodilatation through decreased total arginase activity.
Methods
Subjects
Experimental protocols were approved by the Institutional Review Board at The Pennsylvania State University and conformed to the guidelines set forth by the Declaration of Helsinki. Verbal and written consent were voluntarily obtained from all subjects prior to participation. Nine healthy normocholesterolaemic and nine hyperocholesterolaemic men and women (Table 1) participated in the study consisting of functional in vivo assessment of cutaneous NO-dependent vasodilatation during local skin warming and skin biopsy sampling for in vitro analysis. The hypercholesterolaemic subjects were tested at enrollment and after a 3 month atrovastatin intervention (10 mg daily); the normocholesterolaemic control group was only tested once. The subjects’ age ranged from 40 to 62 years and the groups (hypercholesterolaemic and normocholesterolaemic) were age-matched to account for any possible age-related changes in the local heating response (Minson et al. 2002). Furthermore, the subjects were non-obese, non-smokers, non-diabetic, normally active (neither sedentary nor highly exercise trained), and not currently taking statins or other medications including aspirin, vitamins and antioxidants.
Table 1.
Subject characteristics
| Normocholesterolaemic | Hypercholesterolaemic | Atorvastatin | |
|---|---|---|---|
| Subjects (men, women) | (5, 4) | (6, 3) | |
| Age (years) | 49 ± 2 | 53 ± 3 | |
| Total cholesterol (mg dl−1) | 171 ± 7 | 260 ± 9* | 179 ± 9‡ |
| HDL (mg dl−1) | 60 ± 5 | 51 ± 7 | 55 ± 8 |
| LDL (mg dl−1) | 95 ± 4 | 177 ± 6* | 98 ± 6‡ |
| Triglycerides (mg dl−1) | 86 ± 13 | 139 ± 11* | 134 ± 10 |
| Glucose (mg dl−1) | 93 ± 3 | 94 ± 3 | |
| ADMA (μmol l−1) | 0.43 ± 0.06 | 0.37 ± 0.10 | 0.33 ± 0.08 |
| Oxidized LDL (U l−1) | 64 ± 5 | 136 ± 12* | 89 ± 8‡* |
P < 0.05 difference from the normocholesterolemic group
P < 0.05 difference due to the atorvastatin intervention.
Blood analysis
Serum and plasma samples were obtained at enrollment and after the atorvastatin intervention and stored at −80°C for batched analysis of asymmetrical dimethyl l-arginine (ADMA; Alpco, Salem, NH, USA) and oxLDL (Mercodia, Uppsala, Sweden).
In vivo vasoreactive studies
All protocols were performed in a thermoneutral laboratory with the subject semi-supine and the experimental arm at heart level. Five intradermal microdialysis probes were inserted into the ventral forearm skin for localized delivery of pharmacological agents as previously described (Holowatz et al. 2006; Holowatz & Kenney, 2007). Microdialysis sites were perfused with: (1) 10.0 mmNG.-nitro-l-arginine (l-NAME) to inhibit NO production by NO-synthase; (2) a combination of 5.0 mm (S)-(2-boronoethyl)-l-cysteine-HCl (BEC) and 5.0 mmNω-hydroxy-nor-l-arginine (nor-NOHA) to inhibit arginase (Calbiochem, San Diego, CA, USA); (3) 10.0 mm l-arginine (Sigma) to supplement the substrate for NO synthase and arginase; or (4) 5.0 mm BEC + 5.0 mm nor-NOHA + 10.0 mm l-arginine to inhibit arginase and supplement the substrate for NO-synthase and arginase (FDA investigational drug number 78,954) (Holowatz & Kenney, 2007). A fifth microdialysis site was perfused with only lactated Ringer solution to serve as control. All pharmacological solutions were mixed immediately prior to usage, dissolved in lactated Ringer solution, and sterilized using syringe microfilters (Acrodisc, Pall, Ann Arbor, MI, USA). The concentrations of the pharmacological agents used in this study have been shown to be efficacious in younger, middle-aged and older age groups using the same intradermal microdialysis technique (Holowatz et al. 2006, 2008; Holowatz & Kenney, 2007). Furthermore, the concentrations of the arginase inhibitor cocktail far exceed the Ki for the enzyme isoforms (BEC Ki at pH 7.5 is 0.31 μm; nor-NOHA Ki at pH 7.5 is 1.6 μm) (Ash, 2004).
An index of skin blood flow was measured using integrated laser-Doppler flowmeter probes and local temperature was controlled with a local heater (MoorLAB, Temperature Monitor SH02, Moor Instruments, Millwey (Axminster), UK) placed directly above each microdialysis membrane. This multipoint probe monitored blood flow under an area approximately 2 mm directly over each microdialysis fibre. Arterial blood pressure was measured every 5 min using an automated brachial cuff (Cardiocap) which was verified with brachial auscultation. Cutaneous vascular conductance (CVC) was calculated as laser-Doppler flux divided by mean arterial pressure (MAP).
Local heating protocol
After the resolution of the initial insertion trauma with local skin temperature clamped at 33°C, a standardized local skin warming protocol was performed to induce NO-dependent vasodilatation (Minson et al. 2001). The local heater temperature was increased from 33°C to 42°C at a rate of 0.1°C every second and then clamped at 42°C for the duration of the heating protocol. After skin blood flow reached an established plateau (30–40 min), 10 mm l-NAME was perfused to quantify NO-dependent vasodilatation in all sites. Following a new post-l-NAME stabilization in skin blood flow, local temperature was increased to 43°C and 28 mm sodium nitroprusside (SNP) was perfused to induce maximal cutaneous vasodilatation (CVC) (Johnson et al. 1986; Holowatz et al. 2005). In our previous work and in pilot work this combination of heat and high concentration of SNP has been shown to induce maximal vasodilatation. Higher temperatures (44°C) or increasing concentrations of SNP (50 mm) did not produce a further increase in absolute CVC (Holowatz et al. 2005).
In vitro skin sample analysis
Skin biopsy samples were obtained in the normocholesterolaemic control group and in the hypercholesterolaemic group before and after the oral atrovastatin intervention. Ventral forearm skin samples were obtained on a separate day from the in vivo functional assessment of vasoreactivity and on the opposite arm. The skin was anaesthetized using 2% lidocaine without adrenaline. Using sterile technique two 3 mm diameter skin samples were obtained. Samples were rinsed in lactated Ringer solution and immediately frozen in liquid nitrogen and stored at −80°C until analysis.
Arginase activity assay
Arginase activity was measured by determining levels of urea production as previously described (Berkowitz et al. 2003). Briefly, skin samples were sonicated for 10 min in lysis buffer (50 mmol l−1 Tris-HCl, pH 7.5, 0.1 mmol EDTA, 0.1% Triton X-100, and protease inhibitor) and centrifuged for 30 min at 14,000 g at 4°C. Supernatant (50 μl) was then added to 75 μl of Tris-HCl (50 mmol l−1, pH 7.5) containing 10 mmol l−1 MnCl2, and the mixture was activated by heating for 10 min at 55–60°C. The mixture was incubated with l-arginine (50 μl, 0.5 m, pH 9.7) at 37°C for 1 h, and the reaction was stopped by adding 400 μl of an acid solution (H2SO4–H3PO4–H2O = 1:3:7). For colorimetric determination of urea, α-isonitrosopropiophenone (25 μl, 9% in ethanol) was added, and the mixture was heated at 100°C for 45 min. After the sample was placed in the dark for 10 min at room temperature, the urea concentration was determined spectrophotometrically by measuring absorbance at 550 nm.
Western blot analysis
After centrifugation of skin homogenates twice at 15,000 g at 4°C for 20 min, protein concentration was determined using a Bio-Rad DC protein assay. For Western blot analysis, 25 μg proteins were fractionated by SDS/PAGE and electrotransferred to a nitrocellulose membrane (Hybond-ECL, GE Healthcare). The membranes were blocked for 1 h at room temperature (5% non-fat dry milk, in Tris buffered saline containing 0.1% Tween-20; TBST) and incubated with a primary antibody (arginase I, arginase II or NOS3; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA; 1:1000). Bound antibody was detected with horseradish peroxidase (HRP)-conjugated IgG secondary antibody (1:1000) (Santa Cruz Biotechnology) and visualized using enhanced chemiluminescence. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as loading control.
Data and statistical analysis
All data from the vasoreactive studies were digitized at 40 Hz and stored for offline analysis with signal-processing software (Windaq, Dataq Instruments, Inc., Akron, OH, USA). Skin blood flow data were normalized to a percentage of maximal CVC (%CVCmax), CVC data were averaged for a stable 5 min of baseline, plateau, post l-NAME plateau, and maximal vasodilatation. Due to the transient nature of the local warming response the initial peak and nadir CVC were visually identified as the highest and lowest values and averaged over 10 s. The vasodilatation due to NO was calculated from the difference between the plateau and the post-l-NAME plateau. Because the late plateau phase of the local heating response is primarily dependent on NOS function whereas the early phase has contributions from both sensory nerves and NOS, analysis and further discussion focus on the later phase of the cutaneous vasodilatory response.
Student's unpaired t test was used to determine significant differences between the groups and Student's paired t test was used to determine the effects of the statin intervention on blood characteristics. A one-way ANOVA with Tukey's post hoc test was used to determine differences in arginase activity in the skin samples. A mixed models three-way repeated measures ANOVA was conducted to detect differences in %CVCmax between subject groups and for the statin intervention at the pharmacological treatment sites for the different phases of the local warming response (SAS v. 9.1; SAS Institute Inc., Cary, NC, USA). Specific planned comparisons with the Bonferoni correction were performed when appropriate to determine where differences between groups, statin intervention, and localized drug treatments occurred. The level of significance was set at α= 0.05. Values are presented as means ± SEM.
Results
Subject characteristics are presented in Table 1. At enrollment there was a significant difference between the normocholesterolaemic and hypercholesterolaemic groups for triglycerides, total cholesterol, LDL and oxidized LDL. Three months of atrovastatin therapy decreased total cholesterol, LDL and oxidized LDL in the hypercholesterolaemics, but in the hypercholesterolaemics oxidized LDL continued to be modestly elevated over the normocholesterolaemics (P = 0.014) after the intervention. There was no difference between the groups or with statin therapy for plasma assymetrical dimethyl l-arginine (ADMA; endogenous NOS inhibitor) concentrations.
Representative skin blood flow responses during local heating at the control site are shown in Fig. 1. Figure 1A shows the response in a normocholesterolaemic subject and in a hypercholesterolaemic subject. Figure 1B shows the response in a hypercholesterolaemic subject after 12 weeks of oral atorvastatin. Mean values for the groups for the control sites for the plateau and the plateau after NOS inhibition are depicted on the representative tracings. There were no differences between groups or with the atorvastatin intervention for baseline, initial peak, or the nadir in any of the localized microdialysis treatment sites (all P > 0.05). There were also no differences in absolute maximal CVC with localized microdialysis treatment, between groups, or with the atorvastatin intervention (P > 0.05).
Figure 1. Representative tracings of the skin blood flow response to local skin warming.
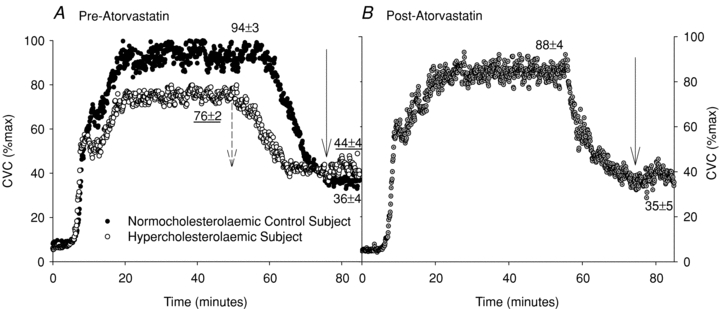
Representative tracings of the skin blood flow response to local skin warming in a normocholesterolaemic control subject and a hyperocholesterolaemic subject (A) and after the oral atorvastatin intervention (B). Relevant means ± SEM group values of the local heating response are also included. NO-dependent vasodilatation was attenuated in hyperocholesterolaemic subjects and was augmented with the atorvastatin intervention. Arrows indicate the reduction in cutaneous vascular conductance (CVC) %max with NOS inhibition with l-NAME.
Figure 2 illustrates the NO-dependent plateau and skin blood flow after NOS inhibition in the normocholesterolaemic and hypercholesterolaemic groups before and after atorvastatin treatment. The reduction in %CVCmax with NOS inhibition is also shown in figure 3. The NOS-dependent plateau in skin blood flow during local heating was attenuated for the hypercholesterolaemic group compared to the normocholesterolaemic (P < 0.001) and was increased after atorvastatin intervention (P < 0.001). %CVCmax after NOS-inhibition was higher in the hypercholesterolaemic group compared to the normocholesterolaemic group and was decreased by the atorvastatin intervention (P = 0.01). The corresponding reduction in %CVCmax with NOS inhibition was less in the hypercholesterolaemic group compared to the normocholesterolaemic group and was increased with the atovastatin intervention (P < 0.001). Localized arginase inhibition (Fig. 2B), l-arginine supplementation (Fig. 2C), or the combined arginase inhibition with l-arginine supplementation (Fig. 2D) augmented the NO-dependent plateau and decreased %CVCmax after NOS inhibition during local heating in the hypercholesterolaemic group (P < 0.001). Further there was a greater reduction in %CVCmax after NOS inhibition with these localized treatments (figure P < 0.001). When compared to the respective control sites these localized treatments did not affect the skin blood flow responses to local heating in the normocholesterolaemic group or in the hypercholesterolaemic group after the statin intervention.
Figure 2. Mean skin blood flow.
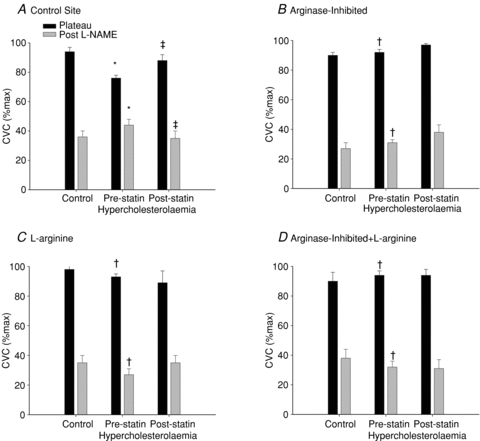
Cutaneous vascular conductance (%max) at the plateau in skin blood flow during local warming and after NOS inhibition with l-NAME in normocholesterolaemic control subjects, hyperocholesterolaemic subjects and after the oral atorvastatin intervention in the control site (A), the arginase-inhibited site (B), the l-arginine supplemented site (C), and combined arginase-inhibited +l-arginine supplemented (D). *P < 0.05 difference from the normocholersterolaemic group; †P < 0.05 difference compared to the control site due to the localized microdialysis drug treatment; ‡P < 0.05 difference due to the atorvastatin intervention.
Figure 3. The reduction in cutaneous vascular conductance with NOS inhibition.
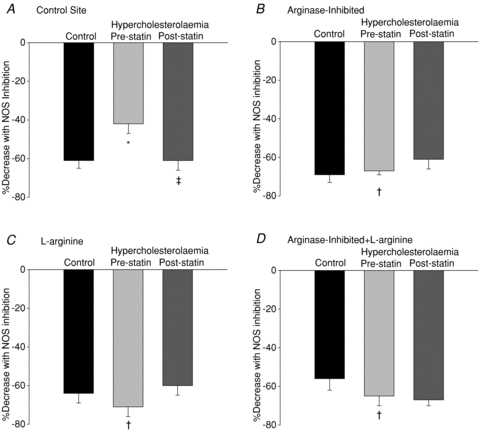
The reduction in cutaneous vascular conductance with NOS inhibition in normocholesterolaemic control subjects, hyperocholesterolaemic subjects and after the oral atorvastatin intervention in the control site (A), the arginase-inhibited site (B), the l-arginine supplemented site (C), and combined arginase inhibited +l-arginine supplemented (D). *P < 0.05 difference from the normocholersterolaemic group; †P < 0.05 difference compared to the control site due to the localized microdialysis drug treatment; ‡P < 0.05 difference due to the atorvastatin intervention.
Figure 4 shows total arginase activity from skin biopsy homogenates from the normocholesterolaemic and hyperocholesterolaemic groups before and after the atorvastatin intervention. Total arginase activity was increased in the hypercholesterolaemic group compared to the normocholesterolaemic group and was decreased with the atrovastatin intervention (P < 0.05). Figure 5 shows arginase I, arginase II and NOS3 protein/GAPDH. Both arginase I and II protein express were increased in the hypercholesterolaemic group compared to the normocholesterolaemic group and did not change with atorvastatin intervention (figure P < 0.05). There was no difference in NOS3 protein between groups or with the atorvastatin intervention.
Figure 4. Arginase activity in skin biopsies.
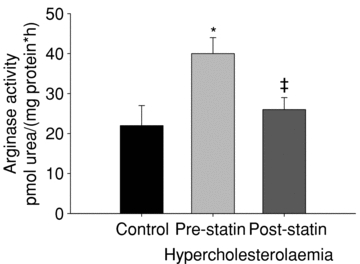
Samples were homogenized in 1× RIPA buffer containing protease inhibitors. Arg assay was performed using α-isonitrosopropiophenone to detect urea produced. Statin treatment restores Arg activity towards that of control. *P < 0.05 difference from the normocholersterolaemic group; ‡P < 0.05 difference due to the atorvastatin intervention.
Figure 5. Arginase I, arginase II and NOS3 expression.
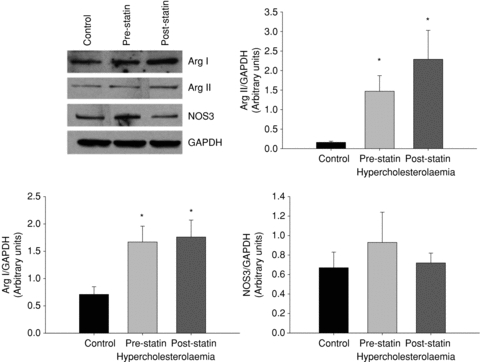
Expression of the three enzymes was determined by Western blotting. GAPDH was used as loading control. Sample blot is shown in first panel. Densitometry analysis was performed using ImageJ software (NIH). *P < 0.05 difference from the normocholersterolaemic group.
Discussion
The major new findings of this study include the following: cutaneous NO-dependent vasodilatation is attenuated in hyperocholesterolaemic humans and is partially due to upregulated arginase activity. In vivo localized arginase inhibition, l-arginine supplementation, or the two combined augments NO-dependent vasodilatation during local warming in hypercholesterolaemic humans before the atorvastatin intervention. Twelve weeks of an oral atorvastatin intervention augments functional NO-dependent vasodilatation in the cutaneous microcirculation by decreasing arginase activity but did not change total arginase protein content. These data suggest that increased arginase activity contribute to cutaneous endothelial dysfunction in preatherosclerotic humans and suggests that one of the mechanisms mediating improved cutaneous vascular function with the atorvastatin intervention is to decrease arginase activity. This decrease in arginase activity may be through a direct effect of lowering oxidized LDL or through one of the pleiotropic effects of statins.
In this study we capitalized on the accessibility of the human cutaneous circulation to examine the underlying mechanisms mediating microvascular dysfunction with hypercholesterolaemia. Others have assessed attenuated cutaneous microvascular reactivity using a variety of vasodilatory stimuli (Binggeli et al. 2003; Stulc et al. 2003; Rossi et al. 2009). Although these studies established a baseline description of cutaneous endothelial dysfunction with hypercholesteroaemia, they did not examine the underlying mechanisms of the microvascular dysfunction. In this study we specifically sought to examine the roles of arginase and freely exchangeable l-arginine pools in attenuated NO-dependent vasodilatation induced by local skin warming (Minson et al. 2001). Both the in vivo functional skin blood flow data and the in vitro analysis of the skin samples indicate that arginase is a possible novel molecular target for increasing NO bioavailability, recoupling NOS, and reversing microvascular dysfunction in hypercholesterolaemic humans. In the present study localized l-arginine supplementation, arginase inhibition or the combination of the two augmented cutaneous NO-dependent vasodilatation. Interestingly, these localized interventions augmented the local heating plateau in skin blood flow and also caused a greater decrease in the %CVCmax after NOS inhibition during local warming (Figs 1 and 2) compared to the control site. These data suggest that in hypercholesterolaemic microvasculature uncoupled NOS3 likely contributes to the attenuation in the established plateau of the local heating response. Furthermore, inhibition of NOS during the local heating response would decrease the production of superoxide from uncoupled NOS. Superoxide has been shown to be a potent vasoconstrictor through activation of the Rho-kinase pathway in the cutaneous microcirculation (Bailey et al. 2004, 2005; Thompson-Torgerson et al. 2007). Thus, inhibition of uncoupled NOS would functionally manifest itself in a release of vasoconstriction or augmented vasodilatation as observed in the post-l-NAME (NOS inhibited) plateau at the control site in the hypercholesterolaemic group (Fig. 2).
Data from the present study examining arginase/NOS uncoupling mechanisms in human subjects is in agreement with the vascular data obtained in various atherosclerotic animal and cell culture models (Ryoo et al. 2006, 2010; Lim et al. 2007). These studies indicate that arginase activity is upregulated by a post-translational mechanism likely to be associated with targeting the activated enzyme to a subcellular location where it can interact with NOS. Moreover, mitochondrial arginase II appears to be the specific isoform upregulated with oxidized LDL-associated endothelial dysfunction (Lim et al. 2007). Ryoo et al. described a novel mechanism for arginase II activation occurring in response to OxLDL activation of the lectin-like OxLDL (LOX-1) receptor. Specifically, activation of LOX-1 induced RhoA Rho-kinase-dependent microtubule depolymerization causing a dissociation of arginase II from the mitochondria to the cytosol where it limits NO synthesis through NOS3 (Ryoo et al. 2006, 2010). In addition to rapid activation there is a concomitant Ox-LDL-mediated increase in arginase II transcription. In the present study both arginase I and II protein content as well as total arginase activity were increased in the samples from hypercholesterolaemic human subjects. Functionally, we could not determine the specific arginase isoform mediating the reduction in NO bioavailability due to the lack of isoform specific inhibitors (Cederbaum et al. 2004; Durante et al. 2007).
In contrast to the atoravasatin-mediated reduction in arginase activity, the intervention did not decrease arginase express in the hyperocholesterolaemic group. This finding further supports that the reduction in arginase activity in the hypercholesteromics may be via a pleiotropic effect and not from the reduction in ox-LDL. This result is consistent with mechanistic data from animal models where the decrease in arginase activity with atorvastatin is through stabilization of the cytoskeleton and likely to be through inhibition of RhoA and Rho-kinase signalling (Ryoo et al. 2010). Furthermore the inhibition of Rho enhances NOS3 phosphoryation and increases NOS3 mRNA stability thereby increasing NO bioavailabilty (Rikitake & Liao, 2005).
In addition to examining the role of arginase in regulating NOS substrate availability in hypercholesterolaemia-induced microvascular dysfunction, we also measured endogenous NOS inhibitors in the plasma. We found no difference in plasma ADMA concentrations between the normocholesterolaemic and hypercholesterolaemic groups. Further, there was no effect of the atorvastatin intervention on plasma ADMA concentrations. These data suggest that hyperchosterolaemia-induced endothelial dysfunction is not mediated by an increase in endogenous NOS inhibitors. NOS3 protein data obtained from the skin samples indicate that there was no difference between the study populations or with the atorvastatin intervention. However, we did not measure NOS phosphorylation or examine dimerized NOS3 as an indicator of NOS uncoupling due to the limited amount of tissue and protein obtained from the skin samples. Additionally, plasma ADMA concentrations may not be reflective of the concentrations of endogenous NOS inhibitors in the endothelial cell microenvironment.
The population examined in the study was a pure hypercholesterolaemic group in the absence of other cardiovascular disease risk factors. Our data demonstrate that microvascular dysfunction is clearly evident in this preclinical population and involves the upregulation of arginase. There are likely to be other mechanisms that contribute to hyperchosterolaemically induced cutaneous microvascular dysfunction including globalized increases in oxidant stress and decreases in critical NOS3 cofactor availability resulting in NOS3 uncoupling. Additional work examining these mechanisms in a more severely atherosclerotic population is necessary. However, these initial translational findings provide a basis for further study.
Limitations
We set out to examine the role of arginase in hypercholesterolaemia-induced cutaneous microvascular dysfunction using both in vivo functional assessments of NO-dependent vasodilatation and an in vitro analysis of skin samples obtained from the same human subjects. Our in vivo and in vitro findings with regard to arginase mechanisms are in agreement with one another and the existing animal literature (Ryoo et al. 2006, 2010; Lim et al. 2007). However, there are limitations with our NOS3 biochemical data because we were not able to successfully measure NOS3 activity in the limited number of skin samples obtained from the subjects. One reason for our inability to accurately measure NOS3 activity in human skin tissue is that there are several endogenous enzyme-independent stores of NO in human skin which increase with ultraviolet light exposure (Mowbray et al. 2009). Therefore, there was a high background concentration and we were unable to detect changes with the limited amount of protein obtained from the skin sample. While it would have been ideal to obtain an indicator of NOS3 activity it was not possible with the small amount of skin tissue.
After finding that NO-dependent vasodilatation was attenuated in the hypercholesterolaemic subjects we planned the statin intervention to determine its effects on functional NO-dependent vasodilatation and in vitro arginase activity. We only performed this intervention in the hypercholesterolaemic group and did not have a placebo control. Ideally we would have also performed the intervention in the normocholesterolaemic group to truly determine if the decrease in arginase activity was a pleiotropic effect of the statin. However, we did not observe functional decrements in NO-dependent vasodilatation in this normocholesterolaemic group and it is unlikely that the statin intervention would have produced any further improvements but it may have decreased arginase activity in the skin biopsy samples.
In summary, hyperchosterolaemia-induced microvascular dysfunction including a reduction in NO-dependent vasodilatation is clearly evident in the human cutaneous circulation. Local pharmacological inhibition of arginase or l-arginine supplementation augments cutaneous NO-dependent vasodilatation. Furthermore, in vitro data indicate that arginase activity is upregulated in hypercholesterolaemic human subjects and can be decreased with 12 weeks of oral atorvastatin therapy, which coincides with a restoration of cutaneous NO-dependent vasodilatory function. Together, these data indicate that arginase is a novel molecular target in the treatment of hypercholesterolaemia and that one of the pleiotropic effects of atorvastatin may be to decrease arginase activity likely through a post-translational mechanism.
Acknowledgments
The authors would like to thank Jane Pierzga for her technical assistance and help with data collection. We would also like to thank James A. Lang, John Jennings, Rebecca Bruning and Anna Stanhewicz for assistance with data collection and Caroline Smith for her editorial assistance with manuscript preparation. This work was supported by NIH R01-HL-089302 and M01-RR-10732.
Glossary
Abbreviations
- ADMA
asymmetrical dimethyl l-arginine
- CVC
cutaneous vascular conductance
- CVCmax
maximum cutaneous vascular conductance
- HC
hyperocholesterolaemic
- LDL
low-density lipoprotein
- NC
normocholes-terolaemic
- NOS
nitric oxide synthase
- oxLDL
oxidized low density lipoprotein
Author contributions
LAH & LS data collection, analysis, interpretation and manuscript preparation; data collection; AW data analysis; DEB & WLK data interpretation and manuscript preparation. In vito analysis of skin samples took place at JHU. In vivo studies took place at PSU. All authors approved the final version of the manuscript.
Disclosures
D. E. Berkowitz is a scientific founder and consultant for Arginetix Inc., a biotechnology company dedicated to the development of therapeutics targeting arginase in diseases in which endothelial dysfunction is an important contributing factor. The remaining authors have nothing to disclose or conflicts of interest to report.
References
- Abularrage CJ, Sidawy AN, Aidinian G, Singh N, Weiswasser JM, Arora S. Evaluation of the microcirculation in vascular disease. J Vasc Surg. 2005;42:574–581. doi: 10.1016/j.jvs.2005.05.019. [DOI] [PubMed] [Google Scholar]
- Ash DE. Structure and function of arginases. J Nutr. 2004;134:2760S–2764S. doi: 10.1093/jn/134.10.2760S. discussion 2765S–2767S. [DOI] [PubMed] [Google Scholar]
- Bailey SR, Eid AH, Mitra S, Flavahan S, Flavahan NA. Rho kinase mediates cold-induced constriction of cutaneous arteries: role of α2C-adrenoceptor translocation. Circ Res. 2004;94:1367–1374. doi: 10.1161/01.RES.0000128407.45014.58. [DOI] [PubMed] [Google Scholar]
- Bailey SR, Mitra S, Flavahan S, Flavahan NA. Reactive oxygen species from smooth muscle mitochondria initiate cold-induced constriction of cutaneous arteries. Am J Physiol Heart Circ Physiol. 2005;289:H243–250. doi: 10.1152/ajpheart.01305.2004. [DOI] [PubMed] [Google Scholar]
- Berkowitz DE, White R, Li D, Minhas KM, Cernetich A, Kim S, Burke S, Shoukas AA, Nyhan D, Champion HC, Hare JM. Arginase reciprocally regulates nitric oxide synthase activity and contributes to endothelial dysfunction in aging blood vessels. Circulation. 2003;108:2000–2006. doi: 10.1161/01.CIR.0000092948.04444.C7. [DOI] [PubMed] [Google Scholar]
- Binggeli C, Spieker LE, Corti R, Sudano I, Stojanovic V, Hayoz D, Luscher TF, Noll G. Statins enhance postischemic hyperemia in the skin circulation of hypercholesterolemic patients: a monitoring test of endothelial dysfunction for clinical practice? J Am Coll Cardiol. 2003;42:71–77. doi: 10.1016/s0735-1097(03)00505-9. [DOI] [PubMed] [Google Scholar]
- Cederbaum SD, Yu H, Grody WW, Kern RM, Yoo P, Iyer RK. Arginases I and II: do their functions overlap? Mol Genet Metab. 2004;81(Suppl 1):S38–44. doi: 10.1016/j.ymgme.2003.10.012. [DOI] [PubMed] [Google Scholar]
- Cracowski JL, Minson CT, Salvat-Melis M, Halliwill JR. Methodological issues in the assessment of skin microvascular endothelial function in humans. Trends Pharmacol Sci. 2006;27:503–508. doi: 10.1016/j.tips.2006.07.008. [DOI] [PubMed] [Google Scholar]
- Debbabi H, Bonnin P, Ducluzeau PH, Leftheriotis G, Levy BI. Noninvasive assessment of endothelial function in the skin microcirculation. Am J Hypertens. 2010;23:541–546. doi: 10.1038/ajh.2010.10. [DOI] [PubMed] [Google Scholar]
- Demougeot C, Prigent-Tessier A, Bagnost T, Andre C, Guillaume Y, Bouhaddi M, Marie C, Berthelot A. Time course of vascular arginase expression and activity in spontaneously hypertensive rats. Life Sciences. 2006;80:1128–1134. doi: 10.1016/j.lfs.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Durante W, Johnson FK, Johnson RA. Arginase: a critical regulator of nitric oxide synthesis and vascular function. Clin Exp Pharmacol Physiol. 2007;34:906–911. doi: 10.1111/j.1440-1681.2007.04638.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holowatz LA. Human cutaneous microvascular ageing: potential insights into underlying physiological mechanisms of endothelial function and dysfunction. J Physiol. 2008;586:3301. doi: 10.1113/jphysiol.2008.157594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holowatz LA, Kenney WL. Up-regulation of arginase activity contributes to attenuated reflex cutaneous vasodilatation in hypertensive humans. J Physiol. 2007;581:863–872. doi: 10.1113/jphysiol.2007.128959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holowatz LA, Thompson CS, Kenney WL. L-Arginine supplementation or arginase inhibition augments reflex cutaneous vasodilatation in aged human skin. J Physiol. 2006;574:573–581. doi: 10.1113/jphysiol.2006.108993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holowatz LA, Thompson CS, Minson CT, Kenney WL. Mechanisms of acetylcholine-mediated vasodilatation in young and aged human skin. J Physiol. 2005;563:965–973. doi: 10.1113/jphysiol.2004.080952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue T, Uchida T, Kamishirado H, Takayanagi K, Morooka S. Antibody against oxidized low density lipoprotein may predict progression or regression of atherosclerotic coronary artery disease. J Am Coll Cardiol. 2001;37:1871–1876. doi: 10.1016/s0735-1097(01)01228-1. [DOI] [PubMed] [Google Scholar]
- Johnson JM, O'Leary DS, Taylor WF, Kosiba W. Effect of local warming on forearm reactive hyperaemia. Clin Physiol. 1986;6:337–346. doi: 10.1111/j.1475-097x.1986.tb00239.x. [DOI] [PubMed] [Google Scholar]
- Kellogg DL, Jr, Zhao JL, Wu Y. Endothelial nitric oxide synthase control mechanisms in the cutaneous vasculature of humans in vivo. Am J Physiol Heart Circ Physiol. 2008;295:H123–129. doi: 10.1152/ajpheart.00082.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim HK, Lim HK, Ryoo S, Benjo A, Schuleri K, Miriel VA, Baraban E, Camara A, Soucy KG, Nyhan D, Shoukas A, Berkowitz D. Mitochondrial arginase II constrains endothelial NOS-3 activity. Am J Physiol Heart Circ Physiol. 2007;293:H3317–H3324. doi: 10.1152/ajpheart.00700.2007. [DOI] [PubMed] [Google Scholar]
- Minson CT, Berry LT, Joyner MJ. Nitric oxide and neurally mediated regulation of skin blood flow during local heating. J Appl Physiol. 2001;91:1619–1626. doi: 10.1152/jappl.2001.91.4.1619. [DOI] [PubMed] [Google Scholar]
- Minson CT, Holowatz LA, Wong BJ, Kenney WL, Wilkins BW. Decreased nitric oxide- and axon reflex-mediated cutaneous vasodilation with age during local heating. J Appl Physiol. 2002;93:1644–1649. doi: 10.1152/japplphysiol.00229.2002. [DOI] [PubMed] [Google Scholar]
- Mowbray M, McLintock S, Weerakoon R, Lomatschinsky N, Jones S, Rossi AG, Weller RB. Enzyme-independent NO stores in human skin: quantification and influence of UV radiation. J Invest Dermatol. 2009;129:834–842. doi: 10.1038/jid.2008.296. [DOI] [PubMed] [Google Scholar]
- Rikitake Y, Liao JK. Rho GTPases, statins, and nitric oxide. Circ Res. 2005;97:1232–1235. doi: 10.1161/01.RES.0000196564.18314.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi M, Carpi A, Di Maria C, Franzoni F, Galetta F, Santoro G. Skin blood flowmotion and microvascular reactivity investigation in hypercholesterolemic patients without clinically manifest arterial diseases. Physiol Res. 2009;58:39–47. doi: 10.33549/physiolres.931351. [DOI] [PubMed] [Google Scholar]
- Ryoo S, Bhunia A, Chang F, Shoukas A, Berkowitz DE, Romer LH. OxLDL-dependent activation of arginase II is dependent on the LOX-1 receptor and downstream RhoA signaling. Atherosclerosis. 2010;214:279–287. doi: 10.1016/j.atherosclerosis.2010.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryoo S, Lemmon CA, Soucy KG, Gupta S, White AR, Nyhan D, Shoukas AA, Romer LH, Berkowitz DE. Oxidized low-density lipoprotein-dependent endothelial arginase II activation contributes to impaired NO signaling. Circ Res. 2006;99:951–960. doi: 10.1161/01.RES.0000247034.24662.b4. [DOI] [PubMed] [Google Scholar]
- Stulc T, Kasalova Z, Prazny M, Vrablik M, Skrha J, Ceska R. Microvascular reactivity in patients with hypercholesterolemia: effect of lipid lowering treatment. Physiol Res. 2003;52:439–445. [PubMed] [Google Scholar]
- Thompson-Torgerson CS, Holowatz LA, Flavahan NA, Kenney WL. Rho kinase-mediated local cold-induced cutaneous vasoconstriction is augmented in aged human skin. Am J Physiol Heart Circ Physiol. 2007;293:H30–36. doi: 10.1152/ajpheart.00152.2007. [DOI] [PubMed] [Google Scholar]
- Toshima S, Hasegawa A, Kurabayashi M, Itabe H, Takano T, Sugano J, Shimamura K, Kimura J, Michishita I, Suzuki T, Nagai R. Circulating oxidized low density lipoprotein levels. A biochemical risk marker for coronary heart disease. Arterioscler Thromb Vasc Biol. 2000;20:2243–2247. doi: 10.1161/01.atv.20.10.2243. [DOI] [PubMed] [Google Scholar]
- Vasankari T, Ahotupa M, Toikka J, Mikkola J, Irjala K, Pasanen P, Neuvonen K, Raitakari O, Viikari J. Oxidized LDL and thickness of carotid intima-media are associated with coronary atherosclerosis in middle-aged men: lower levels of oxidized LDL with statin therapy. Atherosclerosis. 2001;155:403–412. doi: 10.1016/s0021-9150(00)00573-6. [DOI] [PubMed] [Google Scholar]