

Abstract
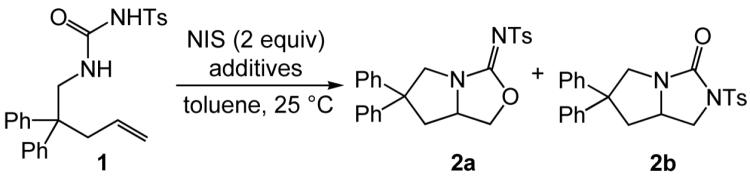
Reaction of N-δ-alkenyl-N’-sulfonyl urea 1 with N-iodosuccinimde (NIS; 2 equiv) and a catalytic amount of AgOTf (20 mol %) at room temperature led to intramolecular alkoxyamination to form bicyclic isourea 2a in 95% isolated yield. In comparison, reaction of 1 with NIS and sodium bicarbonate (1 equiv) at room temperature led to isolation of bicyclic imidazolidin-2-one 2b in 91% yield. These NIS-mediated alkoxyamination and diamination protocols were effective for a range of N-δ-alkenyl-N’-sulfonyl ureas to form the corresponding heterobicyclic compounds in good yield with high chemoselectivity and good to excellent diastereoselectivity.

1. Introduction
Vicinal diamines,1 vicinal amino alcohols,2 and related derivatives are component of a number of naturally occurring and biologically active molecules and find employment as synthetic intermediates, chiral ligands, and chiral auxiliaries. A particularly attractive route to the synthesis of vicinal diamines and amino alcohols is through the direct difunctionalization of alkenes. Early approaches to alkene difunctionalization by Backväll,3 Barluenga,4 Sharpless,5 and others6 employed stoichiometric amounts of a heavy metal complex or salt. The hydroxyamination of alkenes was subsequently rendered catalytic in osmium7 and eventually, both catalytic and enantioselective.8 There has been renewed interest in the direct difunctionalization of alkenes9 and recent efforts in this area have led to the development of effective processes for the copper-mediated diamination of alkenes with sulfamides,10,11 the Pd(II)-12,13 or Ni(II)-catalyzed14 diamination of alkenes with ureas, sulfamides, or guanidines in the presence of a stoichiometric oxidant, the palladium-catalyzed oxidative aminoacetoxylation of alkenes,15 and the Pd(0)- or Cu(I)-catalyzed diamination of alkenes with di-tert-butyldiaziridinone and related reagents.16
A pair of recent reports have demonstrated that the difunctionalization of alkenes with N-sulfonyl ureas can be realized with hypervalent iodine or iodonium reagents in the absence of transition metal catalysts. Michael has reported the intramolecular alkoxyamination of alkenes with N-sulfonyl ureas mediated by PhI=O in the presence of a strong Lewis or Brønsted acid.17 Similarly, Muñiz has reported the IPy2BF4-mediated diamination of alkenes with N-sulfonyl ureas to form bicyclic imidazolidin-2-one derivatives, although high reaction temperature (120 °C) was required to achieve optimal yield.18 In the course of our investigation of a potential gold(I)-catalyzed route to the difunctionalization of alkenes with N-sulfonyl ureas, we instead discovered a pair of complementary procedures for the selective, room temperature intramolecular diamination and alkoxyamination of alkenes with N-sulfonyl ureas mediated by N-iodosuccinimide (NIS). Herein we provide an account of our results in this area.
2. Results and discussion
As part of our ongoing interest in the development of gold(I)-catalyzed processes for the hydroamination of unactivated alkenes,19 we recently reported the room temperature intramolecular hydroamination of N-δ- and N-ε-alkenyl ureas catalyzed by a gold(I) N-heterocyclic carbene complex.20 We noted with interest recent reports that described the utilization of N-iodosuccinimide (NIS) in conjunction with gold(I)-catalysis for the iodofunctionalization of alkynes and allenes.21 On the basis of these precedents, we envisioned a gold(I)-catalyzed, NIS-mediated pathway for the oxidative difunctionalization of an N-δ-alkenyl urea involving electrophilic trapping of the initially formed gold alkyl species I with NIS to form the alkyl iodide II followed by intramolecular halide displacement by the pendant urea (Scheme 1).22
Scheme 1.

In apparent support of the pathway outlined in Scheme 1, reaction of 1-(2,2-diphenylpent-4-enyl)-3-tosylurea (1) with NIS (2 equiv) and a catalytic 1:1 mixture of (IPr)AuCl [IPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidine] and AgOTf (5 mol %) in toluene for 24 h at room temperature led to isolation of isourea 2a in 82% yield (Table 1, entry 1). However, subsequent control experiments revealed that silver, but not gold, was required for the efficient conversion of 1 to 2a. In the absence of metal catalyst, reaction of 1 with NIS (2 equiv) at room temperature was sluggish (~30% conversion in 24 h) and formed bicyclic imidazolidin-2-one 2b as the exclusive product (Table 1, entry 2). In contrast, treatment of 1 with NIS (2 equiv) and a catalytic amount of AgOTf (5 mol %) in toluene at room temperature for 14 h led to complete consumption of 1 with isolation of isourea 2a in 95% yield as the exclusive (>98% of the crude reaction mixture) product (Table 1, entry 3). Silver presumably facilitates the initial C–N formation through π-activation and facilitates C–O bond formation in preference to C–N bond formation in the second ring-closing step by enhancing the electrophilicity of an alkyl iodide intermediate analogous to II, leading to preferential attack by the hard oxygen nucleophile.23
Table 1.
Effect of metal and base additives on the reaction of 1 with NIS
 | ||||
|---|---|---|---|---|
| entry | additive | time (h) | 2a (%)a | 2b (%)a |
| 1 | (IPr)AuCl/AgOTf (5 mol %) | 24 | 82 | – |
| 2 | none | 24 | – | 30 |
| 3 | AgOTf (20 mol %) | 14 | 95 | – b |
| 4 | NaHCO3 (1 equiv) | 2 | – c | 91 |
Isolated yield of >95% purity.
Compound 2b constituted <5% of the crude reaction mixture.
Compound 2a constituted <5% of the crude reaction mixture.
The presence of a metal-free reaction pathway for the conversion of 1 to imidazolidin-2-one 2b was, in retrospect, not surprising as C–N bond formation in the electrophilic cyclization of unsaturated carboxamide derivatives has been demonstrated.24 One approach to the realization of selective C–N bond formation in these transformations is through employment of base in conjunction with an acidic carboxamide derivative such as an N-sulfonyl carboxamide.25 We therefore considered that employment of a base in the reaction of 1 with NIS might facilitate the C–N bond forming processes in the conversion of 1 to 2b. This was indeed found to be the case, and reaction of 1 with NIS in the presence of sodium bicarbonate (1 equiv) at room temperature for 2 h led to formation of a >95:5 mixture of 2b and 2a, from which 2b was isolated in 91% yield (Table 1, entry 4).
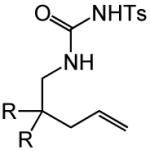
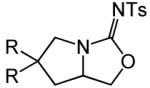
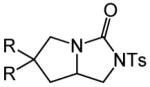
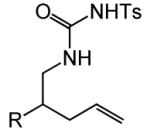
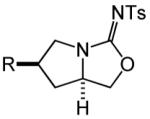
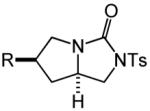
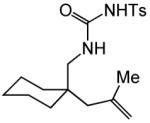
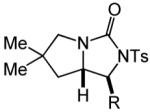
Having identified efficient routes to the selective conversion of 1 to either 2a or 2b, we probed the scope and generality of the NIS-mediated intramolecular alkoxyamination and diamination of alkenes with N-tosylureas (Table 2). N-δ-alkenyl ureas 3 and 4 that possessed gem-dialkyl substitution at the β-position of the alkenyl chain and the unsubstituted N-δ-alkenyl urea 5 underwent efficient intramolecular alkoxyamination and diamination to form the corresponding heterocycles 6-8 in >85% isolated yield (Table 3, entries 1-6). N-δ Alkenyl ureas 9-11 that possessed a single alkyl or phenyl group at the β-position of the alkenyl chain also underwent intramolecular alkoxyamination and diamination to form the corresponding heterocycles 12-14 in >70% isolated yield with up to 9:1 dr with predominant formation of the diastereomer possessing a trans arrangement of the exocyclic hydrocarbyl group and bridgehead hydrogen atom (Table 2, entries 7–12). N-δ-Alkenyl urea 15 that possessed a methyl substituent at the internal olefinic carbon atom underwent intramolecular alkoxyamination and diamination to form heterocycles 16 in good yield (Table 2, entries 13 and 14). N-δ-Alkenyl ureas 17 and 18 that possessed a methyl or phenyl substituent at the terminal olefinic carbon atom underwent intramolecular alkoxyamination to form heterocycles 19a and 20a, respectively in >70 % yield with selective formation of the diastereomer possessing a cis arrangement of the exocyclic alkyl group and bridgehead hydrogen atom (Table 2, entries 15 and 16). N-δ-Alkenyl ureas 17 and 18 also underwent NIS-mediated diamination to form bicyclic imidazolidin-2-ones 19b and 20b, respectively, albeit with diminished yield and/or diastereoselectivity relative to alkoxyamination (Table 2, entries 17 and 18). The N-ω-alkenyl urea 21 underwent efficient NIS-mediated alkoxyamination to form isourea 22a in 83% yield (Table 2, entry 19), but failed to undergo efficient diamination. In contrast, the N-allylaniline derived urea 23 underwent effective diamination to form imidazolidin-2-one 24b in 82% yield (Table 2, entry 20), but failed to undergo effective alkoxyamination.
Table 2.
Intramolecular alkoxyamination and diamination of γ- and δ-alkenyl ureas mediated by NIS (2 equiv) in toluene at 25 °C
| entry | substrate | additivec | time (h) | isourea product | yielda | urea product | yielda |
|---|---|---|---|---|---|---|---|
|
|
|
|||||
| 1 | R = Me (3) | A | 2 | 6a | 94% | ||
| 2 | R = š(CH2)5− (4) | A | 2 | 7a | 93% | ||
| 3 | R = H (5) | A | 3 | 8a | 86% | ||
| 4 | R = Me (3) | B | 1 | 6b | 97% | ||
| 5 | R = š(CH2)5− (4) | C | 1 | 7b | 85% | ||
| 6 | R = H (5) | B | 3 | 8b | 93% | ||
|
|
|
|||||
| 7 | R = Ph (9) | A | 2 | 12a | 72% (5:1) | ||
| 8 | R = i-Pr (10) | B | 3 | 13a | 94% (9:1) | ||
| 9 | R = Et (11) | A | 2 | 14a | 82% (4:1) | ||
| 10 | R = Ph (9) | B | 1 | 12b | 92% (8:1) | ||
| 11 | R = i-Pr (10) | A | 1 | 13b | 88% (9:1) | ||
| 12 | R = Et (11) | B | 2 | 14b | 82% (5:1) | ||
|
|
|
|||||
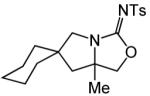
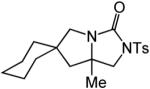
| 13 | 15 | A | 1 | 16a | 95% | ||
| 14 | B | 1 | 16b | 88% | |||
|
|
|
|||||
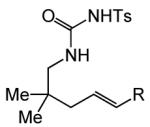
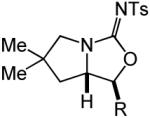
| 15 | R = Me (17) | A | 24 | 19a | 72% (>20:1) | ||
| 16 | R = Ph (18) | A | 3 | 20a | 83% (>20:1) | ||
| 17 | R = Me (17) | B | 2 | 19b | 84% (4.5:1) | ||
| 18 | R = Ph (18) | B | 3 | 20b | 69% (>20:1) | ||
| 19 |
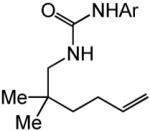
|
A | 24 |
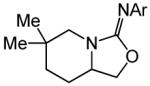
|
83% | ||
| 21(Ar=4-C6H4NO2) | 22a | ||||||
| 20 |
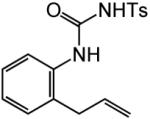
|
B | 16 |
|
82% | ||
| 23 | 24b |
Additives: A = AgOTf (20 mol %); B = NaHCO3 (1 equiv); C = NEt3 (1 equiv).
Isolated yields of >95% purity. Diastereomeric purity shown in parentheses.
3. Summary
We have developed an effective method for the intramolecular diamination of N-alkenyl ureas with NIS catalyzed by AgOTf to form bicyclic imidazolidin-2-ones and for the intramolecular alkoxyamination of N-alkenyl urea with NIS in the presence of a sodium bicarbonate or triethylamine. These processes are characterized by high chemoselectivity, good yield and in many cases, excellent diastereoselectivity.
4. Experimental
4.1. General Methods
Catalytic reactions were performed in sealed glass tubes under an atmosphere of dry nitrogen unless noted otherwise. NMR spectra were obtained on a Varian spectrometer operating at 400 MHz for 1H NMR and 100 MHz for 13C NMR in CDCl3 unless noted otherwise. IR spectra were obtained on a Bomen MB-100 FT IR spectrometer. Gas chromatography was performed on a Hewlett-Packard 5890 gas chromatograph equipped with a 25 m polydimethylsiloxane capillary column. Flash column chromatography was performed employing 200-400 mesh silica gel (EM). Thin layer chromatography (TLC) was performed on silica gel 60 F254. Elemental analyses were performed by Complete Analysis Laboratories (Parsippany, NJ). p-Toluenesulfonyl isocyanate (Acros) was used as received. Tosyl ureas 1,18 3 - 5,18 9,16 15,18 17,18 18,18 and 2311 and p-nitrophenylurea 2126 were synthesized employing published procedures.
4.2. Synthesis of N-Alkenyl Ureas
4.2.1. 1-(2-Isopropyl-4-pentenyl)-3-tosylurea (10)
p-Toluenesulfonyl isocyanate (0.21 mL, 1.4 mmol) was added to a solution of 2-isopropyl-4-pentenylamine11 (0.3 g, 1.4 mmol) in CH2Cl2 (10 mL) at 0 °C and the resulting mixture was stirred overnight at room temperature. The solvent was evaporated under vacuum and the resulting oily residue was chromatographed to give 10 in 87% yield. TLC (hexanes–EtOAc = 1:1): Rf = 0.4. 1H NMR: δ 9.57 (s, 1 H), 7.75 (d, J = 8.0 Hz, 2 H), 7.27 (d, J = 8.0 Hz, 2 H), 6.53 (t, J = 6.0 Hz, 1 H), 5.69 (m, 1 H), 5.03-4.98 (m, 2 H), 3.17 (m, 1 H), 2.41 (s, 3 H), 2.08 (m, 1 H), 1.84 (m, 1 H), 1.59 (m, 1 H), 1.38 (m, 1 H), 0.85 (d, J = 1.5 Hz, 3 H), 0.84 (d, J = 1.5 Hz, 3 H). 13C{1H} NMR: δ 152.4, 144.6, 136.95, 136.92, 129.8, 126.9, 116.5, 44.0, 41.1, 33.4, 28.3, 21.6, 19.5, 19.1. IR (neat, cm−1): 3343, 2958, 1656, 1553, 1464, 1346, 1163, 891, 664. Anal. calcd (found) for C27H28N2O: H, 7.46 (7.43); C, 59.23 (59.25).
4.2.2. 1-(2-Ethyl-4-pentenyl)-3-tosylurea (11)
Reaction of 2-ethyl-4-pentenylamine27 with p-toluenesulfonyl isocyanate employing a procedure similar to that used to synthesize 10 gave 11 in 82% yield. TLC (hexanes–EtOAc = 3:1): Rf = 0.7. 1H NMR: δ 9.54 (s, 1 H), 7.76 (d, J = 8.0 Hz, 2 H), 7.24 (d, J = 8.0 Hz, 2 H), 6.51 (t, J = 5.6 Hz, 1 H), 5.66 (m, 1 H), 4.98 (d, J = 5.2 Hz, 1 H), 4.95 (s, 1 H), 3.12 (t, J = 5.2 Hz, 1 H), 2.38 (s, 3 H), 1.94 (m, 2 H), 1.48 (m, 1 H), 1.20 (m, 2 H), 0.82 (t, J = 7.2 Hz, 3 H). 13C{1H} NMR: δ 152.4, 144.4, 136.7, 135.9, 129.6, 126.9, 116.5, 42.6, 39.1, 35.5, 23.7, 21.4, 10.8. IR (neat, cm−1): 3336, 3100, 1654, 1547, 1347, 1162, 1089, 893, 813, 664. Anal. calcd (found) for C27H28N2O: H, 7.14 (7.21); C, 58.04 (58.05).
4.3. Synthesis of bicyclic isoureas
4.3.1. Compound 2a17
A suspension of 1 (43 mg, 0.10 mmol), NIS (45 mg, 0.20 mmol) and AgOTf (1.3 mg, 4.7 × 10−3 mmol) in toluene (1.0 mL) was stirred for 14 h at room temperature. The crude reaction mixture was loaded directly onto a silica gel column and chromatographed (hexanes–EtOAc = 1:1) to give 2 (42 mg, 95%). TLC (hexanes–EtOAc = 1:1): Rf = 0.3. 1H NMR: δ 7.85 (d, J = 8.5 Hz, 2 H), 7.32-7.14 (m, 12 H), 4.70 (t, J = 9.0 Hz, 1 H), 4.35 (dd, J = 7.0, 9.0 Hz, 1 H), 4.31 (d, J = 11.0 Hz, 1 H), 4.16 (m, 1 H), 3.90 (d, J = 11.0 Hz, 1 H), 2.59 (dd, J = 5.0, 11.5 Hz, 1 H), 2.41 (dd, J = 10.0, 11.0 Hz, 1 H), 2.40 (s, 3 H). 13C{1H} NMR: δ 160.5, 144.9, 144.6, 142.5, 139.4, 129.2, 128.7, 128.6, 127.2, 127.1, 126.9, 126.7, 126.4, 73.3, 58.7, 58.2, 57.2, 43.1, 21.6.
Remaining bicyclic isoureas were synthesized employing procedures similar to that used to synthesize 2a. The 1H and 13C NMR spectra of known compounds 6a-8a,17 12a,18 19a,18 and 20a18 were identical to reported spectra.
4.3.2. Compound 13a
TLC (hexanes–EtOAc = 1:1): Rf = 0.2. 1H NMR: δ 7.77 (d, J = 8.4 Hz, 2 H), 7.17 (d, J = 8.4 Hz, 2 H), 4.58 (t, J = 8.0, 1 H), 4.26 (dd, J = 5.2, 9.2 Hz, 1 H), 4.01 (m, 1 H), 3.33 (dd, J = 8.8, 11.2 Hz, 1 H), 3.18 (dd, J = 8.8, 11.2 Hz, 1 H), 2.33 (s, 3 H), 2.12-2.02 (m, 2 H), 1.52-1.43 (m, 1 H), 1.12 (q, J = 10.8 Hz, 1 H), 0.82 (d, J = 6.8 Hz, 3 H). 13C{1H} NMR: δ 160.4, 142.4, 139.9, 129.1, 129.0, 127.03, 126.99, 72.9, 60.9, 60.8, 50.6, 49.4, 36.1, 32.4, 21.5, 21.2. IR (neat, cm−1): 2956, 1596, 1426, 1277, 1147, 857, 682614. Anal. calcd (found) for C20H22N4O5S: H, 6.88 (6.86); C, 59.60 (59.50).
4.3.3. Compound 14a
TLC (hexanes–EtOAc = 1:1): Rf = 0.3. 1H NMR: δ 7.79 (d, J = 8.0 Hz, 2 H), 7.20 (d, J = 8.0 Hz, 2 H), 4.62 (t, J = 9.2, 1 H), 4.28 (dd, J = 6.0, 9.2 Hz, 1 H), 4.06 (m, 1 H), 3.37 (dd, J = 8.8, 10.8 Hz, 1 H), 3.16 (dd, J = 8.8, 10.8 Hz, 1 H), 2.36 (s, 3 H), 2.30 (m, 1 H), 2.13 (m, 1 H), 1.38 (m, 2 H), 1.11 (q, J = 11.2 Hz, 1 H), 0.86 (t, J = 7.2 Hz, 3 H). 13C{1H} NMR: δ 160.5, 142.3, 139.8, 129.0, 126.9, 73.1, 60.6, 51.6, 43.4, 37.4, 26.8, 21.4, 12.5. IR (neat, cm−1): 2961, 1585, 1440, 1284, 1152, 1080, 859, 823, 666, 613. Anal. calcd (found) for C20H22N4O5S: H, 5.15 (4.96); C, 55.80 (55.67)
4.3.4. Compound 16a
TLC (hexanes–EtOAc = 1:1): Rf = 0.3. 1H NMR: δ 7.79 (d, J = 8.4 Hz, 2 H), 7.20 (d, J = 8.4 Hz, 2 H), 4.27 (dd, J = 8.8, 14.4 Hz, 2 H), 3.77 (d, J = 12.4 Hz, 1 H), 2.81 (d, J = 12.4 Hz, 1 H), 2.36 (s, 3 H), 1.67 (d, J = 12.0 Hz, 1 H), 1.58 (d, J = 13.2 Hz, 1 H), 1.47-1.27 (m, 8 H), 1.32 (s, 3 H), 1.15-1.03 (m, 2 H). 13C{1H} NMR: δ 161.5, 142.2, 139.8, 128.9, 126.9, 81.1, 66.2, 56.4, 50.2, 45.9, 37.5, 36.5, 26.8, 25.3, 23.8, 22.9, 21.5. IR (neat, cm−1): 2925, 1595, 1311, 1156, 816, 663. Anal. calcd (found) for C20H22N4O5S: H, 5.15 (4.96); C, 55.80 (55.67).
4.3.5. Compound 22a
TLC (hexanes–EtOAc = 3:1): Rf = 0.6. 1H NMR: δ 8.13 (d, J = 8.0 Hz, 2 H), 7.38 (d, J = 8.0 Hz, 2 H), 7.31-7.18 (m, 10 H), 4.90 (d, J = 14.0, 1 H), 4.56 (m, 1 H), 3.79 (m, 2 H), 3.05 (d, J = 14.0 Hz, 1 H), 2.75 (d, J = 13.6 Hz, 1 H), 2.37 (dt, J = 2.8, 13.6 Hz, 1 H), 1.91 (dd, J = 2.8, 13.2 Hz, 1 H), 1.25 (m, 1 H). 13C{1H} NMR: δ 155.0, 152.6, 146.5, 143.8, 142.0, 128.6, 128.5, 127.7, 126.7, 126.5, 126.3, 124.7, 123.8, 71.5, 55.1, 50.0, 46.0, 33.8, 26.1. IR (neat, cm−1): 1648, 1567, 1495, 1316, 1265, 1002, 859, 697. HRMS calcd (found) for C25H23N3O3 (M+): 413.1739 (413.1756).
4.4. Syntheses of bicyclic ureas
4.4.1. Synthesis of compound 2b
A suspension of 1 (43 mg, 0.10 mmol), NIS (45 mg, 0.20 mmol), and NaHCO3 (8.4 mg, 0.10 mmol) in toluene (1.0 mL) was stirred at room temperature for 2 h. The crude reaction mixture was loaded directly onto a silica gel column and chromatographed (SiO2; hexanes–EtOAc = 3:1) to give 2b (40 mg, 91%). TLC (hexanes–EtOAc = 3:1): Rf = 0.4. 1H NMR: δ 7.92 (d, J = 8.0 Hz, 2 H), 7.34 (d, J = 8.0 Hz, 2 H), 7.31-7.12 (m, 10 H), 4.09 (d, J = 7.0 Hz, 1 H), 4.07 (d, J = 9.5 Hz, 1 H), 3.93 (m, 1 H), 3.88 (d, J = 11.5 Hz, 1 H), 3.65 (dd, J = 5.5, 9.5 Hz, 1 H), 2.57 (dd, J = 5.0, 11.5 Hz, 1 H), 2.47 (s, 3 H), 2.26 (dd, J = 9.5, 12.0 Hz, 1 H). 13C{1H} NMR: δ 156.3, 145.5, 145.3, 144.8, 135.0, 129.7, 128.6, 128.1, 126.8, 126.78, 126.5, 56.9, 56.2, 54.1, 48.0, 43.7, 21.7.
Remaining imidizolidin-2-ones were synthesized employing procedures similar to that used to synthesize 2b. The 1H and 13C NMR spectra of known compounds 6b-8b,18 16b,18 19b,17 20b,18 and 24b12 were identical to reported spectra.
4.4.2. Compound 12b
TLC (hexanes–EtOAc = 3:1): Rf = 0.3. 1H NMR: δ 7.92 (d, J = 10.0 Hz, 2 H), 7.32 (d, J = 10.0 Hz, 2 H), 7.26-7.17 (m, 3 H), 7.06 (d, J = 8.5 Hz, 1 H), 4.02 (dd, J = 10.0, 12.0 Hz, 1 H), 3.90-3.81 (m, 2 H), 3.53-3.44 (m, 3 H), 2.42 (s, 3 H), 2.34 (m, 1 H), 1.47 (dd, J = 13.0, 28.5 Hz, 1 H). 13C{1H} NMR: δ 156.4, 144.9, 141.0, 135.1, 129.7, 128.7, 128.0, 127.0, 126.9, 56.4, 52.0, 46.9, 45.5, 40.1, 21.7. IR (neat, cm−1): 2963, 1725, 1359, 1166, 1091, 759, 702, 662. Anal. calcd (found) for C20H22N4O5S: H, 5.15 (4.96); C, 55.80 (55.67).
4.4.3. Compound 13b
TLC (hexanes–EtOAc = 1:1): Rf = 0.7. 1H NMR: δ 7.84 (d, J = 8.5 Hz, 2 H), 7.25 (d, J = 8.5 Hz, 2 H), 3.90 (dd, J = 9.0, 11.0 Hz, 1 H), 3.66 (m, 2 H), 3.10 (dd, J = 3.0, 8.5 Hz, 2 H), 2.36 (s, 3 H), 1.92 (m, 2 H), 1.36 (m, 1 H), 0.95 (dd, J = 11.5, 22.0 Hz, 1 H), 0.78 (d, J = 7.0 Hz, 3 H). 13C{1H} NMR: δ 156.2, 144.7, 135.1, 129.6, 127.9, 56.1, 49.3, 47.9, 47.1, 36.2, 32.5, 21.6, 21.2, 21.1. IR (neat, cm−1): 2964, 1726, 1387, 1356, 1168, 1095, 821, 662. Anal. calcd (found) for C13H15N3O3: H, 6.88 (6.92); C, 59.60 (59.51).
4.4.4. Compound 14b
TLC (hexanes–EtOAc = 3:1): Rf = 0.7. 1H NMR: δ 7.89 (d, J = 8.0 Hz, 2 H), 7.30 (d, J = 8.0 Hz, 2 H), 3.95 (dd, J = 8.5, 9.5 Hz, 1 H), 3.70 (m, 1 H), 3.18 (dd, J = 8.5, 11.5 Hz, 1 H), 3.08 (dd, J = 8.0, 11.5 Hz, 1 H), 2.41 (s, 3 H), 2.22-2.14 (m, 1 H), 2.11-2.06 (m, 1 H), 1.33 (m, 2 H), 0.98 (dd, J = 10.5, 22.0 Hz, 1 H), 0.84 (t, J = 8.0 Hz, 3 H). 13C{1H} NMR: δ 156.4, 144.8, 135.1, 129.7, 128.0, 55.9, 50.4, 47.3, 42.1, 37.6, 27.1, 21.7, 12.6. IR (neat, cm−1): 2962, 1726, 1358, 1165, 1092, 757, 662. Anal. calcd (found) for C20H22N4O5S: H, 5.15 (4.96); C, 55.80 (55.67).
4.5. Assignment of Product Relative Configuration
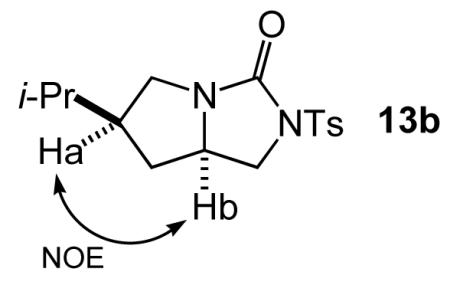
The relative configuration of 13b was established by the presence of a strong cross peak relating tertiary proton Ha to bridgehead proton Hb in the 1H-1H NOESY spectrum. The relative configurations of compounds 12b, 14b, and 12a-14a were assigned based on analogy to 13b. The relative configurations of compounds 19a, 19b, 20a, and 20b were assigned from the published spectra.17,18
Acknowledgments
Acknowledgment made to the NIH (GM-080422) for support of this research.
References and notes
- 1).Lucet D, Gall TL, Mioskowski C. Angew. Chem. 1998;110:2724. doi: 10.1002/(SICI)1521-3773(19981016)37:19<2580::AID-ANIE2580>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 1998;37:2580. [Google Scholar]
- 2).Bergmeier SC. Tetrahedron. 2000;56:2561. [Google Scholar]
- 3).Backväll J-E. Tetrahedron Lett. 1978;19:163. [Google Scholar]
- 4).(a) Aranda VG, Barluenga J, Aznar F. Synthesis. 1974:504. [Google Scholar]; (b) Barluenga J, Aznar F, de Mattos MCS, Kover WB, Garcia-Granda S, Pérez-Carreño E. J. Org. Chem. 1991;56:2930. [Google Scholar]
- 5).(a) Chong AO, Oshima K, Sharpless KB. J. Am. Chem. Soc. 1977;99:3420. [Google Scholar]; (b) Sharpless KB, Patrick DW, Truesdale LK, Biller SA. J. Am. Chem. Soc. 1975;97:2305. [Google Scholar]; (c) Patrick DW, Truesdale LK, Biller SA, Sharpless KB. J. Org. Chem. 1978;43:2628. [Google Scholar]
- 6).Becker PN, White MA, Bergman RG. J. Am. Chem. Soc. 1980;102:5676. [Google Scholar]
- 7).Sharpless KB, Chong AO, Oshima K. J. Org. Chem. 1976;41:177. [Google Scholar]; (b) Herranz E, Biller SA, Sharpless KB. J. Am. Chem. Soc. 1978;100:3596. [Google Scholar]; (c) Herranz E, Sharpless KB. J. Org. Chem. 1978;43:2544. [Google Scholar]
- 8).(a) Li G, Chang H-T, Sharpless KB. Angew. Chem., Int. Ed. Engl. 1996;35:451. [Google Scholar]; (b) Li G, Angert HH, Sharpless KB. Angew. Chem., Int. Ed. Engl. 1996;35:2813. [Google Scholar]; (c) Bodkin JA, McLeod MD. J. Chem. Soc., Perkin Trans. 1. 2002:2733. [Google Scholar]; (d) Donohoe TJ, Johnson PD, Pye R. J. Org. Biomol. Chem. 2003;1:2025. doi: 10.1039/b305189g. [DOI] [PubMed] [Google Scholar]
- 9).de Figueiredo RM. Angew. Chem. Int. Ed. 2009;48:1190. doi: 10.1002/anie.200804362. [DOI] [PubMed] [Google Scholar]
- 10).Zabawa TP, Kasi D, Chemler SR. J. Am. Chem. Soc. 2005;127:11250. doi: 10.1021/ja053335v. [DOI] [PubMed] [Google Scholar]
- 11).Zawaba TP, Chelmer SR. Org. Lett. 2007;9:2035. [Google Scholar]
- 12).Streuff J, Hövelmann CH, Nieger M, Muñiz K. J. Am. Chem. Soc. 2005;127:14586. doi: 10.1021/ja055190y. [DOI] [PubMed] [Google Scholar]
- 13).(a) Bar GLJ, Lloyd-Jones GC, Booker-Milburn KI. J. Am. Chem. Soc. 2005;127:7308. doi: 10.1021/ja051181d. [DOI] [PubMed] [Google Scholar]; (b) Muñiz K, Hövelmann CH, Streuff J. J. Am. Chem. Soc. 2008;130:763. doi: 10.1021/ja075041a. [DOI] [PubMed] [Google Scholar]; (c) Muñiz K, Streuff J, Chávez P, Hövelmann CH. Chem. Asian J. 2008;3:1248. doi: 10.1002/asia.200800148. [DOI] [PubMed] [Google Scholar]; (d) Muñiz K. J. Am. Chem. Soc. 2007;129:14542. doi: 10.1021/ja075655f. [DOI] [PubMed] [Google Scholar]; (e) Hövelmann CH, Streuff J, Brelot L, Muñiz K. Chem. Commun. 2008:2334. doi: 10.1039/b719479j. [DOI] [PubMed] [Google Scholar]
- 14).Muñiz K, Streuff J, Hövelmann CH, Núñez A. Angew. Chem. Int. Ed. 2007;46:7125. doi: 10.1002/anie.200702160. [DOI] [PubMed] [Google Scholar]
- 15).(a) Liu G, Stahl SS. J. Am. Chem. Soc. 2006;128:7179. doi: 10.1021/ja061706h. [DOI] [PubMed] [Google Scholar]; (b) Alexanian EJ, Sorensen EJ. J. Am. Chem. Soc. 2005;127:7690. doi: 10.1021/ja051406k. [DOI] [PubMed] [Google Scholar]; (c) Desai LV, Sanford MS. Angew. Chem., Int. Ed. 2007;46:5737. doi: 10.1002/anie.200701454. [DOI] [PubMed] [Google Scholar]
- 16).(a) Du H, Zhao B, Shi Y. J. Am. Chem. Soc. 2007;129:762. doi: 10.1021/ja0680562. [DOI] [PubMed] [Google Scholar]; (b) Du H, Yuan W, Zhao B, Shi Y. J. Am. Chem. Soc. 2007;129:11688. doi: 10.1021/ja074698t. [DOI] [PubMed] [Google Scholar]; (c) Xu L, Du H, Shi Y. J. Org. Chem. 2007;72:7038. doi: 10.1021/jo0709394. [DOI] [PubMed] [Google Scholar]; (d) Xu L, Shi Y. J. Org. Chem. 2008;73:749. doi: 10.1021/jo702167u. [DOI] [PubMed] [Google Scholar]; (e) Zhao B, Du H, Shi Y. Org. Lett. 2008;10:1087. doi: 10.1021/ol702974s. [DOI] [PubMed] [Google Scholar]
- 17).Cochran BM, Michael FE. Org. Lett. 2008;10:5039. doi: 10.1021/ol8022165. [DOI] [PubMed] [Google Scholar]
- 18).Muñiz K, Hövelmann CH, Campos-Gómez E, Barluenga J, González JM, Streuff J, Nieger M. Chem. Asian J. 2008;3:776–788. doi: 10.1002/asia.200700373. [DOI] [PubMed] [Google Scholar]
- 19).(a) Han X, Widenhoefer RA. Angew. Chem. Int. Ed. 2006;45:1747. doi: 10.1002/anie.200600052. [DOI] [PubMed] [Google Scholar]; (b) Bender CF, Widenhoefer RA. Chem. Commun. 2006:4143. doi: 10.1039/b608638a. [DOI] [PubMed] [Google Scholar]; (c) Bender CF, Widenhoefer RA. Chem. Commun. 2008:2741. doi: 10.1039/b804081h. [DOI] [PubMed] [Google Scholar]; (d) Zhang Z, Lee SD, Widenhoefer RA. J. Am. Chem. Soc. 2009;131:5372. doi: 10.1021/ja9001162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20).Bender CF, Widenhoefer RA. Org. Lett. 2006;8:5303. doi: 10.1021/ol062107i. [DOI] [PubMed] [Google Scholar]
- 21).(a) Buzas A, Gagosz F. Org. Lett. 2006;8:515. doi: 10.1021/ol053100o. [DOI] [PubMed] [Google Scholar]; (b) Buzas A, Istrate F, Gagosz F. Org. Lett. 2006;8:1957. doi: 10.1021/ol0606839. [DOI] [PubMed] [Google Scholar]; (c) Buzas A, Gagosz F. Synlett. 2006;17:2727. [Google Scholar]; (d) Crone B, Kirsch SF. J. Org. Chem. 2007;72:5435. doi: 10.1021/jo070695n. [DOI] [PubMed] [Google Scholar]; (e) Kirsch SF, Binder JT, Crone B, Duschek A, Haug TT, Liébert C, Menz H. Angew. Chem. Int. Ed. 2007;46:2310. doi: 10.1002/anie.200604544. [DOI] [PubMed] [Google Scholar]; (f) Yu M, Zhang G, Zhang L. Org. Lett. 2007;9:2147. doi: 10.1021/ol070637o. [DOI] [PubMed] [Google Scholar]; (g) Yu M, Zhang G, Zhang L. Tetrahedron. 2009;65:1846. [Google Scholar]; (h) Menz H, Binder JT, Crone B, Duschek A, Haug TT, Kirsch SF, Klahn P, Liébert C. Tetrahedron. 2009;65:1880. [Google Scholar]; (i) Buzas A, Istrate F, Gagosz F. Tetrahedron. 2009;65:1889. [Google Scholar]; (j) Poonoth M, Krause N. Adv. Synth. Catal. 2009;351:117. [Google Scholar]; (k) Gockel B, Krause N. Eur. J. Org. Chem. 2010:311. [Google Scholar]
- 22).Muñiz has recently demonstrated the validity of such an approach employing iodosobenzene diacetate as the stoichiometric oxidant: Iglesias A, Muñiz K. Chem. Eur. J. 2009;15:10563. doi: 10.1002/chem.200901199.
- 23).Rudakov ES, Kozhevnikov IV, Zamashchikov VV. Russ. Chem. Rev. 1974;43:305. [Google Scholar]
- 24).Robin S, Rousseau G. Tetrahedron. 1998;54:13681. [Google Scholar]
- 25).(a) Fujita M, Kitagawa O, Suzuki T, Taguchi T. J. Org. Chem. 1997;62:7330. doi: 10.1021/jo970898j. [DOI] [PubMed] [Google Scholar]; (b) Kitagawa O, Fujita M, Li M, Taguchi T. Tetrahedron Lett. 1997;38:615. [Google Scholar]; (c) Biloski AJ, Wood RD, Ganem B. J. Am. Chem. Soc. 1982;104:3233. [Google Scholar]; (d) Rajendra G, Miller MJ. Tetrahedron Lett. 1985;26:5385. [Google Scholar]; (e) Rajendra G, Miller MJ. J. Org. Chem. 1987;52:4471. [Google Scholar]; (f) Rajendra G, Miller MJ. Tetrahedron Lett. 1987;28:6257. [Google Scholar]; (f) Bertele E, Boos H, Dunitz JD, Elsinger F, Eschenmoser A, Felner I, Gribi HP, Gschwend H, Meyer EF, Pesaro M, Scheffold R. Angew. Chem. 1964;76:393. [Google Scholar]; (g) Boeckman RK, Connell BT. J. Am. Chem. Soc. 1995;117:12368. [Google Scholar]
- 26).Li H, Widenhoefer RA. Org. Lett. 2009;11:2671. doi: 10.1021/ol900730w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27).Keiji T, Akio T, Sakae U. J. Chem. Soc., Perkin Trans. 1. 1986;10:1837. [Google Scholar]