

Abstract
Interest in peptides incorporating boronic acid moieties is increasing due to their potential as therapeutics/diagnostics for a variety of diseases such as cancer. The utility of peptide boronic acids may be expanded with access to vast libraries that can be deconvoluted rapidly and economically. Unfortunately, current detection protocols using mass spectrometry are laborious and confounded by boronic acid trimerization, which requires time consuming analysis of dehydration products. These issues are exacerbated when the peptide sequence is unknown, as with de novo sequencing, and especially when multiple boronic acid moieties are present. Thus, a rapid, reliable and simple method for peptide identification is of utmost importance. Herein, we report the identification and sequencing of linear and branched peptide boronic acids containing up to five boronic acid groups by matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS). Protocols for preparation of pinacol boronic esters were adapted for efficient MALDI analysis of peptides. Additionally, a novel peptide boronic acid detection strategy was developed in which 2,5-dihydroxybenzoic acid (DHB) served as both matrix and derivatizing agent in a convenient, in situ, on-plate esterification. Finally, we demonstrate that DHB-modified peptide boronic acids from a single bead can be analyzed by MALDI-MSMS analysis, validating our approach for the identification and sequencing of branched peptide boronic acid libraries.
Keywords: MALDI-MS, peptide sequencing, matrix, deconvolution, high throughput screen
INTRODUCTION
An increasing number of boron containing peptides are being investigated for their potential use in the treatment of various diseases. For example, peptides incorporating boronic acids have been utilized to target the proteasome,1,2 HIV-1 protease,3,4 thrombin,5,6 and human ClpXP.7 Additionally, a wide variety of artificial boron containing lectins (boronolectins) are being developed, taking advantage of the strong complexation of boronic acids with 1,2- and 1,3-diols.8 These boronolectins may prove capable of mediating biological processes such as immune response,9,10 fertilization,11 embryonic development,12 and apoptosis.13 Peptides, proteins, and small molecules that feature boronic acid functionality have been utilized as fluorescent reporter groups,14 sensors for glucose responsive insulin release,15 and for detection of cancer related glycoproteins.16 As the utility of organoboron compounds continue to expand both in number and scope, analytical techniques such as mass spectrometry are increasingly essential for compound characterization and structure elucidation. Boronic acid derivatives have been traditionally analyzed using a wide variety of gas-phas ionization and desorption/ionization, including direct insertion probe electron ionization (DIP/EI),17 electron capture chemical ionization (ECCI),18 liquid secondary ionization (LSI),18,19 fast atom bombardment (FAB),20 and electrospray ionization (ESI). However, there are limited examples in the literature concerning the analysis of boronic acids via MALDI-MS.16,21,22
It is well-known that free boronic acids undergo thermally induced dehydrations7,23,24 or cyclizations to boroxines20,21,25 via dehydration/trimerization reactions, which can hinder detection by mass spectrometry (Figure 1). When dehydration/boroxine formation interferes with MS data analysis, additional steps such as laborious analysis of cyclization products and/or derivatization to the boronic ester may be required. The former has been identified as undesirable for the characterization of peptides incorporating multiple boronic acid functionalities.26 Conversely, the derivatization of boronic acids with diols and simple sugars to the cyclic boronic esters has proven to reliably eliminate boroxine formation (Figure 2).17,19,27,28 Indeed, Haas et al. demonstrated the successful characterization of small molecular weight boronic ester peptides via positive-ion ammonia chemical ionization (CI) as well as positive-ion LSI-MS.18 With regards to sequencing peptide boronic acid libraries, Hall has shown that under appropriate conditions, boroxine formation is not necessarily detrimental to library deconvolution.25 More recently, Leitner and colleagues reported the arginine-specific labeling technique using 2,3-butanedione (BD) and phenylboronic acid (PBA) and subsequent analysis using MALDI-MS.21 Their work demonstrated that boronic acid-peptide conjugates can be utilized for the identification of peptides based on their mass fingerprint. Finally, Lavigne and Thompson have reported the design and synthesis of a peptide boronolectin (PBL) library which showed promise for the detection of cancer related targets.16 Although MALDI-MS was employed for PBL sequence determination, the boronic acid moiety employed in the library contains an ortho-amino methyl group that can complex to boron, protecting it from trimerization/dehydration processes.
Figure 1.

Thermally induced boroxine formation.
Figure 2.

Boronic acid peptide functionalization with a 1,2-diol.
As part of our long range goal of identifying novel cell permeable molecules that can selectively bind to RNA tertiary structure, branched peptide libraries were initially investigated as RNA ligands.29 As an extension of this approach, branched peptides containing boronic acid functionalities are particularly interesting, in part because of the possibility of forming Lewis acid-base complexes. Thus, the necessity for a combinatorial library of branched peptide boronic acids (i.e., one bead-one structure) required a rapid, efficient and cost-effective method for deconvolution of hit compounds. Herein, we report a convenient strategy to identify and sequence branched peptides containing boronic acid moieties using MALDI-MS. Derivatization of peptide boronic acids with 1,2-diols followed by MALDI-MS allowed the successful detection by MS. Fortunately, we also discovered that using DHB as a matrix efficiently converts the peptide boronic acid to a DHB adduct, which provides high quality spectra without the need for pre-derivatization. Ultimately, it was determined that the use of DHB for analysis is superior than pretreatment with 1,2-diols. Using this protocol, we successfully identified and sequenced a branched peptide containing five boronic acid moieties released by photocleavage from a single Tentagel bead (Figure 3).
Figure 3.

Derivatization of peptide boronic acid with DHB.
METHODS
Materials
MALDI matrices 5-chloro-2-mercaptobenzothiazole (CMBT), 1-chloro-4-hydroxyisoquinoline (CHIQ), and 2,5-dihydroxybenzoic acid (DHB) were purchased from Sigma Aldrich. 3-hydroxypicolinic acid (HPA) and 3,4-diaminobenzophenone (DABP) were purchased from Acros, and 2′,6′-dihydroxyacetophenone (DHAP) was purchased from Fluka. All matrices were greater than 98% purity. Milli-Q (MQ) grade H2O was used for peptide dilution, and HPLC grade acetonitrile was purchased from Fischer Scientific. Unless specified otherwise, all matrices were prepared in a 10 mg mL−1 concentration using a 1:1 (v/v) ACN: H2O mixture. CHIQ and DHB were prepared as saturated solutions using a 1:1 (v/v) ACN: H2O mixture.
Peptide Synthesis
Peptides were synthesized using traditional solid phase peptide synthesis (SPPS) using N-α-Fmoc protected L-amino acids (Novabiochem), HCTU ((2-(6-Chloro-1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium hexafluorophosphate) and DIEA (N,N-diisopropylethylamine) in DMF. Solid phase synthesis was performed on a vacuum manifold (Qiagen) outfitted with 3-way Luerlock stopcocks (Sigma) in either Poly-Prep columns or Econo-Pac polypropylene columns (Bio-Rad). The resin was mixed in solution by bubbling argon during all coupling and washing steps. Beads were washed extensively with DMF between reactions, and the couplings were tested for completeness via Kaiser test.30 FITC labeled peptides were prepared following literature protocol.29 Boron was incorporated into branched peptides as 4-borono-L-phenylalanine (BPA) 26,31 or into linear peptides on the N-terminus.7 FLBP132 was diluted to a concentration of 370 μM for initial optimization studies.
Generalized Procedure for Pinacol Derivatization
A 0.1 M pinacol solution was prepared with a 10 mM NH4OAc solution (pH = 9.5) to ensure the formation of the boronic ester. The alkaline pinacol solution was combined with an aliquot of the peptide in a 1:1 (v/v) ratio and allowed to incubate without shaking for 30 min at room temperature. After the incubation period, the MALDI plate was spotted with the matrix of choice, and the sample was then spotted on top of the matrix without mixing. The sample was allowed to dry and 1 μL of MQ water was spotted on top of the sample and allowed to incubate undisturbed for 30 seconds followed by vacuum aspiration. This washing procedure removes unreacted, excess pinacol.
Optimized Procedure for DHB Derivatization
A saturated solution of DHB was prepared in a 1:1 (v/v) ACN: H2O solution, which was then spotted on the plate. After drying, the sample (1.0 μL, 300 μM) was spotted on top of the matrix and allowed to dry. Another 1 μL of 1:1 (v/v) ACN/H2O was spotted on top of the sample to generate a homogenous solution of the sample and matrix. Samples were then allowed to dry, yielding thin needlelike crystals.
Photocleavage of peptides from the ANP resin and MALDI-TOF sequencing
After solid phase peptide synthesis, the FBPA pinacol group was removed via mixing with excess phenyl boronic acid (1.6 M) overnight. Thereafter, amino acid side-chains were deprotected by a 3-hour treatment with 95:2.5:2.5 trifluoroacetic acid/triisopropylsilane/water (v/v). After deprotection, the resin was washed extensively with DMF, DCM, and MeOH before drying and storage at −20 °C protected from the light. A single bead was placed into a clear non-stick 0.5 mL microfuge tube containing 20 μL of 1:1 (v/v) MeOH:H2O and photocleaved in a foil-lined container by irradiation at 365 nm using a 4 W handheld UV lamp for 1 hour. A small aliquot of the supernatant was utilized for MALDI analysis without any further treatment.
Mass Spectrometry
All spectra were acquired with an Applied Biosystems 4800 MALDI-TOF mass spectrometer equipped with a 355 nm Nd:YAG laser. All MALDI spectra were acquired using an Applied Biosystems 123×81 mm Opti-TOF 384 stainless steel plate (Part # 1016629). All spectra were acquired in positive reflector mode. A fixed laser intensity of 6500 (arbitrary units) was utilized to acquire 500 shots per spectra for MS samples and 1000–2000 shots per spectra for MS-MS samples. All MS-MS spectra were acquired via post source decay using the MS-MS 1kV positive mode with a selected precursor ion window set to a relative 200.000 FWHM (Full Width Half Maximal) Da resolution. All spectra were processed using Applied Biosystems 4000 series explorer software v.3.5.2., and settings for peak detection were restricted to a S/N threshold of 200.
RESULTS AND DISCUSSION
The suitability of MALDI as a desorption/ionization technique to detect branched peptides containing multiple boronic acids was initially explored by examining a fluorescein isothiocyanate (FITC) labeled peptide containing a single boronic acid, FLBP1 (Table 1). The molecular ion peak corresponding to the free boronic acid was not observed using CHCA matrix as expected (entry 1). As it was previously reported that modification with 1,2-diols such as pinacol reliably improves molecular ion detection, we next combined FLBP1 with 0.1 M pinacol using a 5 min incubation time (entry 2). Unfortunately, neither the desired molecular ion peak nor any identifiable fragment ions were observed. This result was surprising as previous studies indicate that abbreviated incubation times (10 min) with excess diol were sufficient for boronic ester formation and detection using LSIMS and ammonia CI.18 It was hypothesized that these results could be attributed to incomplete esterification, ionization interference by excess pinacol or matrix incompatibility. To determine if the absence of the expected molecular ion was due to incomplete esterification, incubation time was increased to 30 min (entry 3), which again failed to detect the expected pinacol protected peptide boronic acid.33 As effective MALDI ionization is highly dependent on effective energy transfer of matrix to analyte, high quantities of competing molecules such as pinacol can interfere with ionization. Therefore, on plate water washes were performed in an effort to remove excess pinacol and reduce any possible ionization interference; again no (M+H)+ ions were detected. We suspected that matrix incompatibility was responsible for ineffective ionization of the molecular ion. Therefore, alternative matrices were surveyed to check for compatibility with the esterification protocol. As HPA is traditionally utilized for DNA detection, it was not unexpected that this matrix did not provide the desired molecular ion peak (entry 4). Although the (M+H)+ ion was not observed for DABP and CMBT, we noticed (M+H)+ minus FITC fragments with excellent relative abundance (entries 5–6). Decreasing the laser intensity to minimize fragmentation of the sensitive FITC moiety did not produce the intact molecular ion. Conversely, CHIQ and DHAP were found to be more efficient at ionization and detection of the (M+H)+ ion without cleaving the FITC label, although 100% relative abundance was not always observed (entries 7–8). Interestingly, an unusual peak at m/z 2163.0 appeared with high intensity and percent abundance when DHB was utilized as matrix (entry 9). This peak was 36.8 Da heavier than the expected molecular ion of the pinacol derivatized peptide and upon closer inspection was determined to correspond to the DHB-boronic acid adduct. As boronic acids have been shown to interact strongly with α-hydroxycarboxylates, this result is not entirely surprising.34 The detection of DHB/boronic acid adducts was found to be reproducible over many experiments and suggested that the pinacol moiety was being exchanged with the matrix. Curiously, although DHB and HPA both contain α-hydroxycarboxylate moities, no HPA modification was observed for any peptide sample.
Table 1.
Optimization and screening of matrix.a
| Entry | Additive | Incubation Time (min) | Matrix | Signal m/z (% relative abundance) |
|---|---|---|---|---|
| 1 | none | - | CHCA | 1304.9 (100), 1342.8 (74.4), 2200.1 (28.9) |
| 2 | 0.1 M pinacol | 5 | CHCA | 1025.9 (21.4), 1028.9 (24.0), 1174.0 (27.4), 1304.8 (77.7), 1342.8 (100) |
| 3 | 0.1 M pinacol | 30 | CHCA | 1304.9 (97.6), 1342.9 (100) |
| 4 | 0.1 M pinacol | 30 | HPA | No Signal |
| 5 | 0.1 M pinacol | 30 | DABP | 1304.8 (100), 1736.2 (86.3),b 2093.3 (48.8) |
| 6 | 0.1 M pinacol | 30 | CMBT | 1304.7 (100), 1736.0 (84.1)b |
| 7 | 0.1 M pinacol | 30 | CHIQ | 2126.2 (100)c |
| 8 | 0.1 M pinacol | 30 | DHAP | 1304.8 (27.0), 2126.2 (100)c |
| 9 a | 0.1 M pinacol | 30 | DHB | 1304.7 (100), 2163.0 (79.2)d |
Sample utilized was FLBP1 [FITC-(FBPARW)(FRW)*VRD]; FBPA = 4-borono-L-phenylalanine;
branching lysine. All samples were subjected to a 10 second on plate water wash before analysis. Signals < 20% abundance not reported.
[(M+H)+ - FITC].
(M+H)+.
(M+DHB+H)+
To investigate whether transesterification with DHB was indeed occurring under these conditions, we performed an analysis of the FLBP1 derivatized peptide with pinacol, ethylene glycol, and glycerol (Table 2). First, we confirmed the formation of all 3 esters with the corresponding diols with CHIQ as matrix (entries 1, 3, and 5). As expected, the (M+H)+ ion along with (M+H)+ minus FITC fragments were observed. To test whether the three boronic ester adducts could be exchanged with DHB during the spotting and crystallization process, aliquots of these esters were then spotted using DHB as matrix followed by MALDI-MS analysis. In every case, the boronic ester samples were detected as the DHB ester regardless of the starting diol (entries 2, 4, and 6). These data provided evidence to support our hypothesis that an in situ transesterification reaction was occurring. Moreover, when DHB was incorporated as the matrix, the aforementioned transesterification products were consistently detected in >95% excess to the original pinacol esters (Figure 4). The success of this procedure provided the impetus to test a possible direct esterification with DHB from a boronic acid sample. Gratifyingly, when FLBP1 was spotted, crystallized and shot on the MALDI instrument, the [FLBP1 DHB + H]+ ion was detected in excellent relative abundance (entry 7). This result is valuable because this method allowed the detection of peptide boronic acids without a “pretreatment” step with a 1,2-diol. Hence, further MALDI experiments were performed using the direct method.
Table 2.
Transesterification of 1,2-diols with DHB.a
| Entry | Diol | MALDI Matrix | Predicted Peak ([(M+diol+H)+- FITC]/(M+diol+H)+) | Signal m/z (% relative abundance) |
|---|---|---|---|---|
| 1 | Ethylene glycol | CHIQ | 1680.0/2069.0 | 1304.4 (100), 1680.0 (28.0), 2068.7 (42.5) |
| 2 | Ethylene glycol | DHB | 1771.9/2161.0 | 1304.6 (100), 1771.8 (48.0), 1813.7 (20.4), 2161.8 (99.2) |
| 3 | Glycerol | CHIQ | 1710.0/2099.0 | 1304.7 (100), 1710.0 (42.3), 2099.0 (58.0) |
| 4 | Glycerol | DHB | 1771.9/2161.0 | 1304.7 (100), 1772.0 (88.8), 2162.0 (89.7) |
| 5 | Pinacol | CHIQ | 1736.0/2125.1 | 1304.8 (85.9), 1736.0 (100), 2093.1 (33.0), 2125.1 (45.6) |
| 6 | Pinacol | DHB | 1771.9/2161.0 | 1304.1 (68.7), 1771.8 (45.2), 2161.9 (100) |
| 7 | - | DHB | 1771.9/2161.0 | 1304.4 (49.9), 1771.6 (34.8), 2162.5 (100) |
Sample utilized was FLBP1. All diols were prepared as 0.1 M solutions, mixed with FLBP1 in a 1:1 (v:v) ratio, and allowed to incubate for 30 min. All samples were subjected to a 10 second on plate water wash before analysis. Signals < 20% abundance not reported.
Figure 4.

Displacement of pinacol protected peptide FLBP1 by matrix to yield the DHB protected peptide.
With the optimal protocol in hand, we investigated the robustness of the method with linear as well as branched peptides containing single and multiple boronic acid residues (Table 3). As the boronic acid moiety was introduced onto peptides as an arylboronic acid (i.e. BPA) in prior samples, it was unclear whether this protocol was sensitive to other boronic acid types. Therefore, several linear peptides conjugated to alkylboronic acids were synthesized and analyzed using DHB (entries 1–3). To our delight, all three peptides were readily ionized and detected as the DHB adduct with 100% relative abundance, suggesting that the method is compatible with both alkyl- and arylboronic acids. We then increased the number of boronic acid moieties to two with a more complicated HPLC-purified peptide BP1. We also successfully detected this sample in 100% abundance as the DHB ester (entry 4). To further test the robustness of the procedure, peptides released from a single Tentagel macrobead (140–170 μm) by photocleavage at 360 nm were analyzed (entries 5–6).29 Indeed, these would be the type of samples that will be obtained from screening libraries of branched peptide boronic acids. Unknowns derived from our biochemical assay may contain multiple boronic acid residues in different positions. Applicability of this protocol to libraries containing multiple boronic acid residues requires the effective identification and sequencing of peptides with a high density of boronic acid groups. Thus, a peptide with five BPA units was synthesized, photocleaved, and successfully detected from a single bead (entry 7). This data suggests that in situ DHB transesterification is compatible with high density boronic acid functionalized peptides, and as such should be compatible with peptide library deconvolution. To the best of our knowledge, this is the first example of the utilization of MALDI-MS for the detection of polyboronic acid peptides as DHB esters.
Table 3.
Detection of DHB ester of peptide boronic acids.
| Entry | Sample | Sequence a | Exact Mass of DHB adduct (g/mol) | Signal m/z (% relative abundance) |
|---|---|---|---|---|
| 1 | LP1 |

|
812.3 | 776.6 (52.4), 790.6 (34.6), 811.5 (30.5), 812.5 (100), 826.5 (68.2) |
| 2 | LP2 |

|
826.4 | 826.2 (100), 840.8 (29.7) |
| 3 | LP3 |
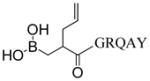
|
852.4 | 408.2 (48.7), 692.4 (73.5), 816.5 (33.2), 851.5 (38.3), 852.4 (100), 866.4 (45.8) |
| 4 | BP1b | (FBPARW)2* HAL | 1769.8 | 1656.1 (12.4), 1769.2 (53.2), 1770.2 (100) |
| 5 | BP2c | (FBPARA)2* VRD | 1588.8 | 1055.1 (74.2), 1322.2 (100), 1589.3 (55.5) |
| 6 | BP3c | (FBPARA)2* HRF | 1658.8 | 953.8 (41.4), 966.8 (55.9), 1066.8 (100), 1124.8 (68.2), 1391.9 (94.5), 1513.0 (36.3), 1659.0 (63.3) |
| 7 | BP4c | (FBPAFBPANW)2* HAFBPAL | 2613.0 | 1647.2 (54.3), 1793.2 (93.4), 2093.4 (30.7), 2305.5 (16.5), 2467.6 (20.1), 2613.6 (25.7) |
FBPA = 4-borono-L-phenylalanine;
branching lysine.
Sample was purified by HPLC.
Analysis of a small aliquot of unpurified peptide sample obtained from one Tentagel macrobead (theoretical yield < 100 pmol).
During the coupling, acidic removal of protecting groups and photocleavage of peptides from the resin, it is important that the boronic acid functionality remain intact, and evidence that boron is present in the peptide is critical. Naturally occurring boron atoms have an isotopic distribution of 1:4 10B:11B. This differential in atomic mass can be a basis for determining whether a boron atom is present in our peptides.18 As demonstrated in Figure 5, the MS spectrum for peptide (RRW)2*HAL was missing the lower molecular weight isotopic peak corresponding to the 10B isotope because this peptides does not contain a boron atom. These signals were clearly present in BP1, whether BP1 was detected as the pinacol or DHB adduct. The data suggests that the boronic acid moiety is stable to the conditions of the synthesis and MALDI characterization.
Figure 5.

Comparison of 10B isotope distribution of A) arginine analogue [(RRW)2*HAL/MW: 1462.9)], B) pinacol ester of peptide BP1 [(FBPARW)2*HAL/MW: 1697.8)], and C) DHB ester of peptide BP1 [(FBPARW)2*HAL/MW: 1769.8)]
MS-MS sequencing of branched peptide boronic acids
To date, sequencing of peptide boronic acids using MS remains challenging. The current method will be valuable, in particular, if sequencing of unknown peptides can be achieved. To determine whether branched peptide boronic acids can be sequenced de novo, BP4 containing five boronic acids was investigated. MS-MS fragmentation was induced on samples prepared using both techniques: a pre-derivatization with pinacol and in situ adduct formation with DHB. The fragmentation patterns observed for both protocols were similar (data not shown). Hence the major advantage of the direct method is the ease and the elimination of a pre-derivatization step with pinacol. A representative MS-MS spectrum observed is shown in Figure 6 (Table 4). Major fragments corresponding to y-ions y7–y5 as well as b-ions b5-b7 were detected in high abundance. Also, minor fragments corresponding to y4, b3, and b4 were detected in low abundance.35 As expected, fragments encompassing the boronic acid residues were detected as DHB adducts. No signal was observed for fragmentation corresponding to the outermost amide bonds y8 and b8 regardless of laser power. Although it is not clear why the y8 and b8 ions were not observed, the suppression or lack of signal is not expected to be as a result of the boronic acid functionality because fragmentation resulting in y7 ion is observed. For example, fragmentation to provide the b8 ion has been observed in other sequencing efforts in our group.35 As our branched peptide boronic acid libraries are synthesized by split and pool techniques and each amino acid position is restricted to four possible variants,29 the information from this experiment was sufficient to unambiguously assign a sequence from a theoretical library of 65,536 members.
Figure 6.

MS/MS of DHB modified peptide BP4.
Table 4.
MS/MS of DHB modified peptide BP4.
| Fragment | Fragment Position | Predicted (M+H)+m/z | Observed (M+H)+m/z |
|---|---|---|---|
|
|
b8 | 2482.8 | Not Observed |
|
|
b7 | 2173.8 | 2173.2 |
|
|
b6 | 2102.7 | 2102.1 |
|
b5 | 1965.7 | 1965.0 |
|
|
y5 | 1694.7 | 1694.1 |
|
|
y6 | 1880.8 | 1880.1 |
|
|
y7 | 1994.8 | 1994.2 |
|
|
y8 | 2303.9 | Not Observed |
CONCLUSIONS
We have developed a simple, rapid and robust on-plate derivatization method for MALDI-MS analysis of linear, branched and FITC labeled peptide boronic acids by using DHB as both the derivatizing agent and matrix. We also demonstrated that trapping the boronic acid moiety as pinacol, ethylene glycol and glycerol adduct is effective for MALDI-MS analysis of peptide boronic acids. However, the major advantage of using DHB is the removal of a laborious, time-consuming and uneconomical derivatization step. When 1,2-diols are used for derivatization, removal of excess reagents by a short water wash is necessary to promote effective ionization and subsequent analysis of the resulting spectrum. Under these circumstances, DABP, CMBT, CHIQ, and DHAP can be used as matrices. Indeed, we discovered that DHB appears universally compatible to the boronic acid moiety, as arylboronic acids (such as 4-borono-Phe) and alkylboronic acids were readily detected. Furthermore, the current method allows the identification and sequencing of peptides containing high density boronic acid groups released from single beads. We expect that the current method will be highly valuable not only in the identification of simple boronic acids but also in the deconvolution of peptide boronic acid libraries used in high throughput screening assays.
Supplementary Material
Acknowledgments
We gratefully acknowledge the Virginia Tech mass spectrometry incubator for the use of MALDI-TOF instrumentation, Dr. Keith Ray for his invaluable support and guidance, and Ken Knott for the synthesis and purification of linear N-terminal boronic acid peptides LP1, LP2, and LP3. This project was supported in part by the department of chemistry at VT, Institute for Critical Technology and Applied Science (ICTAS) and NIH (RO1 GM093834).
Footnotes
SUPPORTING INFORMATION. MALDI-MS spectra of all experiments. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Adams J, Behnke M, Chen S, Cruickshank AA, Dick LR, Grenier L, Klunder JM, Ma YT, Plamondon L, Stein RL. Bioorg Med Chem Lett. 1998;8:333. doi: 10.1016/s0960-894x(98)00029-8. [DOI] [PubMed] [Google Scholar]
- 2.Tsubuki S, Kawasaki H, Saito Y, Miyashita N, Inomata M, Kawashima S. Biochem Biophys Res Commun. 1993;196:1195. doi: 10.1006/bbrc.1993.2378. [DOI] [PubMed] [Google Scholar]
- 3.Prusoff W, Lin TS, Pivazyan A, Sun AS, Birks E. Pharmacol Ther. 1993;60:315. doi: 10.1016/0163-7258(93)90013-4. [DOI] [PubMed] [Google Scholar]
- 4.Pivazyan AD, Matteson DS, Fabry-Asztalos L, Singh RP, Lin PF, Blair W, Guo K, Robinson B, Prusoff WH. Biochem Pharmacol. 2000;60:927. doi: 10.1016/s0006-2952(00)00432-9. [DOI] [PubMed] [Google Scholar]
- 5.Wienand A, Ehrhardt C, Metternich R, Tapparelli C. Bioorg Med Chem. 1999;7:1295. doi: 10.1016/s0968-0896(99)00069-3. [DOI] [PubMed] [Google Scholar]
- 6.Fevig JM, Abelman MM, Brittelli DR, Kettner CA, Knabb RM, Weber PC. Bioorg Med Chem Lett. 1996;6:295. [Google Scholar]
- 7.Knott K, Fishovitz J, Thorpe SB, Lee I, Santos WL. Org Biomol Chem. 2010;8:3451. doi: 10.1039/c004247a. [DOI] [PubMed] [Google Scholar]
- 8.Yan J, Fang H, Wang B. Med Res Rev. 2005;25:490. doi: 10.1002/med.20038. [DOI] [PubMed] [Google Scholar]
- 9.Lis H, Sharon N. Chem Rev. 1998;98:637. doi: 10.1021/cr940413g. [DOI] [PubMed] [Google Scholar]
- 10.Weis WI, Taylor ME, Drickamer K. Immunol Rev. 1998;163:19. doi: 10.1111/j.1600-065x.1998.tb01185.x. [DOI] [PubMed] [Google Scholar]
- 11.Sinowatz F, Topfer-Petersen E, Calvete JJ. Glycosciences. 1997;595 [Google Scholar]
- 12.Clark GF, Oehninger S, Patankar MS, Koistinen R, Dell A, Morris HR, Koistinen H, Seppala M. Hum Reprod. 1996;11:467. doi: 10.1093/humrep/11.3.467. [DOI] [PubMed] [Google Scholar]
- 13.Fukumori T, Takenaka Y, Yoshii T, Kim HR, Hogan V, Inohara H, Kagawa S, Raz A. Cancer Res. 2003;63:8302. [PubMed] [Google Scholar]
- 14.Arimori S, Bell ML, Oh CS, Frimat KA, James TD. Chem Commun (Camb) 2001:1836. [PubMed] [Google Scholar]
- 15.Hoeg-Jensen T, Ridderberg S, Havelund S, Schaffer L, Balschmidt P, Jonassen I, Vedso P, Olesen PH, Markussen J. J Pept Sci. 2005;11:339. doi: 10.1002/psc.624. [DOI] [PubMed] [Google Scholar]
- 16.Zou Y, Broughton DL, Bicker KL, Thompson PR, Lavigne JJ. Chembiochem. 2007;8:2048. doi: 10.1002/cbic.200700221. [DOI] [PubMed] [Google Scholar]
- 17.Longstaff C, Rose ME. Org Mass Spectrom. 1982;17:508. [Google Scholar]
- 18.Haas M, Blom K, Schwartz C. Anal Chem. 1999;71:1574. [Google Scholar]
- 19.Snow RJ, Bachovchin WW, Barton RW, Campbell SJ, Coutts SJ, Freeman DM, Gutheil WG, Kelly TA, Kennedy CA, Krolikowski DA, Leonard SF, Pargellis CA, Tong L, Adams J. Journal of the American Chemical Society. 1994;116:10860. [Google Scholar]
- 20.Kettner C, Mersinger L, Knabb R. J Biol Chem. 1990;265:18289. [PubMed] [Google Scholar]
- 21.Leitner A, Amon S, Rizzi A, Lindner W. Rapid Commun Mass Spectrom. 2007;21:1321. doi: 10.1002/rcm.2967. [DOI] [PubMed] [Google Scholar]
- 22.Lin N, Yan J, Huang Z, Altier C, Li M, Carrasco N, Suyemoto M, Johnston L, Wang S, Wang Q, Fang H, Caton-Williams J, Wang B. Nucleic Acids Res. 2007;35:1222. doi: 10.1093/nar/gkl1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.LeBeau AM, Singh P, Isaacs JT, Denmeade SR. Chem Biol. 2008;15:665. doi: 10.1016/j.chembiol.2008.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sasubilli R, Gutheil WG. J Comb Chem. 2004;6:911. doi: 10.1021/cc049912d. [DOI] [PubMed] [Google Scholar]
- 25.Manku S, Hall D. Aust J Chem. 2007;60:824. [Google Scholar]
- 26.Duggan P, Offerman D. Aust J Chem. 2007;60:829. [Google Scholar]
- 27.Singhawangcha S, Hu LEC, Poole CF, Zlatkis A. J High Resolut Chromatogr Commun. 1978;1:304. [Google Scholar]
- 28.Rose ME, Longstaff C, Dean PDG. J Chromatogr. 1982;249:174. [Google Scholar]
- 29.Bryson DI, Zhang W, Ray WK, Santos WL. Mol Biosyst. 2009;5:1070. doi: 10.1039/b904304g. [DOI] [PubMed] [Google Scholar]
- 30.Kaiser E, Colescott RL, Bossinger CD, Cook PI. Anal Biochem. 1970;34:595. doi: 10.1016/0003-2697(70)90146-6. [DOI] [PubMed] [Google Scholar]
- 31.Malan C, Morin C. J Org Chem. 1998;63 [Google Scholar]
- 32.Details are given in Table 1.
- 33.Using CHCA as a matrix tends to be unpredicatable. For example, depending on the peptide in question 0.1M or 1.0 M pinacol may provide the correct molecular ion peak.
- 34.Gray CW, Jr, Houston TA. J Org Chem. 2002;67:5426. doi: 10.1021/jo025876y. [DOI] [PubMed] [Google Scholar]
- 35.See supplementail information for details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.