

Abstract
Nitroxide-mediated “living” radical polymerization with bisalkoxyamine 2,5,5,8,8,11-hexamethyl-4,9-(1-phenylethoxy)-3,10-diphenyl-4,9-diazadodecane produces polymers of controlled length and narrow molecular weight distributions at temperatures ranging from 70 to 110 °C. Polymerizations were run successfully with styrene (St), tert-butyl acrylate (tBA), and dimethylacrylamide (DMA). EPR measurements of the homolysis of this bisalkoxyamine and monoalkoxyamine 2,2,5-trimethyl-3-(1-phenylethoxy)-4-phenyl-3-azahexane at temperatures ranging from 85 to 105 °C give rate constants for the bisalkoxyamine that are approximately twice as large as those for the monoalkoxyamine. 1H NMR investigations of the decomposition of both alkoxyamines at 125 °C show enhanced decomposition for the bisalkoxyamine. EPR decomposition studies at 120 °C on the corresponding bis- and mononitroxides 2,5,5,8,8,11-hexamethyl-3,10-diphenyl-4,9-diazadodecane-4,9-bisnitroxide and 2,2,5-trimethyl-4-phenyl-3-azahexane-3-nitroxide also show accelerated bisnitroxide decomposition. Low-temperature EPR studies of the bisnitroxide reveal an unusually strong radical–radical interaction, suggesting enhanced stabilization of the intermediate mononitroxide formed during polymerization by interaction with the proximal N-alkoxyamine. The transient mononitroxide is postulated to be stabilized by delocalization of the unpaired electron density over five atoms.
Introduction
Controlled free radical polymerization techniques, such as nitroxide-mediated radical polymerization1–5 (NMRP), atom transfer radical polymerization,6–17 and reversible addition fragmentation transfer,18–24 provide the opportunity to prepare a variety of well-defined macromolecules with designed architectures.25 The radical nature of the intermediates makes these methods very chemoselective, allowing polymerization in the presence of many functionalities without the need for protecting groups. These “living” techniques provide control over the molecular weight of the resulting polymers and maintain low polydispersities.26–28 The use of unimolecular initiators allows for the preparation of a variety of morphologies including linear polymers, graft polymers, and star polymers.29 NMRP forms polymers devoid of metal contaminates. The use of α-hydrogen bearing nitroxides4,5,30–32 allows the formation of block copolymers with a variety of olefin monomers using a single nitroxide as the mediating agent. Despite the great promise of NMRP, most NMRP applications to date require relatively high temperatures: typically between 120 and 135 °C.1
In the past several years significant effort has gone into the development of nitroxide-based initiators that effect controlled polymerization at temperatures lower than the typical conditions of 120 °C. Jérôme et al.33,34 screened a series of nitrones for in situ nitroxide formation for the polymerization of styrene, styrene/acrylonitrile mixtures, and dienes at 110 °C. Additionally, they were successful with the polymerization of tert-butyl methacrylate in water at 80 °C.35 Catala et al.36,37 extensively studied the polymerization of styrene, ethyl acrylate, and n-butyl acrylate at temperatures as low as 90 °C using a stable β-sulfinyl nitroxide. Hawker et al.38 demonstrated that a nitroxide capable of intramolecular H-bonding39 is an efficient way to accelerate the polymerization of styrene and acrylate monomers. Studer et al.31,40–43 synthesized several structurally diverse N-alkoxyamines for use in NMRP with an emphasis on increasing the nitroxide steric bulk and examining the effect of cyclic nitroxide ring size. In some cases, control was achieved at temperatures as low at 70 °C. Charleux has effected polymerization of methyl methacrylate and methacrylic acid by NMRP at 60–90 °C with the addition of small amounts of styrene.44,45 Greci and Bertin have recently developed a new class of nitroxides for the polymerization of methyl methacrylate at 100 °C.46
Recently we reported bisnitroxide-based initiator 2,5,5,8,8,-11-hexamethyl-4,9-(1-phenylethoxy)-3,10-diphenyl-4,9-diaza-dodecane 1 (Figure 1) for use in NMRP.47 This bidirectional initiator effects polymerization in a well-controlled manner at 120 °C; there is a linear correlation between molecular weight and conversion and good control of polydispersity with a variety of monomers. The addition of a second B block is facile, resulting in symmetrical ABA triblock copolymers, with the B block inserted into the center of the macroinitiator. The design of this bisalkoxyamine was based on the parent monodirectional 2,2,5-trimethyl-4-phenyl-3-azahexane-3-nitroxide (TIPNO) initiator 2.4 Interestingly, although the structures of these two initiators are similar, it was observed that polymerizations run with bisalkoxyamine 1 reached significantly higher percent conversions than those run with initiator 2 under identical conditions. Investigations using a macromolecular monodirectional nitroxide initiator demonstrated that nitroxide size is not responsible for this enhanced rate.48 Herein is demonstrated the ability of 1 to carry out low-temperature polymerizations with a selection of monomers. To probe the reason that this initiator is effective at low temperatures, electron paramagnetic resonance (EPR) studies were performed to determine the dissociation rate constants (kd) and the activation energies (Ea) for the homolysis of 1 as compared to 2. In addition, the rates of decomposition of the alkoxyamines were examined by 1H NMR, and the rates of decomposition of the corresponding bis- and mononitroxides 2,5,5,8,8,11-hexamethyl-3,10-diphenyl-4,9-diazadodecane-4,9-bisnitroxide (3) and 2,2,5-trimethyl-4-phenyl-3-azahexane-3-nitroxide (4) were examined by high-temperature EPR. Interestingly, evaluation of the low-temperature EPR spectra of 3 at 130 K revealed a surprisingly large spin–spin interaction between the two nitroxide moieties. These results suggest a new mode of stabilization for the key intermediate mononitroxide.
Figure 1.

Bisalkoxyamine 1, parent TIPNO-based initiator 2, bisnitroxide 3, 2,2,5-trimethyl-4-phenyl-3-azahexane-3-nitroxide (TIPNO) 4, and 2,2,6,6-tetramethyl-1-(1-phenylethoxy)piperidine 5.
Experimental Section
General Materials and Methods
Styrene (St) (99%, Acros Organics), tert-butyl acrylate (tBA) (98%, Aldrich), n-butyl acrylate (nBA) (99+%, Acros Organics), and dimethylacrylamide (DMA) (99%, Aldrich) were distilled under vacuum immediately before use. 2,2,6,6-Tetramethylpiperidinoxy (TEMPO) (98%, Acros Organics), tert-butylbenzene (99%, Alfa Aesar), and p-xylene-d10 (Sigma Aldrich) were used as received. Polymerizations were carried out in sealed ampules. The synthesis and full characterization of 1 and 3 have been previously reported;47 2, 4, and 2,2,6,6-tetramethyl-1-(1-phenylethoxy)piperidine (5) are also reported.4,49 For EPR experiments, nitroxide 3 was purified by flash chromatography (90:10 hexanes/ethyl acetate), and compounds 1–4 were all synthesized within 48 h of use.
Analytical Techniques
NMR spectra were recorded at 500 MHz (Varian) as noted in CDCl3. Gel permeation chromatography (GPC) was performed using a Waters apparatus equipped with five Styragel columns (300 × 4.6 mm, 5 μm bead size), HR 0.5 (pore size: 50 Å, 0–1000 Da), HR 1 (pore size: 100 Å, 100–5000 Da), HR 2 (pore size: 500 Å, 500–20 000 Da), HR 4 (pore size: 10 000 Å, 50–100 000 Da), HR 5E (linear bed, mixed pore sizes, 2000–4 × 106 Da). Tetrahydrofuran (THF) was used as the eluent at a flow rate of 0.35 mL/min at ambient temperature. A refractive index detector was used and the molecular weights were calibrated against seven polystyrene standards ranging from 2000 to 156 000 Da.
EPR Experiments
EPR measurements were taken on a Bruker EMX spectrometer fitted with a variable temperature probe and SHQ cavity. All samples were run in capped EPR tubes in tert-butylbenzene. Freshly synthesized samples were prepared for all experiments. The microwave frequency was 9.44 GHz and power was 0.2 mW. The field was swept 75 G in 10.5 s with a time constant of 41 ms. The field was modulated at 100 kHz at an amplitude of 0.5 G.
For C–O bond homolysis experiments, EPR spectra of bisalkoxyamine 1 (0.601 mM of initiator; 1.20 mM of alkoxyamine functionality) and TIPNO-based initiator 2 (1.20 mM) were recorded every 15 s. Air was used as a radical scavenger.50 The evolution of nitroxide was monitored by calculating the second integral of the EPR signal at each time point. These values were converted to concentrations with the use of standardized calibration curves from freshly prepared solutions of TEMPO (0.603 and 0.308 mM). The experiments were repeated (including the standardized curves) with the temperature fixed at 358, 363, 368, 373, and 378 K. The initial slope51 of a plot of nitroxide concentration vs time was used to determine kd. The derivation of this initial slope method for a bisalkoxyamine is given in the Supporting Information.
For nitroxide decomposition experiments, EPR spectra of bisnitroxide 3 (2.02 mM of initiator; 4.04 mM of nitroxide functionality) were recorded every 30 s, and EPR spectra of mononitroxide 4 (4.03 mM) were recorded every 60 s. These experiments were run at 120 °C.
EPR spectra that were used to determine the interspin distance in 1 were recorded at 130 K (frozen solution). Microwave power was 0.2 mW for full field and 100 mW for half-field spectra. The field was swept 300 G in 83 s with a time constant of 327 ms. The field was modulated at 100 kHz at an amplitude of 1 G.
NMR Studies
Following the procedure of Cameron et al.,32 a solution of bisalkoxyamine initiator 1 in p-xylene-d10 (90 mM of initiator; 180 mM of alkoxyamine functionality) was placed in an NMR tube fitted with a Young’s tap. The tube was degassed with three freeze/pump/thaw cycles and closed under argon. The sealed tube was placed in the NMR cavity preheated to 125 °C. Spectra were recorded every 30 min for 16 h. The three vinyl resonances of styrene at δ 6.74, 5.77, and 5.26 ppm and the methine resonance of the alkoxyamine at approximately δ 4.9 ppm were integrated and used to calculate the relative amounts of styrene (S) and alkoxyamine (A). The same procedure was repeated using TIPNO-based initiator 2 and TEMPO-based initiator 5.
General Polymerization Procedure
A representative example is as follows: a mixture of N-alkoxyamine 1 (18.9 mg, 0.0291 mmol, 1.00 equiv), styrene (2.0 mL, 17.46 mmol, 600.0 equiv: 300 equiv relative to moles of N-alkoxyamine functionality), and 0.5 mL of DMF was degassed in an ampule by three consecutive freeze–pump–thaw cycles and sealed under argon. In addition, bisnitroxide 3 was used in polymerizations with tBA, nBA, and DMA (0.6 mg, 0.002 mmol, 0.05 equiv). The vial was heated to 70 °C until the solution became just slightly viscous, 7 days in this example. After cooling, a small aliquot was taken for a crude 1H NMR spectrum to calculate percent conversion based on integration of the remaining monomer vs polymer peaks. For styrene, the integration from 6.9 to 6.3 ppm (one dd representing 1H in the unreacted monomer and a broad peak representing 2H in the polymerized monomer) was compared to the integration from 5.9 to 5.2 ppm (two dd, representing 2H in the unreacted monomer). A conversion of 45% was observed in this case. The remainder of the polymer sample was dissolved in a minimum amount of CH2-Cl2 (unless otherwise noted below), and the specified ice-chilled precipitation solvent (6–10 mL) was added dropwise. The resulting precipitated polymer was separated by decantation, and volatiles were removed in vacuo. This precipitation procedure was repeated two times to give 800 mg of purified polymer which was analyzed by both 1H NMR and GPC (number-average molecular weight = Mn = 34 500 g/mol, polydispersity index (Mw/Mn) = 1.20, where Mw = weight-average molecular weight). Polymers were precipitated as follows: polystyrene (PS) with methanol, poly(dimethylacrylamide) (PDMA) with hexanes, poly(tert-butyl acrylate) (PtBA) was dissolved in THF and precipitated with 50% aqueous methanol, and poly(n-butyl acrylate) (PnBA) was dissolved in THF and precipitated with methanol.
Time Studies
For polymerization time studies, the desired amounts of initiator, monomer, and free nitroxide were combined to form a homogeneous solution. This mixture was divided evenly among several ampules, and each ampule was sealed under argon after three cycles of freeze–pump–thaw. The ampules were suspended simultaneously in a temperature-controlled oil bath and were removed after various time intervals. 1H NMR spectra of aliquots of the crude polymerization mixtures were taken, and integration of the peaks was used to determine percent conversion and [M]0/[M], the ratio of the initial monomer concentration to the final monomer concentration. After precipitating the polymers, GPC was used to determine the number-average molecular weight Mn,GPC and the polydispersity index, PDI.
ABA Triblock Copolymers
In a typical procedure, 50 mg (0.012 mmol) of the PtBA macroinitiator was combined with 0.500 g (4.80 mmol) of styrene in a 5 mL glass ampule. The ampule was sealed under argon after three cycles of freeze–pump–thaw. It was heated at 70 °C for 6 days. The resulting ABA triblock copolymer was dissolved in THF and precipitated with methanol (Mn = 22 400 g/mol, polydispersity index (Mw/Mn) = 1.29).
Sample Procedure for Thermal Decomposition
A PS sample (50.0 mg, 0.0025 mmol, Mn = 20 300 g/mol) was dissolved in 2 mL of p-xylene and heated to 130 °C for 16 h in a flask fitted with a septum with a needle open to air. The majority of the p-xylene was removed by vacuum distillation. GPC analysis of the concentrated sample gave Mn after cleavage = 11 200 g/mol.
Results and Discussion
Low-Temperature Polymerization Studies
Initial lower temperature studies were run with initiator 1 at 110 °C and 90 °C with styrene, tert-butyl acrylate, and n-butyl acrylate (Supporting Information). Polymerizations with acrylates and acrylamides were carried out with 5% added free binitroxide 3 in order to ensure a sufficient concentration of the persistent nitroxide radical during the first moments of polymerization.4 Polydispersities less than 1.5 and good correlation between the molecular weights calculated from 1H NMR integrations, Mn,NMR, and the experimental molecular weights measured by gel permeation chromatography, Mn,GPC, were observed for polymerization times of 3 h or longer. Following these successful results, polymerizations of styrene, tert-butyl acrylate, and dimethyl acrylamide were run at 70 °C. The results are shown in Table 1.
Table 1.
Bulk Polymerizations of Styrene (St), tert-Butyl Acrylate (tBA), and Dimethylacrylamide (DMA) with Bisalkoxyamine 1 (Figure 1) at 70 °C
| monomera | time (days) | monomer % convb | Mn,NMRc × 10−4 (g/mol) | Mn,GPCd × 10−4 (g/mol) | PDIe |
|---|---|---|---|---|---|
| St | 4 | 30 | 1.96 | 1.81 | 1.31 |
| St | 5 | 45 | 2.90 | 2.68 | 1.23 |
| St | 6 | 45 | 2.90 | 3.04 | 1.21 |
| St | 7 | 45 | 2.90 | 3.45 | 1.20 |
| St | 8 | 56 | 3.59 | 3.51 | 1.20 |
| St | 9 | 69 | 4.40 | 4.24 | 1.18 |
| St | 10 | 61 | 3.88 | 4.09 | 1.19 |
| St | 11 | 65 | 4.14 | 4.61 | 1.18 |
| tBAf | 4 | 10 | 0.86 | 1.15 | 1.54 |
| tBAf | 5 | 10 | 0.86 | 1.06 | 1.57 |
| tBAf | 6 | 11 | 0.94 | 1.22 | 1.58 |
| tBAf | 7 | 16 | 1.33 | 1.34 | 1.55 |
| tBAf | 8 | 17 | 1.38 | 1.52 | 1.58 |
| tBAf | 9 | 20 | 1.59 | 1.78 | 1.60 |
| tBAf | 10 | 17 | 1.37 | 1.97 | 1.55 |
| tBAf | 11 | 24 | 1.95 | 2.23 | 1.64 |
| DMAf | 4 | 41 | 2.50 | 1.69 | 1.44 |
| DMAf | 5 | 38 | 2.30 | 1.55 | 1.44 |
| DMAf | 6 | 47 | 2.84 | 2.15 | 1.42 |
| DMAf | 7 | 55 | 3.31 | 2.17 | 1.49 |
| DMAf | 8 | 57 | 3.46 | 2.04 | 1.52 |
| DMAf | 9 | 69 | 4.17 | 2.43 | 1.47 |
600 equiv used.
Percent conversion calculated by 1H NMR integration of crude polymer.
Molecular weight calculated from percent conversion determined by 1H NMR.
Number-average molecular weight measured by gel permeation chromatography (GPC) and calibrated with polystyrene standards.
Polydispersity index (Mw/Mn) from GPC.
5% of bisnitroxide 3 was added (St = styrene, tBA = tert-butyl acrylate, DMA = dimethylacrylamide).
These results at 70 °C provide evidence for “living” polymerizations. The polymerization of tert-butyl acrylate is a representative example. (Plots for the polymerization of all three monomers can be found in the Supporting Information.) A linear relationship exists between ln([M]0/[M]) and time (Figure 2), indicating that there are no significant termination events during polymerization. In addition, the molecular weights based on 1H NMR percent conversions and the experimental molecular weights obtained by GPC correlate well (Table 1 and Figure 3), suggesting an absence of chain transfer events. The polydispersity indices are very low for polystyrene (PDI = 1.18–1.30); however, somewhat higher polydispersity indices are evident for both tert-butyl acrylate (PDI = 1.54–1.64) and dimethylacrylamide (PDI = 1.42–1.52).
Figure 2.

Polymerization of tert-butyl acrylate with initiator 1 at 70°C. The ratio of the initial monomer concentration [M]0 to the monomer concentration [M] at some later time was calculated from 1H NMR integrations of the crude polymer samples.
Figure 3.

Evolution of Mn,GPC (number-average molecular weight determined by gel permeation chromatography and calibrated with polystyrene standards) and polydispersity index (Mw/Mn) as a function of Mn,NMR (molecular weight calculated from 1H NMR) for the polymerization of tert-butyl acrylate at 70 °C with bidirectional initiator 1.
A proof of “livingness” is the ability to add a second block. Thus, ABA triblock copolymers were prepared at 70 °C with bisalkoxyamine 1. GPC traces for one example are shown in Figure 4, where an internal B block of styrene was added to an A block macroinitiator of poly(tert-butyl acrylate) prepared at 70 °C. There is an increase in molecular weight and a decrease in the polydispersity index of the resultant ABA triblock copolymer.
Figure 4.
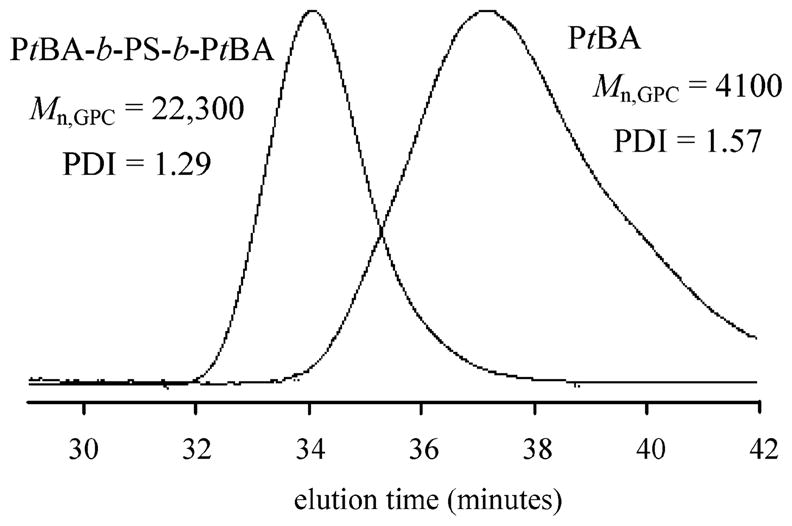
Addition of a polystyrene B block to a poly(tert-butyl acrylate) A block macroinitiator to give an ABA PtBA-b-PS-b-PtBA triblock copolymer.
As demonstrated in the initial report of initiator 1,47 monomer addition occurs at both ends of this symmetrical initiator. Bidirectional growth can be confirmed by several methods.52–58 Because the final polymer contains a central bisnitroxide, thermolytic cleavage of the alkoxyamine C–O bonds under conditions that result in nitroxide decomposition59 yield polymers of half the original molecular weight (Scheme 1). Presumably, the reaction occurs to form terminal olefins on the polymer chains and hydroxylamine functionalities on the bisnitroxide.51,54,60 Under these reaction conditions, the hydroxylamines most likely undergo oxidation to nitroxides and further decomposition.59 Thus, polystyrene and poly(tert-butyl acrylate) samples prepared at 90 and 70 °C were heated to 130 °C in p-xylene overnight to effect cleavage at the center. The results of these thermolytic cleavage reactions confirm symmetrical bidirectional growth (Table 2). An example GPC trace is shown in Figure 5.
Scheme 1.
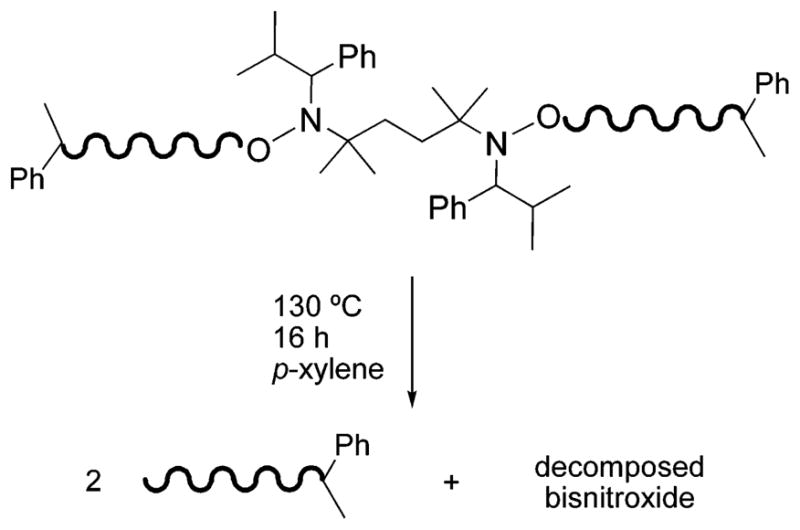
Thermolytic Cleavage of a Polymer Containing an Internal Bisnitroxide
Table 2.
Central Cleavagea of Polymers Prepared with Bisalkoxyamine Initiator 1
| monomer | original |
after cleavage |
||
|---|---|---|---|---|
| Mn,GPCb × 10−4 (g/mol) | PDIc | Mn,GPCb × 10−4 (g/mol) | PDId | |
| Ste | 2.03 | 1.16 | 1.12 | 1.17 |
| tBAe | 2.09 | 1.38 | 1.49 | 1.42 |
| Stf | 0.80 | 1.24 | 0.33 | 1.62 |
| tBAf | 0.78 | 1.43 | 0.52 | 1.63 |
Samples were heated as 1.2 mM solutions in p-xylene for 16 h.
Number-average molar mass measured by gel permeation chromatography (GPC) and calibrated with polystyrene standards.
Polydispersity index (Mw/Mn) from GPC before cleavage.
Polydispersity index (Mw/Mn) from GPC after cleavage.
Original polymers were prepared at 90 °C.
Original polymers were prepared at 70 °C. (St = styrene, tBA = tert-butyl acrylate).
Figure 5.
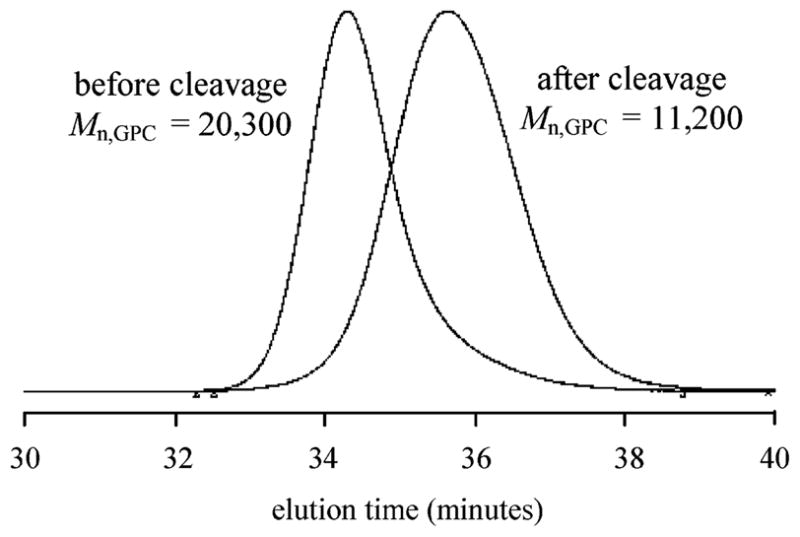
Cleavage of internal bisnitroxide polystyrene sample prepared at 90 °C by heating to 130 °C in p-xylene for 16 h. The resulting polymer is approximately half the original molecular weight as determined by gel permeation chromatography and calibrated with polystyrene standards (original sample: Mn = 20 300 g/mol; after cleavage: Mn = 11 200 g/mol).
As a control, polymerizations with the parent monodirectional TIPNO initiator 2 were also investigated at 70 °C with styrene, tert-butyl acrylate, n-butyl acrylate, and dimethylacrylamide. The polymerization of styrene was successful. However the other monomers either underwent uncontrolled polymerization or with 5% added bisnitroxide did not undergo polymerization at all (Table 3).
Table 3.
Bulk Polymerization with Parent TIPNO-Based Initiator 2 (Figure 1) at 70 °C
| monomera | time (days) | % convb | Mn,NMRc× 10−4 (g/mol) | Mn,GPCd × 10−4 (g/mol) | PDIe |
|---|---|---|---|---|---|
| St | 11 | 46 | 2.96 | 2.98 | 1.24 |
| tBA | 11 | 1 | 0.13 | 0.38 | 1.54 |
| tBAf | 11 | 0 | N/A | N/A | N/A |
| nBA | 11 | 92 | 7.11 | 8.53 | 2.02 |
| nBAf | 11 | 0 | N/A | N/A | N/A |
| DMA | 3 | 83 | 4.99 | 1.22 | 2.46 |
| DMAf | 3 | 68 | 4.10 | 1.85 | 2.16 |
600 equiv used.
Calculated by 1H NMR integration of crude polymers.
Theoretical molecular weight calculated from percent conversion determined by 1H NMR.
Number-average molecular weight measured by gel permeation chromatography (GPC) and calibrated with polystyrene standards.
Polydispersity index (Mw/Mn) from GPC.
5% free bisnitroxide 3 (Figure 1) added. (St = styrene, tBA = tert-butyl acrylate, nBA = n-butyl acrylate).
Dissociation Kinetics
The ability of bidirectional initiator 1 to effect polymerization at lower temperatures than the parent initiator 2 motivated a detailed investigation of the kinetics of bond homolysis. Following the method of Fischer et al.,50 the rate constants for the dissociation of bisalkoxyamine 1 and parent initiator 2 (Scheme 2) were compared at five different temperatures, ranging from 358 to 378 K. Freshly synthesized alkoxyamine initiators 1 and 2 were used in this EPR investigation in order to ensure the purity of the samples. Oxygen was used as a radical scavenger, and tert-butylbenzene was used as the solvent. Because bisalkoxyamine 1 contains twice as many nitroxide moieties as parent initiator 2, a 0.601 mM solution of 1 was used, compared to a 1.20 mM solution of 2. In this way, the concentration of alkoxyamine functionalities was 1.20 mM in both samples. The nitroxides which form during these homolysis experiments decompose at high temperatures;59 therefore, the stable nitroxide TEMPO was used as a calibration standard to convert the second integral of the EPR signal into nitroxide concentration.
Scheme 2.
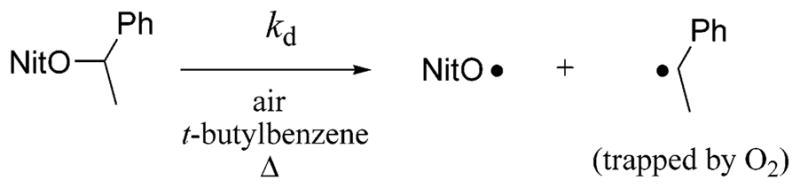
Alkoxyamine Dissociation Reaction Used To Determine kd, where NitO Represents a Nitroxide
As a typical example, Figure 6 shows the concentration of nitroxide vs time when samples of bisalkoxyamine 1 and parent alkoxyamine 2 were heated at 363 K for 1 h. The C–O bond homolysis of bisalkoxyamine 1 is significantly faster than that of parent alkoxyamine 2. Similar results were seen at 358, 368, 373, and 378 K (see Supporting Information).
Figure 6.
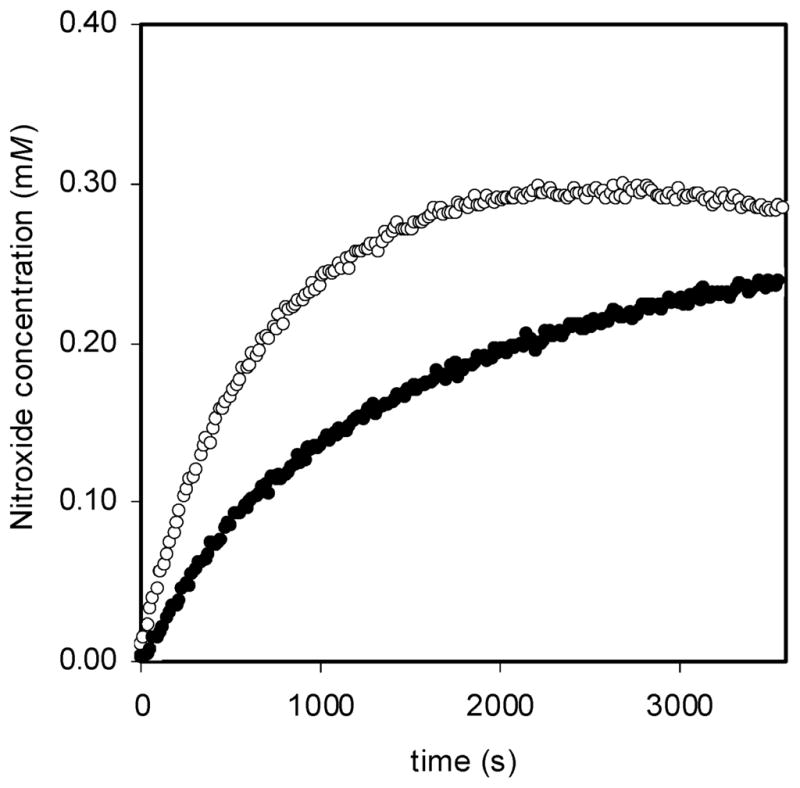
Nitroxide concentration measured by EPR as a function of time for the homolysis of bisalkoxyamine initiator 1 (○) and parent alkoxyamine initiator 2 (●) when heated at 363 K in tert-butylbenzene in the presence of oxygen.
Because the nitroxide that forms during this homolysis undergoes further decomposition, the value of the rate constant kd was calculated from the initial slope of the graph of nitroxide concentration vs time:
| (1) |
where [N] is the concentration of nitroxide functionality and [A]0 is the initial concentration of alkoxyamine functionality. The derivation of eq 1 for a bisalkoxyamine is given in the Supporting Information.
The calculated rate constants (kd), activation energies (Ea), and pre-exponential factors (A) of 1 and 2 are shown in Table 4 along with the literature values for the parent TIPNO initiator 2. The calculated kd values for 2 agree well with the literature values. The calculated activation energies and Arrhenius parameters are very sensitive to temperature. A difference of 1 K in the measured temperature can cause a difference of 5 kJ/mol in the calculated EA and a factor of 10 difference in A. This might explain the discrepancy between the calculated EA and A values for 2 and the literature values. Thus, caution must be used when comparing values measured by different researchers. As seen in Table 4, the dissociation rate constants for bisalkoxyamine 1 are about twice as large as those for parent alkoxyamine 2 in the temperature range studied. This increased rate of C–O bond homolysis contributes to the ability of bisalkoxyamine initiator 1 to mediate polymerizations at faster rates and lower temperatures than parent initiator 2. Fischer has shown through kinetic analysis that the polymerization time of NMRP decreases with increasing kd, and control is maintained as long as kd does not become too large (about 10−1 s−1).61 The improved polymerization capabilities of alkoxyamines with acyclic nitroxides such as TIPNO and SG1 have been attributed in part to their higher kd values (~10−3 s−1) compared to TEMPO-based alkoxyamines (~10−4 s−1).50 Using the measured values of Ea and A and extrapolating to 120 °C gives dissociation rate constants of 5.2 × 10−3 and 2.7 × 10−3 s−1 for alkoxyamines 1 and 2, respectively. The value for 1 is larger than many values found in the literature31,50 but not too large for the polymerization to lose control.
Table 4.
Kinetic Data for the Homolytic Dissociation of Bisalkoxyamine Initiator 1 Compared to Parent Initiator 2 in tert-Butylbenzene
| bisalkoxyamine 1 | monoalkoxyamine 2 | lit. value for 2 | |
|---|---|---|---|
| k358 (s−1) | 1.6 × 10−4 | 8.3 × 10−5 | 6.9 × 10−5a |
| k363 (s−1) | 3.3 × 10−4 | 1.6 × 10−4 | 1.3 × 10−4a |
| k368 (s−1) | 5.2 × 10−4 | 2.3 × 10−4 | 2.2 × 10−4a |
| k373 (s−1) | 9.2 × 10−4 | 4.8 × 10−4 | 4.0 × 10−4a |
| k378 (s−1) | 1.2 × 10−3 | 6.1 × 10−4 | 6.9 × 10−4a |
| Ea (kJ/mol) | 113 | 115 | 130 |
| A (s−1) | 5.3 × 1012 | 4.7 × 1012 | 5.6 × 1014 |
Calculated using reported values51 of activation energy (EA) and Arrhenius constant (A).
1H NMR Alkoxyamine Decomposition Studies
The relative rates of decomposition of bisalkoxyamine 1 and parent initiator 2 in the absence of monomer were determined by the method of Fukuda et al.62 In order to try to simulate the conditions during polymerization, each alkoxyamine was heated in the absence of molecular oxygen. Repeated homolysis and recombination ensues, with eventual decomposition to form styrene and hydroxylamine (Scheme 3).63
Scheme 3.
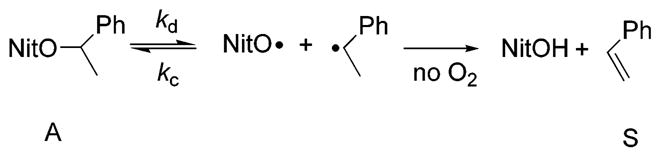
Decomposition of N-Alkoxyamine (A) by Extended Thermolysis To Give Hydroxylamine and Styrene (S), where NitO Represents a Nitroxide
The pseudo-first-order integrated rate law for this decomposition is given by eq 2:
| (2) |
As shown by Fukuda,62,64 the rate constant kdec may be expressed as
| (3) |
where pdec is the probability of decomposition. This equation accounts for the fact that faster rates of homolysis can lead to faster apparent rates of decomposition. Because the decomposition of alkoxyamines into hydroxylamines and alkenes can lead to incomplete monomer conversions and high polydisperisties,65 smaller pdec values are desirable in alkoxyamine initiators.
This reaction is conveniently monitored by 1H NMR spectroscopy by integration of the signals corresponding to the vinylic hydrogens of styrene and the alkoxyamine methine proton at δ 4.90 (CH3CHO) to give the relative amounts of styrene, S, and alkoxyamine, A. Because bisalkoxyamine 1 contains twice as many nitroxide moieties as parent initiator 2, a 90 mM solution of 1 was used, as compared to a 180 mM solution of 2. In this way, the concentration of alkoxyamine functionality was 180 mM in both samples.
Figure 7 shows the disappearance of the alkoxyamine functionality and the evolution of styrene for bisalkoxyamine 1 and parent alkoxyamine 2 at 125 °C. For both 1 and 2, the decrease in alkoxyamine concentration appears to occur more rapidly than the increase in styrene concentration. The total concentration of alkoxyamine and styrene, [A] + [S], does not remain constant. This may indicate that either (1) the alkoxyamine decomposes through an additional mechanism that does not result in the formation of styrene or (2) the styrene that forms undergoes an additional reaction, such as polymerization. No broad polymer peaks were detected in the region 6.2–7.4 ppm in the 1H NMR spectra, indicating that no significant polymerization occurs under these dilute conditions. Analogous decomposition studies using the TEMPO-derived alkoxyamine 5 showed a similar deficit in the mass balance (Supporting Information). However, this apparent loss of material was not reported by Fukuda, et al.62 or Fischer et al.,60 who also studied the decomposition of 5. The results in Figure 7 were reproducible, even at different alkoxyamine concentrations (Supporting Information) and extended T1 relaxation times; thus, the possibility of an alternate decomposition pathway cannot be ruled out.
Figure 7.
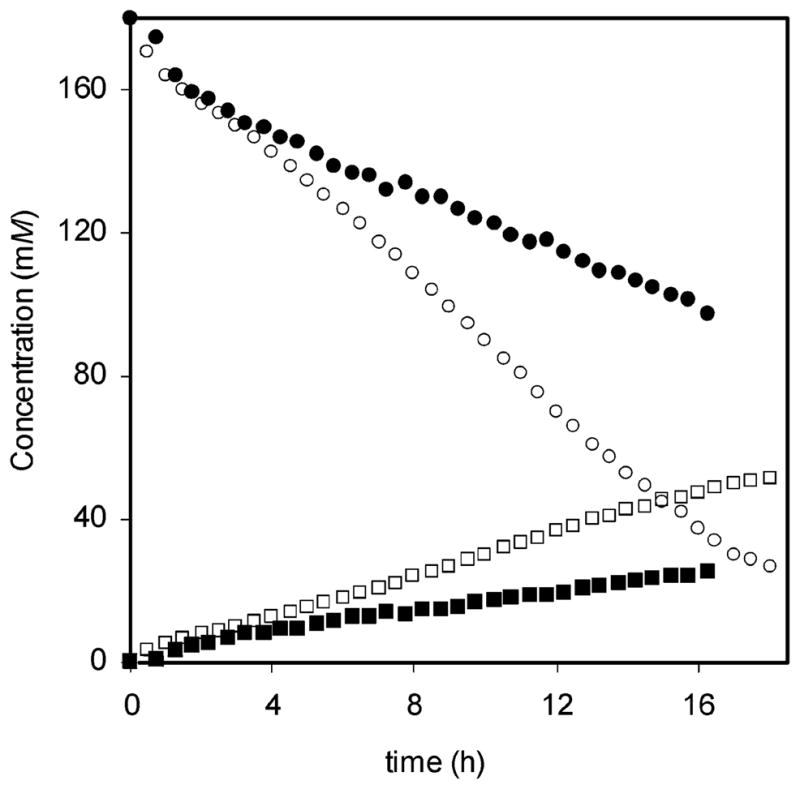
Alkoxyamine (A) and styrene (S) concentrations as a function of time when bisalkoxyamine 1 (○= [A]; □= [S]) and parent alkoxyamine 2 (●= [A]; ■= [S]) were heated at 125 °C in p-xylene-d10 in the absence of oxygen. Concentrations were calculated from 1H NMR integration values.
The kinetics of these decomposition reactions were further analyzed by plotting ln([A] + [S])/[A]) as a function of time (Figure 8, eq 2). If the decompositions are first-order or pseudo-first-order, this plot will be linear. This is the case for parent alkoxyamine 2 and for TEMPO-based initiator 5. Slopes of the best fit lines give kdec values of 3.7 × 10−6 and 1.6 × 10−5 s−1 for 2 and 5, respectively. The kdec value for 2 was the same at initial alkoxyamine concentrations of 90 and 180 mM. The value for 5 correlates well with the literature value of kdec = 4.5 × 10−5 s−1 at 140 °C determined by Fukuda and co-workers.62 However, as is evident in Figure 8, the relationship between ln([A] + [S])/[A]) and time for initiator 1 is not linear. This experiment with initiator 1 was repeated with extended T1 relaxation times (original T1 = 1 s) of 10 and 15 s with no significant change, indicating that the nonlinearity is not a result of increased molecular relaxation times resulting from the absence of molecular oxygen or the increase in temperature.66 The curvature in this plot suggests an alternate decomposition pathway for alkoxyamine 1 (or bisnitroxide 3), rather than the simple mechanism shown in Scheme 3.
Figure 8.
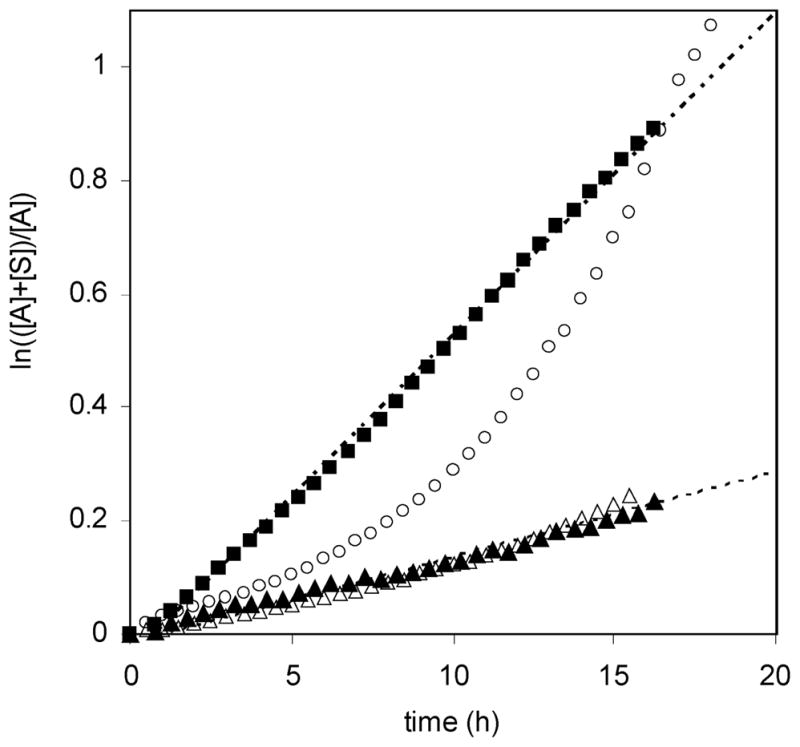
Plot of ln(([A] + [S])/[A]) as a function of time for bisalkoxyamine 1 (○, [A]0 = 180 mM), parent alkoxyamine 2 (▲, [A]0 = 180 mM; △, [A]0 = 90 mM), and TEMPO-based alkoxyamine 5 (■, [A]0 = 90 mM) upon heating at 125 °C in p-xylene-d10 in the absence of oxygen. The concentrations of alkoxyamine functionality [A] and styrene [S] were measured by 1H NMR integration values.
The rate of decomposition of alkoxyamine into hydroxylamine and styrene can be quantified by looking at the rate of formation of styrene, rather than the disappearance of alkoxyamine. This avoids the complication of alkoxyamine decomposition through a different mechanism. An alternative expression of the integrated rate law is
| (4) |
and a plot of this relationship for alkoxyamines 1 and 2 is shown in Figure 9. For both alkoxyamines 1 and 2, this relationship is linear, and slopes of the best-fit lines give kdec values of 5.2 × 10−6 and 2.8 × 10−6 s−1, respectively. This kdec value for 2 is close to the previous value found from eq 2 and Figure 8. Using the measured values of Ea and A, kd at 125 °C was calculated, and eq 3 was used to determine the probability of decomposition pdec (Table 5). These results show that bisalkoxyamine 1 is no more likely to decompose into hydroxylamine and styrene than parent alkoxyamine 2. Their estimated pdec values are both on the order of magnitude of 10−4. In contrast, TEMPO-based alkoxyamine 5 has a pdec value near 10−2, making it ~40 times more likely to decompose to styrene and hydroxylamine, as shown in Scheme 3.
Figure 9.
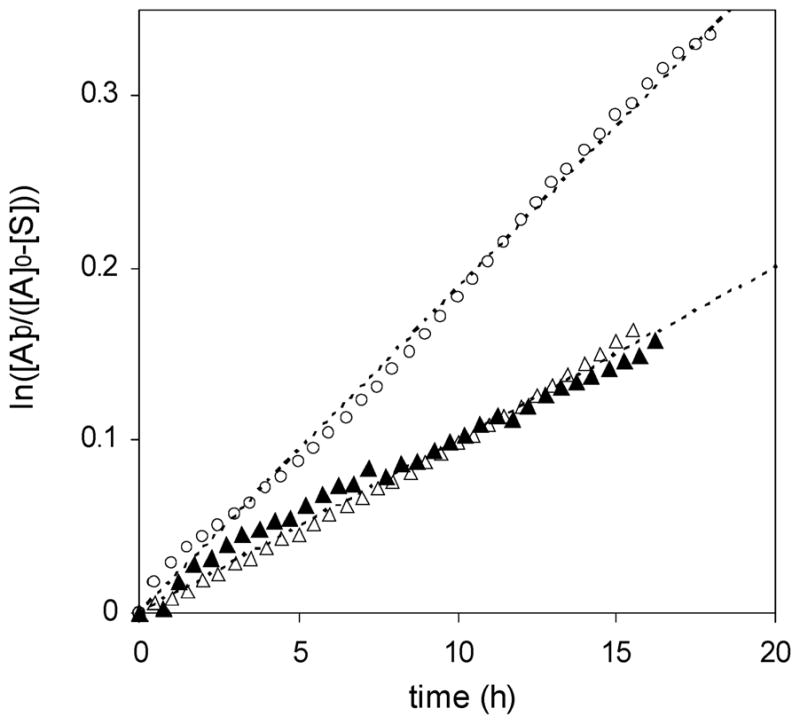
Plot of ln([A]0/([A]0 – [S])) as a function of time for bisalkoxyamine 1 (○, [A]0 = 180 mM) and parent alkoxyamine 2 (▲, [A]0 = 180 mM; △, [A]0 = 90 mM) upon heating at 125 °C in p-xylened10 in the absence of oxygen. The concentration of styrene [S] was measured by 1H NMR integration values, and [A]0 is the initial concentration of alkoxyamine functionality.
Table 5.
Kinetic Data for the Decomposition of Alkoxyamines 1, 2, and 5 at 125 °C in p-Xylene-d10 in the Absence of Oxygen
| alkoxyamine | kdec/s−1 (from eq 2) | kdec/s−1 (from eq 4) | kd/s−1 | pdec |
|---|---|---|---|---|
| 1 | 5.2 × 10−6 | 8.1 × 10−3 | 6.4 × 10−4 | |
| 2 | 3.7 × 10−6 | 2.8 × 10−6 | 4.2 × 10−3 | (8.8–6.6) × 10−4 |
| 5 | 1.6 × 10−5 | 8.0 × 10−4a | 2.0 × 10−2 |
Calculated using reported values50 of EA and A.
EPR Nitroxide Decomposition Studies
The curvature of the plot for bisalkoxaymine 1 in Figure 8 suggests that the decomposition pathway for this species is enhanced. Decomposition to form hydroxylamine and styrene as depicted in Scheme 3 is not the only mode of nitroxide decomposition (Scheme 4). α-Hydrogen bearing nitroxides work well in nitroxide-mediated radical polymerization specifically because of their ability to undergo bimolecular decomposition59,67 at polymerization temperatures, keeping the concentration of the free nitroxide low. Fischer has shown kinetically that the rate of polymerization can be enhanced by the decay of the persistent radical without adversely affecting the polydispersity or control of the molecular weight.68 Additives that cause nitroxide decomposition3 and persistent radicals with built-in decomposition pathways69 have both been used to increase polymerization rates under controlled conditions. As long as nitroxide decomposition does not simultaneously create a “dead” chain (as occurs with hydroxylamine and alkene formation), the concentration of the persistent radical will remain low enough to allow higher monomer conversions in less time.
Scheme 4.
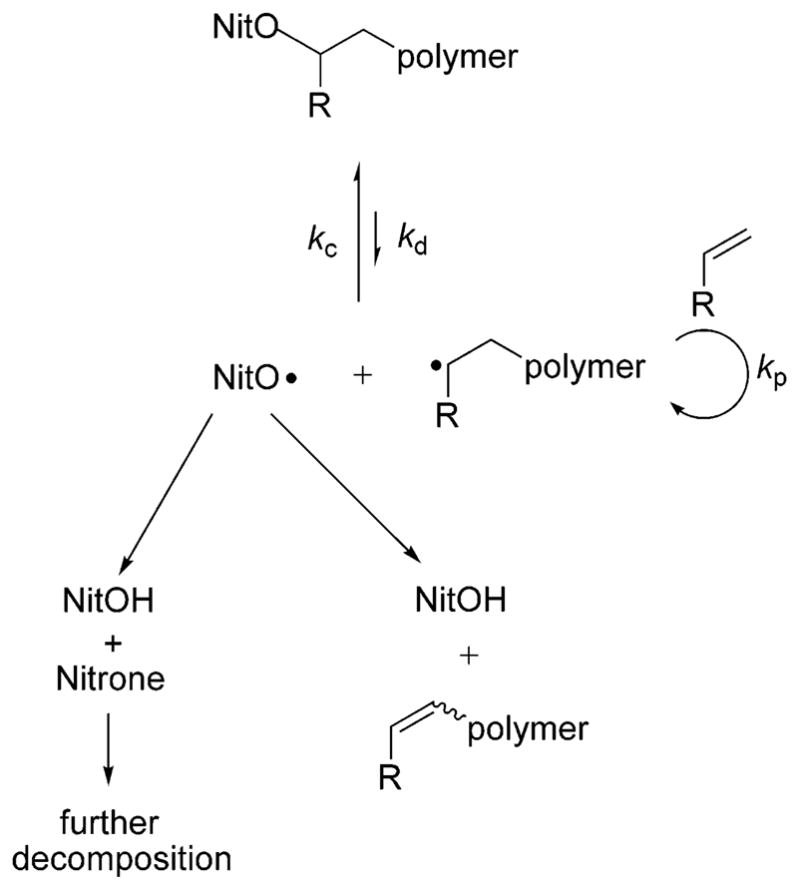
Key Reactions during Nitroxide-Mediated Radical Polymerization, where NitO Represents a Nitroxide
The relative rates of decomposition of bisnitroxide 3, TIPNO mononitroxide 4, and TEMPO were investigated using EPR. Freshly synthesized nitroxide samples in tert-butylbenzene were heated at 120 °C, and the change in nitroxide concentration with time was measured. Because one molecule of bisnitroxide 3 contains two nitroxide moieties, a 2.02 mM solution of 3 was used, compared to 4.03 mM solutions of 4 and TEMPO. In this way, the concentration of nitroxide functionality was 4.0 mM in all samples. As with the homolysis experiments, TEMPO was used as a calibration standard to convert the EPR signal into nitroxide concentration. Figure 10 shows the concentration of nitroxide vs time when samples of bisnitroxide 3, mononitroxide 4, and TEMPO were heated at 120 °C for 2 h. As expected, the stable nitroxide TEMPO does not decompose at 120 °C: its concentration remains 4.0 mM throughout the experiment. In contrast, both nitroxides 3 and 4 undergo an overall decrease in concentration when heated. Bisnitroxide 3 decomposes almost completely within 1 h, while only 20% of mononitroxide 4 decomposes within the same amount of time. Although the decomposition kinetics do not follow a simple first- or second-order rate law, it is evident that bisnitroxide 3 decomposes at a much faster rate than mononitroxide 4. Similar results were obtained when the experiment was repeated at 2.0 mM nitroxide concentration (Supporting Information).
Figure 10.
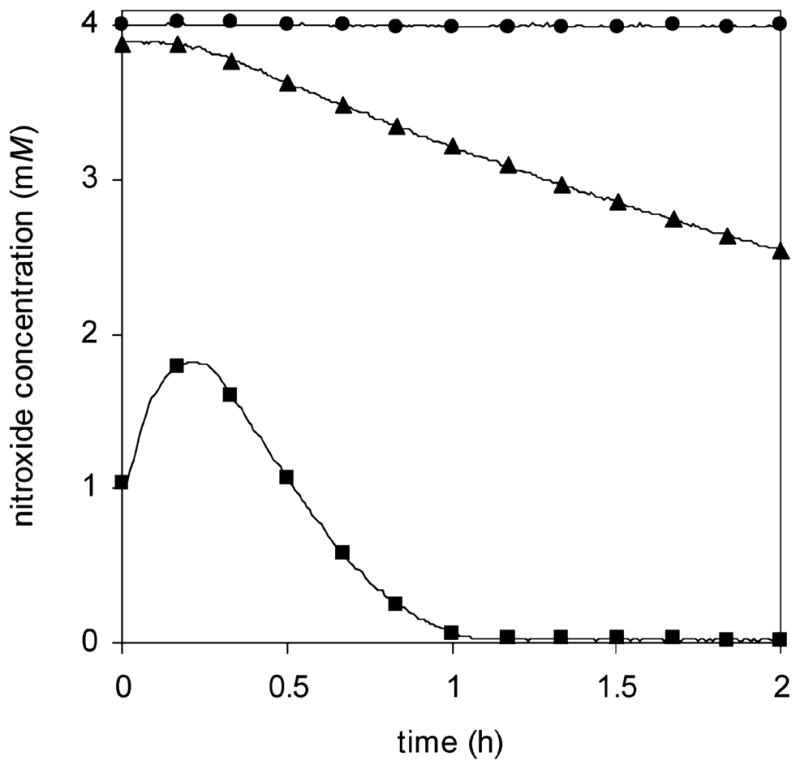
Variation of nitroxide concentration with time upon heating of bisnitroxide 3 (■), mononitroxide 4 (▲), and TEMPO (●) at 120 °C in tert-butylbenzene. Scans were taken every 30 to 60 s; only a small selection of data points are shown for clarity. Nitroxide samples 3 and 4 were synthesized, purified by flash chromatography, and stored in tert-butylbenzene at 0 °C for 1 day prior to use.
An unusual feature of these decomposition experiments is that the initial concentration of nitroxide, as measured by EPR, is not equal to 4.0 mM. For mononitroxide 4, this effect is small: the initial measured concentration is 3.9 mM. However, for bisnitroxide 3, the initial measured concentration is only 1.0 mM. This difference in concentration is too large to be the result of experimental error. As bisnitroxide 3 was heated, its concentration increased from 1.0 to 1.8 mM before beginning to decrease. This unusual behavior is reproducible; a separately synthesized sample of bisnitroxide 3 gave nearly identical results (Supporting Information). We hypothesize that some of the nitroxide may have converted to hydroxylamine prior to running the EPR experiment. Upon heating in the presence of air, the hydroxylamine is oxidized back to nitroxide. This would explain the low concentration of nitroxide at the beginning of the experiment as well as the initial increase in concentration during the first 15 min. The nitroxide concentration of 3 never reaches 4.0 mM because it decomposes at the same time that it is being formed. Nevertheless, even with a smaller initial concentration, bisnitroxide 3 decomposes more rapidly than parent nitroxide 4.
This experiment does not simulate polymerization conditions. All EPR samples were exposed to air, and nitroxide concentrations were somewhat higher than those found in a typical polymerization. Attempts to repeat this experiment in the absence of oxygen (by performing freeze–pump–thaw on the EPR samples) led to nonreproducible results. This may be due to the inability to completely remove all of the oxygen from the sample. Nitroxide concentrations on the order of 1–10−2 mM, which would more closely mimic polymerization conditions,70,71 are more difficult to detect accurately by EPR.
The enhanced decomposition of bisnitroxide 3 may result from an intramolecular disproportionation reaction by single electron transfer59 in the rare event that the bisnitroxide is formed at elevated temperatures. Formation of bisnitroxide 3 during the polymerization is expected to be more significant at extended polymerization times. Regardless of the mechanism of decomposition, bisnitroxide 3 decomposes faster than parent nitroxide 4, and this may contribute to the enhanced rates of polymerization observed for initiator 1.
Low-Temperature EPR Studies
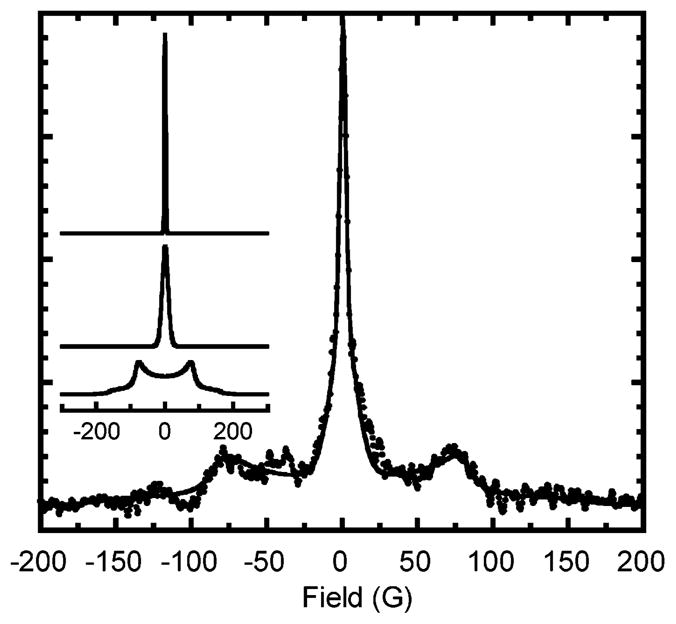
Both bisnitroxide 3 and mononitroxide 4 were examined at low-temperature by EPR.72 Bisnitroxide 3 shows a strong interaction at low-temperature, indicating that the conformation holds both nitroxide moieties close in space. An indication of strong biradical dipolar coupling is a half-field absorption signal as well as line broadening at the full field absorption line shape.73–75 When two radical spins are held in close proximity, the normally forbidden transition (Δm = 2) becomes allowed and observable in the half-field region. The integration of this signal normalized against spin concentration correlates to the interspin distance through the relationship of 1/r6.73 Figure 11 shows the EPR spectra of 3 and 4. As expected, only bisnitroxide 3 exhibited a half field signal. The relative intensity (adjusted for difference in microwave power) is 3.2 × 10−3: using eq 5, the nitroxide–nitroxide distance is 4.7 Å.
Figure 11.
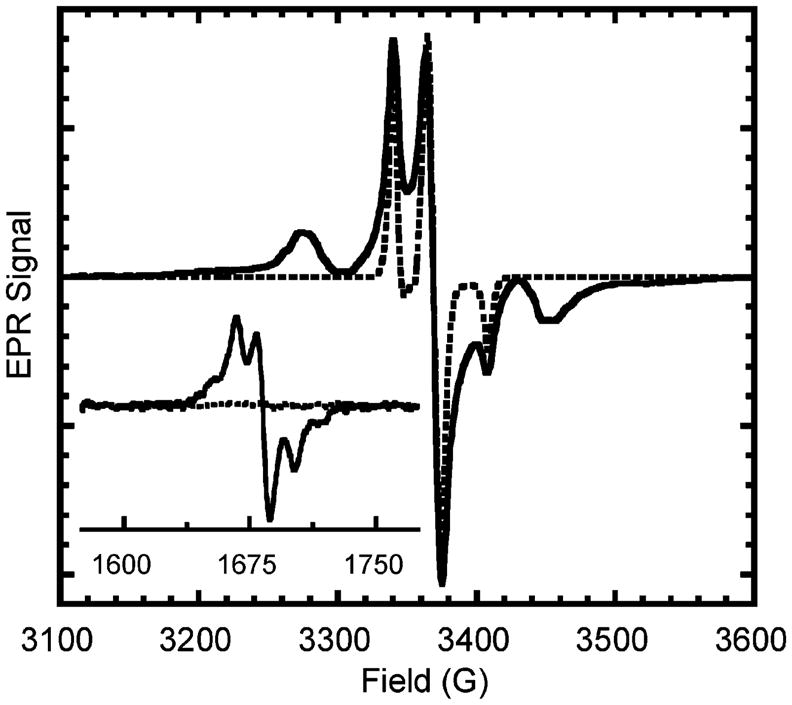
EPR spectra of bisnitroxide 3 (solid line) and TIPNO mononitroxide 4 (dashed line). Spectra peak heights are normalized. Inset shows the half-field transition for 3; the intensity is not to scale.
| (5) |
where A = 19.5 for organic radicals, r is the interspin distance in angstroms, and υ is the spectrometer frequency in GHz.
In addition to the appearance of a half-field signal, dipolar coupling is also revealed by the line shape of the full field EPR spectrum. In the absence of angular correlation between the dipolar tensor and the nitroxide magnetic tensors, the overall line shape can be expressed as the convolution of the EPR spectrum of an isolated spin with a dipolar spectral function. The peak-to-peak width of the dipolar broadening function provides an alternate method to calculate the interspin distance, as given by eqs 6 and 7.75 The dipolar broadening function was determined by deconvolution of the spectrum of bisnitroxide 3 with the spectrum of the mononitroxide TIPNO 4 (representative of an isolated spin). The Fourier transform method of Rabenstein and Shin76 was employed, with the addition of a regularization parameter to limit noise. The resulting broadening function is shown in Figure 12. The majority of the broadening function conforms to the expected “Pake pattern” that results from a pair of spins with strong dipolar coupling, superimposed with two much narrower functions whose structures cannot be resolved. Lack of resolution may result from the smoothing effects of noise suppression, distance distributions, or other factors; however, these narrower broadening functions often conform to a Gaussian line shape.77 The fwhm (full width at half-maximum) of this Gaussian function can then be used to calculate a distance using eq 6.78
Figure 12.
Dipolar broadening function. The dipolar broadening function (dots) was determined by Fourier transform deconvolution of the bisnitroxide EPR spectrum with the mononitroxide TIPNO spectrum. The simulated broadening function (solid line) is the sum of the three functions shown in the inset. They are, from the top, a narrow Gaussian (2.25 G fwhm and 9% of total area), a broader Gaussian (20 G fwhm and 30% of total area), and a “Pake” function (150 G wide and 61% of total area).
| (6) |
where B is the Gaussian fwhm in G and r the distance in Å.
The broadening function could only be well fit with a minimum of three separate functions, each of which corresponds to a population with different interspin distances. The three functions are also shown in Figure 12, with their sum superimposed on the dipolar broadening function. In addition, this simulated broadening function was convoluted with the EPR spectrum of mononitroxide 4 to yield a simulated spectrum of the bisnitroxide 3, shown in Figure 13. The details of the three broadening functions are as follows: first, the narrow central spike was fit with a Gaussian of width 2.25 G. This would be derived from an interspin distance of over 20 Å (eq 6) and is most likely from a small amount (9%) of monoradical impurity, arising from either the incomplete oxidation of both nitroxides during synthesis or the decomposition of one over time. The next part of the broadening function was simulated with a 20 G Gaussian function. This corresponds to a distance of 9.8 Å (eq 6) and is representative of a population (30%) of the bisnitroxide with the two spins at a maximum separation, as depicted by structure 6 (Figure 14). The majority of the broadening function (61%) is well fit with a “Pake pattern” with a width of 150 G between the inner turning points and represents the two spins in close proximity, as depicted by structure 7 (Figure 14). Employing eq 7, a distance of 5.8 Å is calculated.
Figure 13.
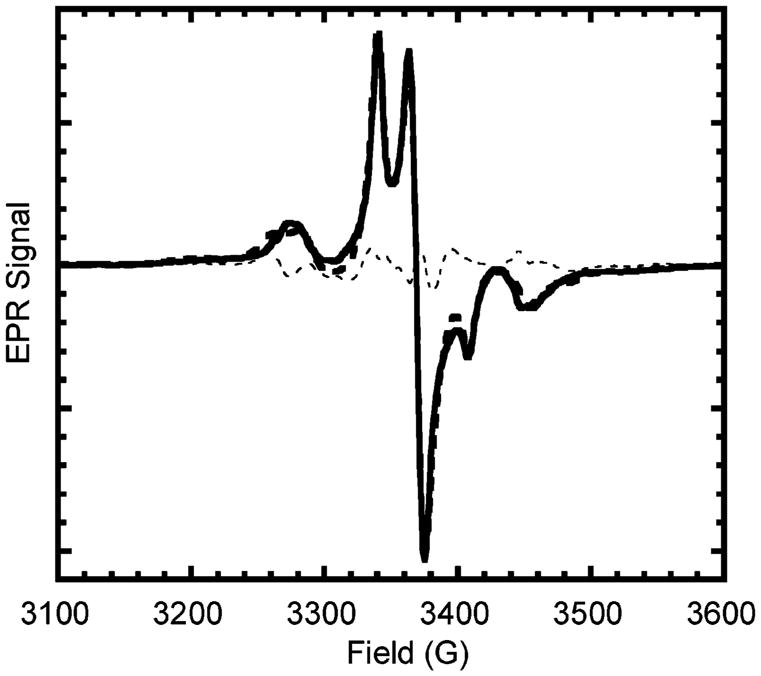
Simulation of experimental spectrum. The experimental EPR spectrum of the bisnitroxide 3 (solid line) was simulated by the convolution of the TIPNO 4 spectrum with the simulated broadening function (dashed line) from Figure 10. Also shown is the residual signal (dotted line).
Figure 14.
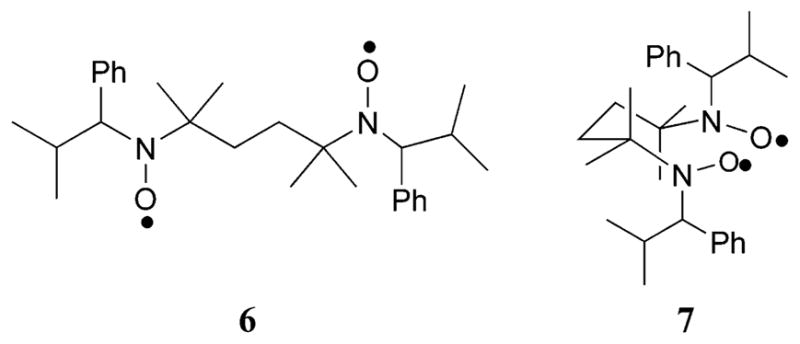
Nonassociated nitroxides with a distance of 9.8 Å (6) and interacting nitroxides with a distance of 5.8 Å (7).
| (7) |
where 2Bpake is the distance between inner turning points in G, g is the g-factor = 2 for nitroxides, B2 = 4635 G/Å3, and r is the distance in Å.
The interspin distances calculated from the half-field method and the deconvolution method corroborate the interaction of the two nitroxides. Previous studies have shown that when both techniques are applied to the same system, the half-field method predicts a shorter interspin distance than deconvolution methods.79 One reason for this is that the half-field signal increases in amplitude with closer distances, while the full field signal broadens (but retains the same area) and decreases in amplitude. Therefore, taken together, these two methods describe a majority population with an inter-spin distance in the range of 4.7–5.8 Å and a smaller population with a distance in the 9–10 Å range.
Implications toward Polymerization
The characteristics of 1 in NMRP cannot be explained by existing effects involving single nitroxides. For example, earlier work from this lab48 showed that macroinitiator 8 (Figure 15), which is sterically similar to the alkoxyamine formed from 1 during polymerization, effects NMRP at the same rate as the monodirectional initiator 2. Similarly, the sterically hindered alkoxyamine 9 (Figure 15) developed by Studer31 effects polymerization of both styrene and n-butyl acrylate more slowly than bisinitiator 1. Thus, it is clear that neither slow diffusion due to the polymeric nature of the nitroxide nor steric congestion around the nitroxide can explain the enhanced rate of homolysis of initiator 1.
Figure 15.

Alkoxyamine initiators structurally similar to 1: polymeric initiator 8 containing a macromolecular nitroxide48 and Studer’s sterically congested neopentyl initiator 9.31
The EPR data show that ~61% of the bisnitroxide exists in a state in which there is significant spin–spin interaction. The bisnitroxide is a mixture of two inseparable diastereomers. It is likely that one diastereomer experiences a closer spin–spin interaction that the other. To account for the unusually short nitroxide–nitroxide distance, a chairlike conformation 7 enhanced by two Thorpe–Ingold gem-dimethyl effects80 places the two nitroxide groups in close proximity. The other diastereomer may adopt an extended conformation 6, in which the nitroxide groups are remote. This correlates with the two nitroxide interspin distances of approximately 5.8 and 9.8 Å derived from the EPR measurements.
The close proximity of the two nitroxide moieties in bisnitroxide 3 may explain the faster rates of C–O bond homolysis and nitroxide decomposition of bisalkoxyamine 1 as compared to parent alkoxyamine 2. We propose that during polymerization, homolysis of the C–O bond is facilitated by stabilization of the resulting mononitroxide through a synergistic interaction from the adjacent N-alkoxyamine. The resulting mixed nitroxide and N-alkoxyamine species 10 (Figure 16) resides in a chairlike conformation where the two N–O moieties are held in close proximity. Resonance structures 10, 11, and 12 can be drawn in which the active carbon of the remaining polymer chain is bonded to either N–O group. This radical can therefore be thought of as being delocalized over five atoms (including the two nitrogen atoms), as depicted in resonance hybrid 13, with the polymer chain being shared equally between both partial nitroxides. This stabilization would lead to a higher kd for C–O bond homolysis, in agreement with the experimental findings presented herein. The near proximity of the nitroxide moieties may also explain the increased rate of nitroxide decomposition. An intramolecular decomposition pathway would lead to faster rates of decomposition, as was observed in the EPR experiments.
Figure 16.

Proposed resonance structures of the key nitroxide intermediate during polymerization.
This proposed nitroxide resonance stabilization implies that dissociation of the first nitroxide from the bisalkoxyamine occurs more easily than dissociation of the second nitroxide. Thus, the kd values in Table 4 refer to the dissociation of the first nitroxide moiety, since the initial slope method (eq 1) was used to calculate kd (see Supporting Information for a detailed derivation).
This unprecedented mode of stabilization suggests a new strategy for the design of nitroxides for use in low-temperature NMRP in which bisnitroxides are conformationally biased or covalently restricted to reside in close proximity. Work along these lines as well as other applications of this strongly interacting bisnitroxide are currently under investigation.
Conclusion
Bidirectional initiator 1 effects the polymerization of styrene, tert-butyl acrylate, n-butyl acrylate, and dimethylacrylamide at temperatures as low as 70 °C. The ability to effect NMRP at temperatures ~50 °C lower than those commonly employed will extend NMRP to applications involving biomolecules and other heat sensitive substrates. Kinetic studies reveal bidirectional initiator 1 undergoes homolytic cleavage ~2 times faster that monodirectional initiator 2. When heated in the absence of oxygen, both 1 and 2 are equally likely to decompose into hydroxylamine and styrene; however, bidirectional initiator 1 exhibits an accelerated decomposition, as seen dramatically in Figure 8. EPR studies similarly show that bisnitroxide 3 decomposes faster than TIPNO nitroxide 4. Low-temperature EPR studies indicate a striking spin–spin interaction for bisnitroxide 3. Chairlike conformation 7 enhanced by two Thorpe–Ingold gem-dimethyl interactions is proposed to impart conformational constraints facilitating this unusually strong spin–spin interaction. The near proximity of the nitroxide moieties apparently leads to an enhanced rate of bond homolysis and thus allows bisalkoxyamine 1 to effect controlled polymerizations at temperatures as low as 70 °C. A new class of proximal bisnitroxides can be envisioned for future advances in NMRP.
Supplementary Material
Acknowledgments
The authors thank Research Corporation (RA0336) for financial support in addition to equipment grants from the NSF (CHE-0342912) (NMR) and DBI-0217922 (to G.M.) and grants from NIH (CA52955) (ESITOFMS) and (GM 65790, to G.M.). J.R. thanks NSF for a Graduate Research Fellowship. We are also grateful to C. Bernasconi for helpful discussions on kinetics and H. Ito for suggested 1H NMR experiments.
Footnotes
Supporting Information Available: Data tables for polymerizations at 90 and 110 °C, kinetic plots and GPC traces for monomer polymerization time studies, data table for ABA triblock copolymers, concentration vs time plots and Arrhenius plot for the thermal homolysis EPR experiments, concentration vs time plots from the 1H NMR and EPR decomposition experiments, and derivation of eq 1. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.Hawker CJ, Bosman AW, Harth E. Chem Rev. 2001;101:3661–3688. doi: 10.1021/cr990119u. [DOI] [PubMed] [Google Scholar]
- 2.Solomon DH, Rizzardo E, Cacioli P. 135280. Eur Pat Appl. 1985
- 3.Georges MK, Veregin RPN, Kazmaier PM, Hamer GK, Saban M. Macromolecules. 1994;27:7228–7229. [Google Scholar]
- 4.Benoit D, Chaplinski V, Braslau R, Hawker CJ. J Am Chem Soc. 1999;121:3904–3920. [Google Scholar]
- 5.Benoit D, Grimaldi S, Robin S, Finet JP, Tordo P, Gnanou Y. J Am Chem Soc. 2000;122:5929–5939. [Google Scholar]
- 6.Pintauer T, Matyjaszewski K. Coord Chem Rev. 2005;249:1155–1184. [Google Scholar]
- 7.Matyjaszewski K, Xia JH. Chem Rev. 2001;101:2921–2990. doi: 10.1021/cr940534g. [DOI] [PubMed] [Google Scholar]
- 8.Kamigaito M, Ando T, Sawamoto M. Chem Rev. 2001;101:3689–3745. doi: 10.1021/cr9901182. [DOI] [PubMed] [Google Scholar]
- 9.Patten TE, Xia JH, Abernathy T, Matyjaszewski K. Science. 1996;272:866–868. doi: 10.1126/science.272.5263.866. [DOI] [PubMed] [Google Scholar]
- 10.Kato M, Kamigaito M, Sawamoto M, Higashimura T. Macromolecules. 1995;28:1721–1723. [Google Scholar]
- 11.Percec V, Barboiu B. Macromolecules. 1995;28:7970–7972. [Google Scholar]
- 12.Granel C, Dubois P, Jerome R, Teyssie P. Macromolecules. 1996;29:8576–8582. [Google Scholar]
- 13.Wayland BB, Basickes L, Mukerjee S, Wei ML, Fryd M. Macromolecules. 1997;30:8109–8112. [Google Scholar]
- 14.Ando T, Kamigaito M, Sawamoto M. Macromolecules. 1998;31:6708–6711. doi: 10.1021/ma980320r. [DOI] [PubMed] [Google Scholar]
- 15.Collins JE, Fraser CL. Macromolecules. 1998;31:6715–6717. [Google Scholar]
- 16.Percec V, Barboiu B, van der Sluis M. Macromolecules. 1998;31:4053–4056. [Google Scholar]
- 17.Moineau G, Granel C, Dubois P, Jerome R, Teyssie P. Macromolecules. 1998;31:542–544. [Google Scholar]
- 18.Moad G, Rizzardo E, Thang SH. Aust J Chem. 2005;58:379–410. [Google Scholar]
- 19.Lowe AB, McCormick CL. Prog Polym Sci. 2007;32:283–351. [Google Scholar]
- 20.Chiefari J, Chong YK, Ercole F, Krstina J, Jeffery J, Le TPT, Mayadunne RTA, Meijs GF, Moad CL, Moad G, Rizzardo E, Thang SH. Macromolecules. 1998;31:5559–5562. [Google Scholar]
- 21.Mayadunne RTA, Rizzardo E, Chiefari J, Krstina J, Moad G, Postma A, Thang SH. Macromolecules. 2000;33:243–245. [Google Scholar]
- 22.Mayadunne RTA, Rizzardo E, Chiefari J, Chong YK, Moad G, Thang SH. Macromolecules. 1999;32:6977–6980. [Google Scholar]
- 23.Perrier S, Takolpuckdee P. J Polym Sci, Part A: Polym Chem. 2005;43:5347–5393. [Google Scholar]
- 24.Barner-Kowollik C, Buback M, Charleux B, Coote ML, Drache M, Fukuda T, Goto A, Klumperman B, Lowe AB, McLeary JB, Moad G, Monteiro MJ, Sanderson RD, Tonge MP, Vana P. J Polym Sci, Part A: Polym Chem. 2006;44:5809–5831. [Google Scholar]
- 25.For one example, see: Bosman AW, Vestberg R, Heumann A, Frechet JMJ, Hawker CJ. J Am Chem Soc. 2003;2125:2715–2728. doi: 10.1021/ja028392s.
- 26.Solomon DH. J Polym Sci, Part A: Polym Chem. 2005;43:5748–5764. [Google Scholar]
- 27.Shipp DA. J Macromol Sci, Polym Rev. 2005;C45:171–194. [Google Scholar]
- 28.Braunecker WA, Matyjaszewski K. Prog Polym Sci. 2007;32:93–146. [Google Scholar]
- 29.Qiu J, Charleux B, Matyjaszewski K. Prog Polym Sci. 2001;26:2083–2134. [Google Scholar]
- 30.Rodlert M, Harth E, Rees I, Hawker CJ. J Polym Sci, Part A: Polym Chem. 2000;38:4749–4763. [Google Scholar]
- 31.Studer A, Harms K, Knoop C, Muller C, Schulte T. Macromolecules. 2004;37:27–34. [Google Scholar]
- 32.Lagrille O, Cameron NR, Lovell PA, Blanchard R, Goeta AE, Koch R. J Polym Sci, Part A: Polym Chem. 2006;44:1926–1940. [Google Scholar]
- 33.Detrembleur C, Sciannamea V, Koulic C, Claes M, Hoebeke M, Jerome R. Macromolecules. 2002;35:7214–7223. [Google Scholar]
- 34.Sciannamea V, Guerrero-Sanchez A, Schubert US, Catala JM, Jerome R, Detrembleur C. Polymer. 2005;46:9632–9641. [Google Scholar]
- 35.Detrembleur C, Teyssie P, Jerome R. Macromolecules. 2002;35:1611–1621. [Google Scholar]
- 36.Drockenmuller E, Catala JM. Macromolecules. 2002;35:2461–2466. [Google Scholar]
- 37.Drockenmuller E, Lamps JP, Catala JM. Macromolecules. 2004;37:2076–2083. [Google Scholar]
- 38.Harth E, van Horn B, Hawker CJ. Chem Commun. 2001:823–824. [Google Scholar]
- 39.Studer A. Angew Chem, Int Ed. 2000;39:1108–1111. doi: 10.1002/(sici)1521-3773(20000317)39:6<1108::aid-anie1108>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 40.Schulte T, Studer A. Macromolecules. 2003;36:3078–3084. [Google Scholar]
- 41.Knoop CA, Studer A. J Am Chem Soc. 2003;125:16327–16333. doi: 10.1021/ja037948o. [DOI] [PubMed] [Google Scholar]
- 42.Wetter C, Gierlich J, Knoop CA, Muller C, Schulte T, Studer A. Chem– Eur J. 2004;10:1156–1166. doi: 10.1002/chem.200305427. [DOI] [PubMed] [Google Scholar]
- 43.Siegenthaler KO, Studer A. Macromolecules. 2006;39:1347–1352. [Google Scholar]
- 44.Dire C, Charleux B, Magnet S, Couvreur L. Macromolecules. 2007;40:1897–1903. [Google Scholar]
- 45.Nicolas J, Dire C, Mueller L, Belleney J, Charleux B, Marque SRA, Bertin D, Magnet S, Couvreur L. Macromolecules. 2006;39:8274–8282. [Google Scholar]
- 46.Guillaneuf Y, Gigmes D, Marque SRA, Astolfi P, Greci L, Tordo P, Bertin D. Macromolecules. 2007;40:3108–3114. [Google Scholar]
- 47.Hill NL, Braslau R. Macromolecules. 2005;38:9066–9074. [Google Scholar]
- 48.Ruehl J, Braslau R. J Polym Sci, Part A: Polym Chem. 2007;45:2015–2025. [Google Scholar]
- 49.Braslau R, Tsimelzon A, Gewandter J. Org Lett. 2004;6:2233–2235. doi: 10.1021/ol049271v. [DOI] [PubMed] [Google Scholar]
- 50.Marque S, Le Mercier C, Tordo P, Fischer H. Macromolecules. 2000;33:4403–4410. [Google Scholar]
- 51.Huang WL, Chiarelli R, Charleux B, Rassat A, Vairon JP. Macromolecules. 2002;35:2305–2317. [Google Scholar]
- 52.Higaki Y, Otsuka H, Takahara A. Macromolecules. 2004;37:1696–1701. [Google Scholar]
- 53.Hawker CJ, Barclay GG, Dao JL. J Am Chem Soc. 1996;118:11467–11471. [Google Scholar]
- 54.Lizotte JR, Anderson SG, Long TE. J Polym Sci, Part A: Polym Chem. 2004;42:1547–1556. [Google Scholar]
- 55.Buston JEH, Coldham I, Mulholland KR. J Chem Soc Perkin Trans. 1999;1:2327–2334. [Google Scholar]
- 56.Liu YB, Maden A, Murray WV. Tetrahedron. 2002;58:3159–3170. [Google Scholar]
- 57.O’Bryan G, Braslau R. Macromolecules. 2006;39:9010–9017. [Google Scholar]
- 58.Lee S, Zhao ZS. Tetrahedron Lett. 1999;40:7921–7924. [Google Scholar]
- 59.Nilsen A, Braslau R. J Polym Sci, Part A: Polym Chem. 2006;44:697–717. [Google Scholar]
- 60.Ananchenko GS, Fischer H. J Polym Sci, Part A: Polym Chem. 2001;39:3604–3621. [Google Scholar]
- 61.Fischer H. Macromolecules. 1997;30:5666–5672. [Google Scholar]
- 62.Goto A, Kwak Y, Yoshikawa C, Tsujii Y, Sugiura Y, Fukuda T. Macromolecules. 2002;35:3520–3525. [Google Scholar]
- 63.It should be noted that the mechanism of decomposition may not go through the homolytic cleavage shown in Scheme 2. Alternative mechanisms, particularly for ester functionalized initiators, cannot be ruled out, such as nonradical ionic elimination or a concerted mechanism. For phenethyl-functionalized initiators, however, most evidence is in support of radical intermediates.63,67
- 64.Ohno K, Tsujii Y, Fukuda T. Macromolecules. 1997;30:2503–2506. [Google Scholar]
- 65.Souaille M, Fischer H. Macromolecules. 2001;34:2830–2838. [Google Scholar]
- 66.Ito H, Dalby C, Pomerantz A, Sherwood M, Sato R, Sooriyakumaran R, Guy K, Breyta G. Macromolecules. 2000;33:5080–5089. [Google Scholar]
- 67.Bowman DF, Gillan T, Ingold KU. J Am Chem Soc. 1971;93:6555–6561. [Google Scholar]
- 68.Souaille M, Fischer H. Macromolecules. 2002;35:248–261. [Google Scholar]
- 69.Steenbock M, Klapper M, Mullen K. Acta Polym. 1998;49:376–378. [Google Scholar]
- 70.Fukuda T, Terauchi T, Goto A, Ohno K, Tsujii Y, Miyamoto T, Kobatake S, Yamada B. Macromolecules. 1996;29:6393–6398. [Google Scholar]
- 71.Fischer H. J Polym Sci, Part A: Polym Chem. 1999;37:1885–1901. [Google Scholar]
- 72.For an extensive database of bisnitroxides and the EPR spectra see: Organic Cation Radicals and Polyradicals. Index of Substances for Volumes II/1 and II/9, Landolt-Börnstein - Group II Molecules and Radicals. 9d2. Springer-Verlag; Berlin: 1980.
- 73.Eaton SS, Eaton GR. J Am Chem Soc. 1982;104:5002–5003. [Google Scholar]
- 74.van der Waals JH, de Groot MS. Mol Phys. 1959;2:333–340. [Google Scholar]
- 75.Slichter CP. Principles of Magnetic Resonance. 3. Springer-Verlag; New York: 1996. p. 669. [Google Scholar]
- 76.Rabenstein MD, Shin YK. Proc Natl Acad Sci USA. 1995;92:8239–8243. doi: 10.1073/pnas.92.18.8239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ottemann KM, Thorgeirsson TE, Kolodziej AF, Shin YK, Koshland DE. Biochemistry. 1998;37:7062–7069. doi: 10.1021/bi980305e. [DOI] [PubMed] [Google Scholar]
- 78.Walter ED, Sebby KB, Usselman RJ, Singel DJ, Cloninger MJ. J Phys Chem B. 2005;109:21532–21538. doi: 10.1021/jp0515683. [DOI] [PubMed] [Google Scholar]
- 79.Persson M, Harbridge JR, Hammarstrom P, Mitri R, Martensson LG, Carlsson U, Eaton GR, Eaton SS. Biophys J. 2001;80:2886–2897. doi: 10.1016/S0006-3495(01)76254-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jung ME, Gervay J. Tetrahedron Lett. 1988;29:2429–2432. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.