

Abstract
Background
Autoimmune mechanisms, particularly through generation of auto-antibodies, may contribute to the pathophysiology of idiopathic dilated cardiomyopathy (iDCM). The precise role of cellular autoimmune responses to cardiac-specific antigens has not been well described in humans. The purpose of this study was to characterize the cellular autoimmune response to cardiac troponin I (cTnI), specifically, the release of cytokines by peripheral blood mononuclear cells (PBMCs), in subjects with iDCM and healthy controls.
Methods and Results
We performed enzyme-linked immunospot (ELISpot) assays on PBMCs isolated from subjects with iDCM and healthy controls to examine the ex vivo interferon-gamma (IFN-γ) and interleukin-10 (IL-10) production in response to cTnI exposure. Thirty-five consecutive subjects with iDCM (mean age 53 ±11 years, 60% male, LVEF 23 ±7%) and 26 controls (mean age 46 ±13 years, 46% male) were prospectively enrolled. IFNγ production in response to cTnI did not differ between the groups (26 ±49 versus 38 ±53 number of secreting cells, respectively, p = 0.1). In contrast, subjects with iDCM showed significantly higher IL-10 responses to cTnI compared with controls (386 ±428 versus 152 ±162 number of secreting cells, respectively, p<0.05). Among iDCM subjects, heightened IL-10 response to cTnI was associated with reduced systemic inflammation and lower prevalence of advanced diastolic dysfunction when compared to those with normal IL-10 response to cTnI.
Conclusions
Our preliminary findings suggest that a heightened cellular autoimmune IL-10 response to cTnI is detectable in a subset of patients with iDCM, which may be associated with reduced systemic levels of hsCRP and lower prevalence of advanced diastolic dysfunction.
Introduction
Despite the lack of definitive etiology for a large majority of patients with idiopathic dilated cardiomyopathy (iDCM), the presence of an inflammatory state is evident(1). Breakdown in self-tolerance is a typical characteristic of autoimmune diseases; hence, detectable responses to cardiac-specific antigens are supportive of an ongoing autoimmune process in patients with iDCM. In most cases, the generation of cardiotoxic autoantibodies is proposed as the primary pathogenic mechanism(2-6). However, cellular autoimmunity, particularly specific to cardiac antigens, is less well described. There is indirect evidence of cell-mediated immunity on histologic examination of endomyocardial biopsies from patients with iDCM, which consistently demonstrate CD4+ and CD8+ infiltrates(7). Pronounced T1-helper cell activation has also been observed in patients with both ischemic and idiopathic cardiomyopathies in proportion to disease severity(8).
In a murine model of viral myocarditis, a latent phase associated with infiltrating immune T lymphocytes and subsequent cell-mediated response plays pivotal roles in the ongoing destruction of cells(9), particularly in response to cardiac troponin I (cTnI)(3). Detection of subclinical levels of circulating cTnI in the setting of heart failure is not uncommon(10). As evidence for such cardiac-specific cellular autoimmunity response is lacking in the human failing heart, the objective of this study is to characterize the cellular autoimmune responses to cTnI by detecting specific release of cytokines (interferon-gamma [IFN-γ] as pro-inflammatory; interleukin-10 [IL-10] as anti-inflammatory) ex vivo by peripheral blood mononuclear cells (PBMCs) isolated from subjects with iDCM and healthy controls. We hypothesized that iDCM patients may exhibit a greater propensity to generate a pro-inflammatory cytokine response to cTnI than healthy controls.
Methods
Study population
Thirty-five consecutive subjects with iDCM and 26 controls subjects were prospectively enrolled after informed consent was obtained. Idiopathic dilated cardiomyopathy was defined as a clinical diagnosis of heart failure secondary to left ventricular dilation and systolic dysfunction for at least 6 months in the absence of congenital, valvular or coronary artery disease, or any systemic disease or toxins known to cause cardiomyopathy. Significant coronary artery disease was excluded in subjects with iDCM by coronary angiography within 5 years of enrollment. At the time of entry into the study, all subjects with iDCM were receiving standard medical therapies according to guideline recommendations and were clinically stable (without change in dose or medication regimen) for at least 3 months. Functional capacity was determined by self-reported New York Heart Association (NYHA) functional class as assessed by a physician. Healthy controls were identified by advertisements followed by careful medical history and physical examination including a screening echocardiography by a cardiologist (W.T.) to rule out any evidence of cardiac disease. Exclusion criteria for the entire study population included current pregnancy or pregnancy within the year preceding enrollment, use of immunomodulatory medications within the 6 month period preceding enrollment, or any condition resulting in an immunocompromised state.
Within the iDCM cohort, we reviewed transthoracic echocardiograms performed within 3 months of enrollment, and performed offline measurements by a single experienced research sonographer (A.B.) of cardiac structure and function blinded to the assay measurements, including mitral Doppler inflow patterns. Overall severity of diastolic function was assessed based on integrated assessment of all available echocardiographic parameters at the time of evaluation.
High-sensitivity C-reactive protein (hsCRP) was measured in EDTA-plasma samples using the Abbott Architect ci8200 platform (Abbott Laboratories, Abbott Park IL). All blood samples were drawn at the same time-point for each subject. Informed consent was obtained from each individual and the study was approved by the Institutional Review Board at the Cleveland Clinic.
Cell Isolation and Preparation
PBMCs were isolated from whole blood utilizing Ficoll density gradient centrifugation, counted, washed and resuspended in complete media (RPMI-1640 [Cleveland Clinic, Central Services Media Lab] with 10% newborn bovine serum [Life Technologies, Gaithersburg, MD], 100 U/ml penicillin/streptomycin, 2mM L-glutamine, 25mM HEPES) containing 10% DMSO at a concentration of 10×106 cells/mL. PBMCs were then frozen in 1mL aliquots at a controlled rate of -1°C/min until -80°C and later transferred to a liquid nitrogen freezer until analysis.
Enzyme-linked immunospot (ELISpot) protocol
Thawed PBMCs suspensions were immediately diluted 10-fold with complete media containing 20U/ml DnaseI (Sigma-Aldrich, St. Louis, MO). After washing twice & resuspending in DNaseI-media, the cells were placed in 37°C for post-thaw recovery. After 60 minutes, cells were then washed, resuspended in complete media, and counted prior to ELISpot culture. The ELISpot protocol used in this study has been described in detail previously(11). Briefly, multiscreen plates (Millipore, Billerica, MA) were coated with cytokine capture monoclonal antibody overnight at 4°C, either with anti-IL10 or anti-IFNγ (BD Biosciences, San Diego, CA). After washing & blocking plates (1%BSA [fraction V; Sigma-Aldrich, St. Louis, MO] in PBS), PBMCs were cultured for 48 hours in 200μl of either media, tetanus toxoid (1:100; EMD Chemicals Inc, Gibbstown, NJ), anti-human CD3 (5μg/ml; (BD Biosciences, San Diego, CA), or recombinant human cTnI (50ug/ml; Life Diagnostics, Inc, West Chester PA). All antigens/controls were tested in triplicate. Cells were cultured at 200,000 - 300,000/well with results normalized to the number of cytokine secreting cells/300,000 cells. Complete culture media consisted of RPMI-1640 supplemented with 100 IU/ml penicillin/streptomycin, 2mM L-glutamine, 25mM HEPES, and 10% newborn bovine serum. After the plates were incubated for 24 hours and then washed (with PBS followed by PBS/0.05%Tween), secondary biotinylated anti-cytokine antibody was added at 100ul/well and incubated overnight. Plates were then washed with PBS/Tween-20 (Fisher, Fairlawn, NJ), and streptavidin-HRP (DAKO, Carpenteria, CA) was added for 2 hours at room temperature. Plates were washed in PBS and spot color was developed by adding amino-ethyl-carbazole (AEC, Sigma-Aldrich, St. Louis, MO) substrate diluted in acetate buffer containing 30% H2O2. Plates were then washed with diH2O to stop the reaction. After drying, images of the wells were acquired and spots enumerated by ImmunoSpot Series 1 ELISpot analyzer (Cellular Technology, Cleveland, OH). Digital images were analyzed for the presence of areas in which the color density exceeds the background by a factor calculated from deducting the actual color from the background value. For each subject, the individual background value (mean spot number in media wells, +3 standard deviations) was subtracted from the mean spot number obtained in the presence of each stimulating agent. Positive responses were defined as wells giving responses greater than the mean +3 standard deviations of un-stimulated wells.
Statistical Methods
Categorical variables were summarized as proportions and frequencies. Continuous variables were summarized as mean ± standard deviation if normally distributed, or median and inter-quartile range (IQR) if non-normally distributed. Normality was assessed by the Shapiro–Wilk W-test. The Wilcoxon Rank test was used to compare the cytokine responses between patients with iDCM and controls. The Spearman rank coefficient was used to measure associations between the cytokine production and continuous clinical variables. All p-values reported are from two-sided tests and a p-value <0.05 was considered statistically significant. The statistical analyses were performed using JMP 5.1, SAS 9.1.3 (SAS Institute, Cary, NC, USA).
Results
Subject Characteristics
Baseline characteristics of the iDCM and control groups are presented in Table 1. The self-reported mean duration of disease for the iDCM group was 4.9±4.8 years, with 60% experiencing symptoms for over two years. As expected, the iDCM group was older, more likely to be receiving cardiac medication, and demonstrated higher circulating levels of hsCRP than that of healthy controls. Of note, within the iDCM group about 60% of subjects were already treated on statin therapy. The majority of the iDCM group had mild heart failure symptoms (74% NYHA class 1 or II).
Table 1.
Clinical characteristics of patients with idiopathic dilated cardiomyopathy compared with control group
| Control subjects (n = 26) | Patients with idiopathic dilated cardiomyopathy (n = 35) | |||
|---|---|---|---|---|
| Total | Normal IL-10 response (n = 22) | Heightened IL-10 response (n =13) | ||
| Age (years) | 46 ±13 | 53 ± 11 * | 51 ±13 | 57 ± 7 |
| Male (%) | 46 | 60 | 63 | 54 |
| Hypertension (%) | 31 | 37 | 45 | 23 |
| Diabetes Mellitus (%) | 0 | 22 * | 27 | 15 |
| Dyslipidemia (%) | 31 | 46 | 41 | 54 |
| Atrial fibrillation (%) | 0 | 34 * | 27 | 46 |
| History of smoking (%) | 8 | 11 | 13 | 8 |
| Systolic blood pressure (mm Hg) | 115 ± 11 | 111 ± 19 | 117 ± 18 | 101 ± 16 † |
| BMI | 27 ± 5 | 37 ± 33 * | 41 ± 42 | 29 ± 3 |
| NYHA Class (I-II/III, %) | N/A | 74/26 | 72/28 | 77/23 |
| CRT/ICD (%) | N/A | 54 | 55 | 50 |
| Serum Creatinine (mg/dL) | 0.8 ± 0.2 | 1.0 ± 0.26* | 0.9 ± 0.2 | 1.0 ± 0.3 |
| hsCRP (mg/L) | 1.8 ± 1.8 | 6.0 ± 8.9* | 8.3 ± 10.5 | 2.0 ± 2.0 † |
| ACEI/ARB (%) | 19 | 86* | 81 | 92 |
| B-blocker (%) | 11 | 89* | 91 | 92 |
| Aldosterone inhibitor (%) | 4 | 37* | 32 | 46 |
| Statin (%) | 15 | 60* | 64 | 54 |
| Diuretic (%) | 4 | 60* | 52 | 80 |
Abbreviations: IL-10 – interleukin 10, BMI – body mass index, NYHA –New York Heart Association, CRT – Cardiac resynchronization therapy, ICD – Implantable cardiac defibrillator, hsCRP – high sensitivity C reactive protein, ACEI – angiotensin converting enzyme inhibitor, ARB – angiotensin receptor blocker, B-blocker – beta blocker.
p value < 0.05 when comparing control subjects and overall patient group.
p value < 0.05 when comparing normal and heightened response groups in patients with iDCM.
Cytokine Response to cTnI between iDCM and Control
Our first goal was to determine if cytokine responses to cTnI differ between iDCM and controls. In our study cohort, we observed similar levels of IFN-γ production by PBMCs in response to cTnI between iDCM and healthy controls (26 ± 49 vs. 38 ± 53 number of secreting cells, p = 0.1, Figure 1). However, we observed significantly higher IL-10 production by PBMCs in response to cTnI compared to control group (386 ± 428 vs. 152 ± 162 number of secreting cells, p<0.05, Figure 1). Correspondingly, the ratios of IL-10/ IFN-γ responses to cTnI were also higher in iDCM than in healthy controls (140 ± 259 vs 12 ± 26, p<0.05, Figure 1). Interestingly, there was no correlation between IL-10 production and IFN-γ production in the iDCM group (r = 0.32, p = 0.06). To confirm the validity and the antigen-specific nature of the assay results, there were detectable cytokine responses but no differences between groups in response to other control antigens or media.
Figure 1. Cytokine Response to Cardiac Troponin I in Idiopathic Dilated Cardiomyopathy and Healthy Controls.
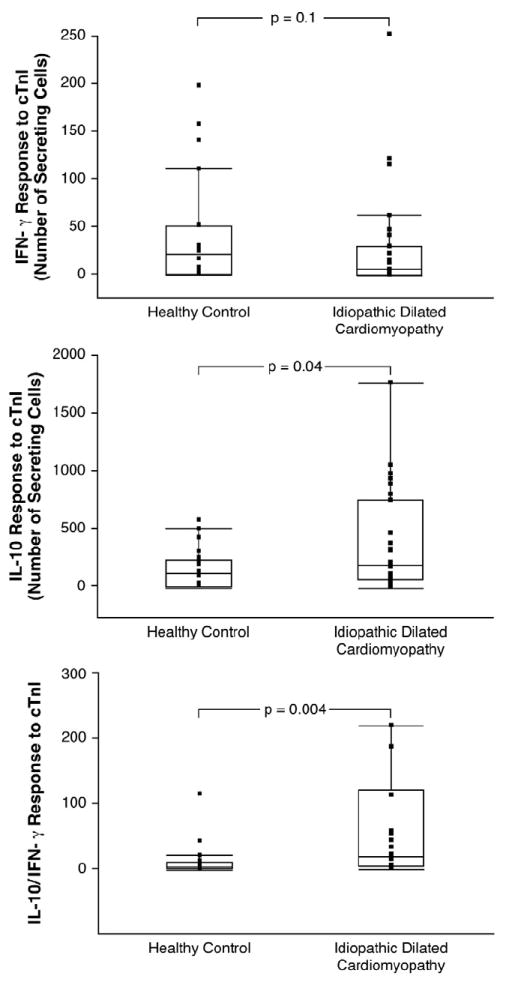
Interferon-gamma (IFN-γ, upper panel), interleukin-10 (IL-10, middle panel), and ratio of IL-10 to IFN-γ (lower panel) production by peripheral blood mononuclear cells (PBMCs) in response to cardiac troponin I in patients with idiopathic dilated cardiomyopathy compared with controls.
Interleukin-10 Responses to cTnI and Clinical Phenotype
We then focused on characterizing the clinical phenotype of IL-10 response to cTnI, particularly in the iDCM cohort to gain insights into underlying anti-inflammatory autoimmune mechanisms of iDCM. We arbitrarily stratified the iDCM cohort according to their IL-10 response to cTnI as “heightened” (≥99th percentile of healthy controls) or “normal” (<99th percentile of healthy controls). Patients with higher IL-10 responses to cTnI did not differ in baseline characteristics or medication use except for a lower systemic blood pressure as shown in Table 1. In particular, there were no differences in age and gender between these two subsets. There was no difference in functional capacity as determined by NYHA class, p = 0.2. We found those with heightened IL-10 responses to cTnI demonstrated significantly lower hsCRP levels than those with normal responses (2.1 ± 2.1 vs. 8.4 ±10.5 mg/L, p = 0.04, Figure 2). Furthermore, patients with heightened IL-10 response demonstrated less advanced diastolic dysfunction (Figure 3 and Tables 2a and 2b). These observations were similar when IL-10/IFN-γ was considered (Table 2b). Similar findings were noted if the iDCM cohort was stratified according to median IL-10 responses. However, those with higher IFN-γ responses to cTnI did not demonstrate higher hsCRP levels compared to those with lower IFN-γ responses to cTnI, even though they correlated with worsening parameters of diastolic dysfunction (Table 2b).
Figure 2. Interleukin-10 response to Cardiac Troponin I and Systemic Inflammation.
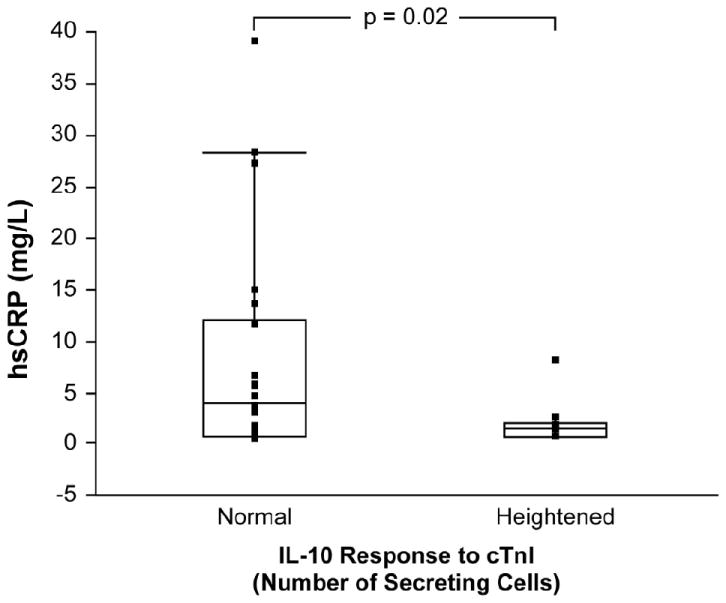
High-sensitivity C-reactive protein (hsCRP) measurements in patients with interleukin-10 (IL-10) response to cardiac troponin I (cTnI) ≥99th percentile of values of control group compared to those with IL-10 response <99th percentile.
Figure 3. Interleukin-10 response to Cardiac Troponin I and Advanced Diastolic dysfunction.
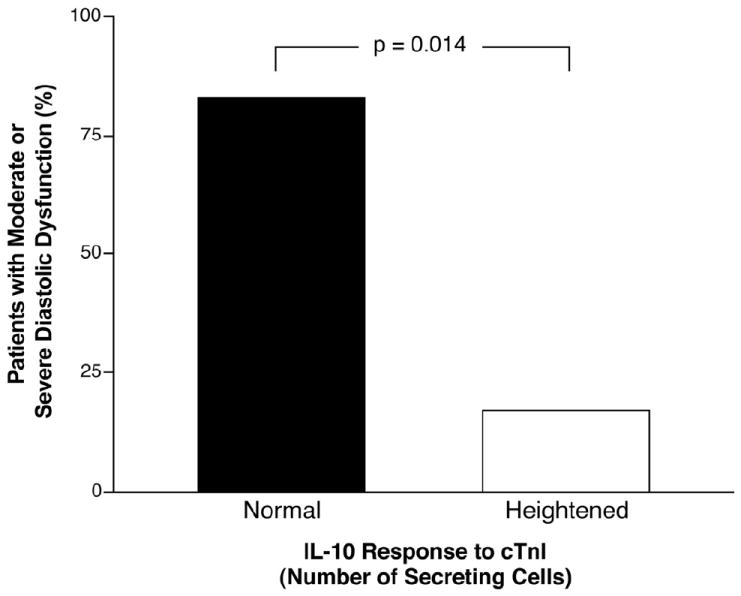
Prevalence of moderate (Stage II) or severe (Stage III) diastolic dysfunction in patients with interleukin-10 (IL-10) response to cardiac troponin I (cTnI) ≥99th percentile of values of control group compared to those with IL-10 response <99th percentile.
Table 2.
a. Echocardiographic characteristics with cytokine response to cardiac troponin I in patients with idiopathic dilated cardiomyopathy
| Normal IL-10 response (n = 22) | Heightened IL-10 response (n =13) | |
|---|---|---|
| LVEF (%) (n=35) | 23 ± 7 | 23 ± 6 |
| LVEDd (cm) (n=34) | 6.5 ± 1.0 | 6.3 ± 0.9 |
| MV DT (mS) (n=30) | 182 ± 48 | 238 ±74* |
| MV E/A (n=32) | 1.4 ± 0.9 | 0.8 ± 0.3* |
| PV S (cm/s) (n=29) | 46 ± 13 | 48 ± 12 |
| PV S/D (n=29) | 1.2 ± 0.4 | 1.3 ± 0.6 |
| b. Univariable correlation analysis of echocardiographic findings with cytokine response to cardiac troponin I in patients with idiopathic dilated cardiomyopathy | |||
| Correlation with Cytokine Responses to cTnI by ELISpot (Spearman’s r) | |||
| IL-10 response | IFN-γ response | IL-10/IFN-γ response | |
| LVEF (%) | -0.03 | - 0.12 | 0.01 |
| LVEDd (cm) | 0.11 | - 0.03 | 0.06 |
| MV DT (mS) | 0.37* | 0.12 | 0.14 |
| MV E/A | - 0.56* | 0.15 | -0.64* |
| PV S (cm/s) | 0.01 | - 0.56* | 0.55* |
| PV S/D | 0.15 | - 0.54* | 0.61* |
Values are mean ± standard deviation unless otherwise stated.
p value < 0.05 when comparing normal and heightened IL -10 response groups.
Abbreviations: IL-10 – interleukin 10, LVEF – left ventricular ejection fraction, LVEDd – left ventricular end diastolic diameter, MV DT – mitral valve deceleration time, MV E/A – ratio of mitral valve inflow peak E (early diastolic) and A (late diastolic) velocities, PV S– pulmonary venous peak systolic velocity, PV S/D – ratio of pulmonary venous peak systolic (S) velocity to peak anterograde diastolic (D) velocities.
Abbreviations: ELISpot - enzyme-linked immunospot assay, IL-10 – interleukin 10, IFN-γ – interferon-gamma, LVEF – left ventricular ejection fraction, LVEDd – left ventricular end diastolic diameter, MV DT – mitral valve deceleration time, MV E/A – ratio of mitral valve inflow peak E (early diastolic) and A (late diastolic) velocities, PV S– pulmonary venous peak systolic velocity, PV S/D – ratio of pulmonary venous peak systolic (S) velocity to peak anterograde diastolic (D) velocities. cTnI – cardiac troponin I.
p value < 0.05
Discussion
We report for the first time that a subset of patients with iDCM may generate a heightened (but independent) anti-inflammatory cellular autoimmunity response to a cardiac-specific antigen (namely cTnI) in peripheral blood. Furthermore, we have established that such IL-10 production in response to cTnI corresponds to a global reduction in systemic levels of hsCRP and more favorable echocardiographic features. Although we originally hypothesized that subjects with iDCM may likely exhibit a pro-inflammatory cellular autoimmune response towards cTnI, such a potentially pathogenic response was no more likely than that found in healthy individuals. Instead, it is the imbalance of pro- and anti-inflammatory responses against cardiac-specific antigens that dictated the ultimate degrees of systemic inflammation and cardiac phenotype. Taken together, our preliminary data suggest that an endogenous immune tolerance mechanism (in the form of immune regulation as a response to exposure to cardiac antigens) may be operational, and may influence the degree of systemic inflammation as well as myocardial pathology (in the form of diastolic dysfunction). These preliminary findings may support the “autoimmune hypothesis,” which suggests that an imbalance of protective and deleterious autoimmune responses may contribute to downstream myocardial pathology. A larger study is needed with more comprehensive evaluation of protective cellular immune responses to cardiac-specific antigens such as cTnI to confirm these preliminary findings, and strategies to modulate such responses are potentially enticing.
Cardiac troponins, as an intracellular self-antigen that is released in the bloodstream during acute myocardial infarction, myocarditis, or other forms of cardiac damage leading to dysfunction, has been considered a marker (or downstream product) of myocardial necrosis rather than a source of self-antigen (or mediator of disease progression). We have recently reported in stable cardiac patients that the presence of detectable cTnI may identify a substantial group of patients with heightened risk of adverse long-term events, independent of traditional risk factors, hsCRP, and creatinine clearance(12). We further identified that such “subclinical myocardial damage” was associated with a systemic inflammatory and oxidative stress state(12). Hence, circulating cardiac troponins may serve as self-antigens that can induce specific autoimmune responses that can influence disease progression in a manner similar to other immune-mediated diseases (e.g. hepatitis, inflammatory bowel disease, multiple sclerosis). This postulate has recently been supported by several mechanistic studies, the majority describing humoral response to cTnI in the generation of autoantibodies(3-5). A recent human study confirmed that the absence of auto-antibodies against cTnI following acute myocardial infarction was associated with less post-infarct cardiac remodeling. In fact, there is a clear association between duration and degree of exposure to circulating cardiac troponin and development of autoantibodies(5). However, in patients with heart failure, there is still limited understanding of the prevalence of detectable cTnI using the latest high-sensitivity cTnI assays and its relationship with autoimmune responses to cTnI.
The role of cell-mediated immunity in the pathogenesis of iDCM has not been clearly defined to date, but there is evidence to suggest that it plays an important part especially through the production of cytokines. Cytokines have been identified as playing an important pathogenic role in developing heart failure with well-documented elevation of pro-inflammatory cytokines and diminution of anti-inflammatory cytokines in HF associated with both hemodynamic and structural changes in the heart(13-16). Levels of pro-inflammatory cytokines have been associated with functional status and disease severity(17). Furthermore, in a group of patients with iDCM that responded to immunotherapy, there was profound enhancement of IL-10, an important regulatory anti-inflammatory cytokine(18). There has also been evidence of differential levels of IL-10 based on etiology of HF that has been suggested as indirect evidence of autoimmune pathogenesis in iDCM(15). Our data indicated that IFN-γ responses to cTnI were similar between iDCM patients versus healthy controls, but a heightened IL-10 response to cTnI was observed in patients with iDCM. Since IL-10 inhibits the formation of reactive oxygen species and enhances the production of nitric oxide, these findings suggest a potential protective immune pathway induced by exposure to cTnI in patients with iDCM. This paradox was confirmed by our observation that systemic inflammation (hsCRP) was reduced in those demonstrating high IL-10 response to cTnI. It is therefore conceivable that increased IL-10 production may attenuate the inflammatory process that leads to cardiac remodeling and fibrosis, a process that can be manifested as diastolic dysfunction. In fact, these results corroborate results from a recent animal study that found that left ventricular cardiac remodeling related to ischemia and reperfusion can be reduced by intra-nasal administration of troponin peptides leading to modulation of the host cellular immune response with increased IL-10 production and decreased IFN-γ production(19). Tolerance has also been induced in experimental autoimmune myocarditis by intranasal exposure of animals with cardiac myosin(20). Therefore, it is conceivable that a localized rather than a global anti-inflammatory strategy via the induction of protective autoimmune responses towards cardiac-specific antigens via the induction of mucosal immunity may provide cardio-protection from adverse cardiac remodeling.
These preliminary data are by no means definitive, and warrants further investigations and needs to be reproduced. Several important questions still need to be addressed before further therapeutic development in this area can be achieved. For example, it is unclear what cellular phenotypes were responsible for the IL-10 production, and what factors may lead to enhanced IL-10 response to cTnI. Our novel observations imply that the recognition of a self-antigen potentially by adaptive T regulatory cells, often activated by experimental presentation of antigens by oral or nasal routes, can lead to generation of a protective autoimmune response and result in reduced inflammatory consequences. Previous animal studies have suggested T-regulatory cells and mast cells often produce IL-10(21), and thus may be recruited to limit inflammatory injury. Among the known cell types that have been examined in the literature for cardiac dysfunction, TH17 subsets (producing interleukin-17) may also play an important role in modulating inflammatory processes in myocarditis(22-24). We still have a lack of knowledge as to the time course of the development of autoimmune responses to cardiac troponins as well as its balance with humoral autoimmunity. Furthermore, we do not know whether such a cardiac-specific cellular autoimmune response is reproducible in a particular individual, whether it can be modulated with medical therapy, and whether it has important prognostic value in the clinical setting. Such questions will require a larger, confirmatory study involving a variety of subjects with sequential measurements following an index event.
Study Limitations
This is a single-center pilot study, and we acknowledge important limitations regarding the cross-sectional nature of the case-control comparison and our small sample size and the limited number of cytokines tested. Our echocardiographic evaluation relied on studies performed within 3 months of study enrollment thus lacking direct association, and more advanced measurements of diastolic function such as tissue Doppler imaging or strain imaging or adjunctive assessment such as concomitant natriuretic peptide levels were not obtained. Hence, we acknowledge that the qualitative assessment of severity of diastolic dysfunction is complex and may be subject to bias. Although the majority of chronic stable iDCM subjects were well-treated and had mild symptoms, they were likely at different stages of their disease states and therefore may manifest differential autoimmune responses. As the definition of iDCM was a clinical one, endomyocardial biopsy was not performed to confirm the diagnosis, and therefore we cannot rule out the presence of chronic myocarditis which may elicit a different autoimmune response profile. Furthermore, insufficient samples were available to measure natriuretic peptide levels or other cytokine responses, or to analyze with fluorescence activated cell sorting and intracellular staining techniques.
As in many human studies, wide variability of responses can occur due to the complexity of the autoimmune responses, and the presence of cardiac-specific autoimmune responses does not necessarily guarantee a similar and consistent finding in vivo. Although our results are suggestive of an active cell-mediated immune response in iDCM, we still cannot identify whether an autoimmune process is a primary process in the etiology of this condition or a secondary effect after another initiating process. We also do not know precisely what other cytokines may be involved, or whether such cellular responses are simply a reflection of systemic cellular responses to any antigen. Nevertheless, the lack of differences in other common antigens and media served as a control to demonstrate a cardiac-specific nature of our findings in patients with iDCM. The prospects of modulating protective immune mechanisms via vaccination with cardiac-specific antigens for at-risk individuals are enticing, and may warrant further investigations in this area.
Conclusion
There appears to be some indication that some patients with iDCM may demonstrate a release of protective IL-10, potentially via a cellular autoimmune response to circulating cTnI. Our preliminary findings suggested that the presence of such IL-10 response to cTnI is associated with reduced systemic levels of hsCRP and a lower prevalence of advanced diastolic dysfunction.
Acknowledgments
Acknowledgment of grants and other support: This work was supported in part by the National Institutes of Health Clinical and Translational Science Award (CTSA UL1-RR024989) and the Cleveland Clinic Research Programs Committee (RPC) Funding Award.
Footnotes
Disclosures Dr. Tang has previously received research grant support from Abbott Laboratories. Other authors have no conflict of interest to disclose for this study.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lappe JM, Pelfrey CM, Tang WH. Recent insights into the role of autoimmunity in idiopathic dilated cardiomyopathy. J Card Fail. 2008;14:521–30. doi: 10.1016/j.cardfail.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Caforio AL, Mahon NG, Baig MK, et al. Prospective familial assessment in dilated cardiomyopathy: cardiac autoantibodies predict disease development in asymptomatic relatives. Circulation. 2007;115:76–83. doi: 10.1161/CIRCULATIONAHA.106.641472. [DOI] [PubMed] [Google Scholar]
- 3.Goser S, Andrassy M, Buss SJ, et al. Cardiac troponin I but not cardiac troponin T induces severe autoimmune inflammation in the myocardium. Circulation. 2006;114:1693–702. doi: 10.1161/CIRCULATIONAHA.106.635664. [DOI] [PubMed] [Google Scholar]
- 4.Kaya Z, Goser S, Buss SJ, et al. Identification of cardiac troponin I sequence motifs leading to heart failure by induction of myocardial inflammation and fibrosis. Circulation. 2008;118:2063–72. doi: 10.1161/CIRCULATIONAHA.108.788711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leuschner F, Li J, Goser S, et al. Absence of auto-antibodies against cardiac troponin I predicts improvement of left ventricular function after acute myocardial infarction. Eur Heart J. 2008;29:1949–55. doi: 10.1093/eurheartj/ehn268. [DOI] [PubMed] [Google Scholar]
- 6.Okazaki T, Tanaka Y, Nishio R, et al. Autoantibodies against cardiac troponin I are responsible for dilated cardiomyopathy in PD-1-deficient mice. Nat Med. 2003;9:1477–83. doi: 10.1038/nm955. [DOI] [PubMed] [Google Scholar]
- 7.Kuhl U, Noutsias M, Seeberg B, Schultheiss HP. Immunohistological evidence for a chronic intramyocardial inflammatory process in dilated cardiomyopathy. Heart. 1996;75:295–300. doi: 10.1136/hrt.75.3.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fukunaga T, Soejima H, Irie A, et al. Relation between CD4+ T-cell activation and severity of chronic heart failure secondary to ischemic or idiopathic dilated cardiomyopathy. Am J Cardiol. 2007;100:483–8. doi: 10.1016/j.amjcard.2007.03.052. [DOI] [PubMed] [Google Scholar]
- 9.Kishimoto C, Kuribayashi K, Masuda T, Tomioka N, Kawai C. Immunologic behavior of lymphocytes in experimental viral myocarditis: significance of T lymphocytes in the severity of myocarditis and silent myocarditis in BALB/c-nu/nu mice. Circulation. 1985;71:1247–54. doi: 10.1161/01.cir.71.6.1247. [DOI] [PubMed] [Google Scholar]
- 10.Kociol RD, Pang PS, Gheorghiade M, Fonarow GC, O’Connor CM, Felker GM. Troponin elevation in heart failure prevalence, mechanisms, and clinical implications. J Am Coll Cardiol. 2010;56:1071–8. doi: 10.1016/j.jacc.2010.06.016. [DOI] [PubMed] [Google Scholar]
- 11.Pelfrey CM, Rudick RA, Cotleur AC, Lee JC, Tary-Lehmann M, Lehmann PV. Quantification of self-recognition in multiple sclerosis by single-cell analysis of cytokine production. J Immunol. 2000;165:1641–51. doi: 10.4049/jimmunol.165.3.1641. [DOI] [PubMed] [Google Scholar]
- 12.Tang WH, Wu Y, Nicholls SJ, et al. Subclinical myocardial necrosis and cardiovascular risk in stable patients undergoing elective cardiac evaluation. Arterioscler Thromb Vasc Biol. 2010;30:634–40. doi: 10.1161/ATVBAHA.109.201210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aukrust P, Ueland T, Lien E, et al. Cytokine network in congestive heart failure secondary to ischemic or idiopathic dilated cardiomyopathy. Am J Cardiol. 1999;83:376–82. doi: 10.1016/s0002-9149(98)00872-8. [DOI] [PubMed] [Google Scholar]
- 14.Levine B, Kalman J, Mayer L, Fillit HM, Packer M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Engl J Med. 1990;323:236–41. doi: 10.1056/NEJM199007263230405. [DOI] [PubMed] [Google Scholar]
- 15.Lindberg E, Magnusson Y, Karason K, Andersson B. Lower levels of the host protective IL-10 in DCM--a feature of autoimmune pathogenesis? Autoimmunity. 2008;41:478–83. doi: 10.1080/08916930802031645. [DOI] [PubMed] [Google Scholar]
- 16.Seta Y, Shan K, Bozkurt B, Oral H, Mann DL. Basic mechanisms in heart failure: the cytokine hypothesis. J Card Fail. 1996;2:243–9. doi: 10.1016/s1071-9164(96)80047-9. [DOI] [PubMed] [Google Scholar]
- 17.Torre-Amione G, Kapadia S, Benedict C, Oral H, Young JB, Mann DL. Proinflammatory cytokine levels in patients with depressed left ventricular ejection fraction: a report from the Studies of Left Ventricular Dysfunction (SOLVD) J Am Coll Cardiol. 1996;27:1201–6. doi: 10.1016/0735-1097(95)00589-7. [DOI] [PubMed] [Google Scholar]
- 18.Gullestad L, Aass H, Fjeld JG, et al. Immunomodulating therapy with intravenous immunoglobulin in patients with chronic heart failure. Circulation. 2001;103:220–5. doi: 10.1161/01.cir.103.2.220. [DOI] [PubMed] [Google Scholar]
- 19.Frenkel D, Pachori AS, Zhang L, et al. Nasal vaccination with troponin reduces troponin specific T-cell responses and improves heart function in myocardial ischemia-reperfusion injury. Int Immunol. 2009;21:817–29. doi: 10.1093/intimm/dxp051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Y, Afanasyeva M, Hill SL, Kaya Z, Rose NR. Nasal administration of cardiac myosin suppresses autoimmune myocarditis in mice. J Am Coll Cardiol. 2000;36:1992–9. doi: 10.1016/s0735-1097(00)00939-6. [DOI] [PubMed] [Google Scholar]
- 21.Huber SA, Feldman AM, Sartini D. Coxsackievirus B3 induces T regulatory cells, which inhibit cardiomyopathy in tumor necrosis factor-alpha transgenic mice. Circ Res. 2006;99:1109–16. doi: 10.1161/01.RES.0000249405.13536.49. [DOI] [PubMed] [Google Scholar]
- 22.Baldeviano GC, Barin JG, Talor MV, et al. Interleukin-17A is dispensable for myocarditis but essential for the progression to dilated cardiomyopathy. Circ Res. 2010;106:1646–55. doi: 10.1161/CIRCRESAHA.109.213157. [DOI] [PubMed] [Google Scholar]
- 23.Chang H, Hanawa H, Yoshida T, et al. Alteration of IL-17 related protein expressions in experimental autoimmune myocarditis and inhibition of IL-17 by IL-10-Ig fusion gene transfer. Circ J. 2008;72:813–9. doi: 10.1253/circj.72.813. [DOI] [PubMed] [Google Scholar]
- 24.Li N, Bian H, Zhang J, Li X, Ji X, Zhang Y. The Th17/Treg imbalance exists in patients with heart failure with normal ejection fraction and heart failure with reduced ejection fraction. Clin Chim Acta. 2010;411:1963–8. doi: 10.1016/j.cca.2010.08.013. [DOI] [PubMed] [Google Scholar]