

Abstract
Observational and experimental studies continue to support the association of infection and infection-stimulated inflammation with development of cardiovascular disease (CVD) including atherosclerosis and thrombosis. Microvesicles (MV) are heterogeneous populations of sealed membrane-derived vesicles shed into circulation by activated mammalian cells and/or pathogenic microbes that may represent an interface between bacterial/microbial infection and increased risk of CVD. This review evaluates how MV act to modulate and intersect immunological and inflammatory responses to infection with particular attention to progression of CVD. While infection-related stimuli provoke release of MV from blood and vascular cells, MV express phosphatidylserine (PS) and other procoagulant factors on their surface which initiate and amplify blood coagulation. In addition, MV mediate cell-cell adhesion which may stimulate production of pro-inflammatory cytokines in vascular cells, which in turn aggravate progression of CVD and propagate atherothrombosis. MV transfer membrane receptors, RNA and proteins among cells, and present auto-antigens from their cells of origin to proximal or remote target cells. Because MV harbor cell surface proteins and contain cytoplasmic components of the parent cell, they mediate biological messages and play a pivotal role in the crossroad between infection-stimulated inflammation and cardiovascular diseases.
Keywords: atherosclerosis, infection, inflammation, leukocytes, platelets
Introduction
Both chronic and acute infection, especially when provoked by gram-negative bacteria, increase risk of cardiovascular events including instability of atherosclerotic plaques and formation of microvascular thrombi (1–3). Increased risk of thrombosis does not seem to be specific for one type of infection but rather seems to be a universal potential consequence of infection regardless of the organ system affected: dental infections including periodontitis (4), infection of the urinary tract (5) and upper airway (5, 6) or systemic sepsis (7). In addition, exposure to infections and their subsequent inflammatory responses may contribute to increased incidence of venous thromboembolism with hospitalization (8).
In addition to risk for thrombosis, gram-negative bacteria, for example, Chlamydia pneumoniae (C. pneumoniae) that causes upper respiratory tract infection, may exacerbate coronary artery disease (CAD) as C. pneumoniae has been cultured from atherosclerotic plaque. Also found in monocytes, macrophages and foam cells, C. pneumoniae seems to be ubiquitous in all populations with CAD and contributes to the pathobiology of atherosclerotic plaque by causing inflammation and endothelial dysfunction (9). Other microbes may also participate in development of vascular disease processes as 26 different microbes including Staphylococcus, Streptococcus, Escherichia coli, and Propionibacterium acnes, have been cultured from calcific aortic aneurysms (10). However, mechanisms by which these microbes and their byproducts initiate atherosclerotic injury, facilitate its progression, or merely colonize preexisting atheromas to modify disease progression remain controversial (9). One mechanism that has emerged to link infection-stimulated inflammation with altered thrombotic potential of the blood and atherogenic processes is that infection increases production of activated cell membrane-derived pro-inflammatory and procoagulant microvesicles (MV) from invading microbes and host cells which then activate other host cellular and soluble components of the cardiovascular system (10–15). This review will focus first on the generation of MV during infection and then focus on direct and indirect impact of MV on the onset and progression of CVD. Understanding how infection contributes to production and propagation of MV may suggest possible new early diagnostic and prognostic tests and therapeutic targets for prevention and treatment to reduce progression of CVD.
Definition and properties
MV are heterogeneous populations of activated cell membrane-derived sealed vesicles, ranging from 20 nm to 1μm, shed from the cell surface in response to activation and apoptosis (16–20). The terms ‘microvesicles’ and ‘microparticles’ have been used interchangeably in the literature. But a distinction is warranted as microparticles and microvesicles may overlap in size, but a distinct double membranous border distinguishes MV from other similar size microparticles present in the blood including lipoproteins, protein aggregates, and protein-mineral aggregates (10, 21–24). The literature is further confused by the term “ectosome” which refers to “membrane released vesicles with right-side-out oriented vesicles with cytoplasmic content” (25).
Because MV and ectosomes are sealed membrane vesicles, they bear antigens of their cellular origin and also may retain proteins, receptors and some functional properties of their parent cell (18, 25, 26). Thus, they facilitate transcellular communication throughout the circulation by delivering bioactive molecules and even genetic information when they come in contact with or bind to other cells in the circulation or the blood vessels (27). Elevated levels of circulating platelet-, monocyte- or endothelial-derived MV are associated with inflammation and CVD (11, 28–32). Both mammalian cells and gram-negative bacteria and other microbial pathogens release MV (Table 1)(26). Thus, infectious agents could promote formation of MV from host cells; conversely, host-cell-derived MV could provide a feedback loop to either exacerbate or reduce infection, inflammation, vascular dysfunction and thrombotic processes at sites distant from the site of infection.
Table 1.
Comparison between pathogen-derived and mammalian cell-derived MV
| Microbe-derived MV | Mammalian cell-derived MV | |
|---|---|---|
| Origin | bacteria, fungus, protozoan pathogens | all mammalian cells |
| Size | 20–250 nm | 100 nm – 1 um |
| Formation | budding of outer membrane and vesiculation | cell membrane blebbing and vesiculation |
| Release | all stages of growth and activation | cell activation and apoptosis |
| Content | RNA, DNA, enzymes, LPS | RNA, protein and membrane receptors |
| Function | deliver of virulent factors and resistance to antibiotics; pro-inflammatory; immuno-stimulatory, or anti-tumor depending on target host cells | transfer of signaling molecules and functional genetic information; remove cellular waste; thrombogenic; pro-inflammatory and immuno-stimulatory depending upon stimuli to cell of origin |
Mammalian cells
MV are released during physiological processes from certain types of cells and during pathological processes from every type of mammalian cells. For example, only platelet-derived MV are observed in blood of healthy humans and mice and are released following activation of platelets by a variety of stimuli including adrenaline, ADP, thrombin, collagen, Ca2+ ionophore A23187, complement and shear stress (33, 34). In general, formation of MV may begin within minutes after addition of an agonist, with the rise of intracellular calcium, which in platelets can be inhibited by calcium chelators (EDTA or EGTA) commonly used as anticoagulants (35, 36). Increases in cytosolic calcium initiate kinase mediated reorganization of the cytoskeleton including myosin light-chain kinase (37), Rho-associated kinases (38) and proteases like calpain (39, 40).
However, formation of MV is not a uniform process; release of MV differs quantitatively and phenotypically among stimuli. For example, endothelial cells release phenotypically different MV following exposure to the pro-inflammatory cytokine, tumor necrosis factor α (TNFα) which initiated receptor mediated intracellular signaling as compared to exposure to media stripped of growth factors which induced apoptosis (19). Endothelial cell-derived MV generated by plasminogen activator inhibitor -1 (PAI-1) and those produced by TNFα have overlapping but distinct protein compositions, with some proteins distinctively and exclusively related to one stimulus (41). Early studies which analyzed purified MV by SDS-PAGE revealed a pattern of limited complexity (42). However, proteomic analysis of the composition of MV from CEM T-lymphocytic cells following stimulation with phytohemagglutinin for 72 h or actinomycin for 18 h support the idea that the spectrum of proteins found in MV is influenced in part by the type of stimulus which stimulates their formation (43).
Confocal and electron microscopic images of MV formation provide evidence for selective arrangement and sorting of certain proteins. Thus, translocation of cellular molecules into MV might not be random (44). Additional work is needed to fully appreciate differential signaling systems induced by various stimuli and to determine if phenotypic assessment of MV might provide useful information reflecting the nature of cell activation and injury as a diagnostic or prognostic tool or treatment options (45).
Upon release, MV transport their biologically active contents including lipid, protein, RNA and cell surface receptors among cells (18). Specific surface receptors and cellular markers can be used to identify the origin and physiological status of the parent cell (Table 2). By identifying the cellular origin and establishing a reference range of blood borne MV in healthy individuals, it will be possible to understand which subpopulation may contribute to specific pathogenic processes.
Table 2.
Surface markers identifying cellular origin of blood borne microvesicles
| Cell type | Surface marker |
|---|---|
| Polymorphonuclear cells and neutrophils | selectins and integrins complement regulators HLA-1 |
| Lymphocytes | CD4, CD3, CD8 or CD 19 |
| Platelets | GPIIb/IIIa (CD41/CD61), GPIX (CD42a) |
| Endothelial cells | CD31/-CD42a, CD62E or CD146 |
Gram-negative Bacteria and MV
Bacterial derived MV are made up of an outer membrane which engulfs periplasmic components (46, 47) and which serves as a delivery system of genetic material, protein, and lipopolysaccharide (LPS) (47–50), and promotes adherence of the bacterium to infect host cells (51) or other bacteria (47). MV formation may be a functional requirement for bacteria as DNA packaged within MV can be protected from degradation by soluble DNase (50, 52). Resistance to antibiotics can be acquired by the recipient bacteria by integrating MV from the parental bacteria species (47).
MV released by bacteria may contribute to infection-induced cardiovascular diseases like coronary artery diseases since virulence genes, LPS and other toxins and enzymes carried by MV can be absorbed by host cells (53). MV from certain species of bacteria like P. gingivalis (a gram-negative bacteria related to chronic periodontitis) activate platelets (54, 55), suggesting a possible mechanism by which bacteria-derived MV increase risk of thrombosis. However, even though bacterial-derived MV can be detected in infected human tissues (56), no studies have evaluated how bacteria-derived MV lead to onset and progression of CVD. In addition, most information regarding production of bacterial MV was obtained during normal growth of bacteria in vitro. Little is known about mechanisms underlying production and release of bacterial derived MV, or about what and how stimuli regulate their production and composition by the host in vivo (49, 54, 57–59).
Host processes triggered by bacterial infection
Direct response of mammalian cells to LPS-receptor activation
Bacteria activate host cells mainly through the binding of LPS, the major pathogenic molecule of outer membrane component of gram-negative bacteria to toll-like receptor 4 (TLR4), a transmembrane glycoprotein present on platelets, leukocytes, lymphocytes, and endothelium (60–62). In pigs, infusion of LPS (10 mg/kg/ per hr) and in mice a single intra-peritoneal injection of endotoxin (7.5 mg/kg), increased platelet-derived MV within 3 and 6 hours, respectively (63–66). Experiments like these, while providing support for the concept that infection stimulates production of platelet-derived MV, should be interpreted with caution as measurement of a MV from a single cellular origin does not provide information about generation of MV from other cell types or interactions among MV of different cellular origin or interactions of MV with other blood cells. In addition, depending on the dose of LPS or type of bacterial infection, receptors other than TLR4 may be activated. Therefore, complement activation or cytokine-mediated mechanisms in vascular cells complicate unraveling mechanisms of LPS induced production of host MV (67–69).
In vitro studies can be used to identify effects of LPS on blood elements and cells of the vascular wall. In diluted blood which prevents cell-cell interactions, addition of LPS (500 ng or 1 μg/mL) increased the number of TF-bearing MV and MV from platelets after 4 hours of incubation (70). Incubation of human monocytes with LPS (5 μg/mL for 5 hours) increased the total number of MV, TF-bearing MV and those bearing phosphatidylserine (PS) and other adhesion molecules (42, 71). MV formation visualized by fluorescence microscopy occurred in leukocytes and endothelial cells after incubation of LPS (5 μg/mL) for 90 min (72). Thus, LPS stimulates MV production from circulating blood cells and cells of the vascular wall.
In unpublished work by our group, incubation of whole blood with LPS for one hour at a dose which stimulated TLR-4 receptors did not increase the total number of MV. However, leukocyte-derived MV were associated with platelets which subsequently altered the reactivity status of the platelets (Figure 1). These data provide evidence as have other studies that MV generated by one cell type in response to LPS alter reactivity of other cell types within the circulation (69, 73–76). Although existing data provide evidence that LPS induces MV release from isolated cell or cells in vitro, more studies are needed to understand how other bacterial proteins affect MV production from host cells in vivo. In particular, studies are needed to determine whether and how LPS, as a natural component of bacteria and a content of bacteria-derived MV, affects composition of infected mammalian cell-derived MV distinct from other stimuli.
Figure 1.
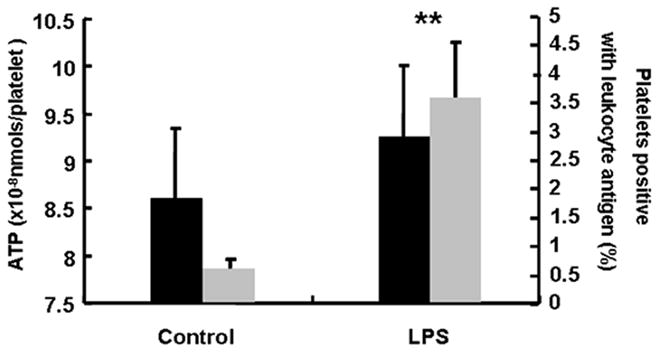
ATP secretion in response to thrombin (0.1 U/mL) (black bars) and platelets associated with leukocyte-derived MV (gray bars) after whole blood from mice was incubated with LPS (5 μg/mL) or saline (control) for 1h. P<0.01 vs. Control.
Indirect response of mammalian cells to infection-associated processes
Mammalian cells release MV in response to a variety of stimuli associated with infection, many of which are considered conventional biomarkers for CVD including inflammatory cytokines, lipids and lipidperoxidation, oxidative stress and apoptosis. These stimuli are also important participants in the defense mechanisms and pathogenesis of infection.
Pro-inflammatory agents
Release of cytokines provoked in the host cells by invading micro-organisms may in turn stimulate release of MV from non-infected cells (9). For example, pro-inflammatory cytokines such as TNFα or interleukin-1 (IL-1) stimulate MV production from endothelial cells and monocytes which contain tissue factor (TF) and have high procoagulant activity (75–77). Activated complement component (C5a) also increases the release of cellular MV from endothelia and platelets (78). Leukocyte-derived MV significantly increased after 1h incubation with 10μM fMLP(formylmethionyl-leucyl-phenylalanine, a formylated tripeptide originally isolated from bacterial filtrates) (79).
C-reactive protein (CRP), which promotes agglutination and bacterial phagocytosis, is an acute-phase response protein produced by the liver in response to infection as well as by cytokines IL-6, IL-1, and TNFα (80). CRP is elevated in individuals at risk for recurrent myocardial infarction, coronary heart disease or stroke and may predict the risk of future cardiovascular events (81). However, circulating CRP is used as a biomarker of inflammation but not as a specific biomarker linking the inflammation to an acute or chronic infection in CVD.
Oxidative stress and metabolic failure
Oxidative stress, characterized by disruption of oxidant and antioxidant systems within cells, increases with infection, inflammation and immunological disorders (82, 83). Reactive oxygen species (ROS) damage cellular membrane structures leading to release of MV (39, 84, 85). Oxidative stress is a common mechanism underlying cardiovascular diseases (86, 87), and diverse therapeutic interventions such as antioxidants can impede or delay the onset and progression of cardiovascular diseases (88, 89).
Oxidative stress markers independently increase the total number of MV and MV derived from platelets (90). Energetic failure within platelets is also associated with increased shedding of thrombogenic MV, which may result from decreases in activity of enzymes of the mitochondrial electron transport chain (91). Oxidative stress also causes membrane phospholipid rearrangement and PS-bearing MV to be shed from red blood cells (92).
Oxidative stress in endothelial cells increases release of MV expressing vascular cell adhesion molecular-1 (VCAM-1) (93). Endothelium-derived MV released in response to oxidative stress stimulated neutrophil adhesion via platelet-activating factor (PAF) receptors (84). However, the exact mechanism of how oxidative stress and metabolic failure lead to MV release is not clear, but chronic treatment with antioxidants, such as vitamin C or carvedilol, decreases circulating endothelial MV in patients with heart failure and exhibits a protective effect (94, 95).
Monocyte-derived MV released during apoptosis induce production of superoxide anion in endothelial cells (96), suggesting that production of reactive oxygen species are both the result of and stimulus for release of MV and thus, represent a mechanism of positive feedback to cellular injury. Reactive oxygen species promote a procoagulant phenotype through up-regulation of TF expression and activity in MV (97).
Apoptosis
Release of membrane blebs or MV are an early feature of apoptosis activated by various stressors including bacterial infection, cytokines and oxidative stress (83, 98). Release of MV may be a protective mechanism against intracellular stress as MV containing caspase 3 prevent intracellular accumulation of the enzyme which could allow escape from apoptosis and thus, contribute to cell survival (99).
Mechanisms leading to release of MV upon apoptosis are only partly understood. Loss of tumor suppressor gene p53 leads to an increased release of TF-bearing MV (100). MV are not released from MCF-7 cells lacking caspase-3 but release is restored by transfection with functional caspase-3 (101). These results implicate both p53 and caspase-3 in formation of MV. As with oxidative stress, MV released as a result of apoptosis can stimulate apoptosis in other cells as platelet-derived MV containing functional active caspase-3 induce apoptosis of human macrophages (102).
Contribution of MV to cardiovascular disease
CVD, in particular, atherosclerosis is described as an inflammatory disease (103). However, the source of inflammation is usually attributed to disruptions in metabolism related to altered lipid metabolism. The preceding discussion expands this view to include processes associated with infection and immunity. Indeed, pro-inflammatory cytokines are independent risk factors for CVD, and in persons with chronic infections, inflammatory responses increase risk of atherosclerosis (104). The number and type of MV generated during infection, inflammation, and thrombotic complications suggest that MV may provide an interface between infection, initiation and amplification of cardiovascular diseases (Figure 2).
Figure 2.
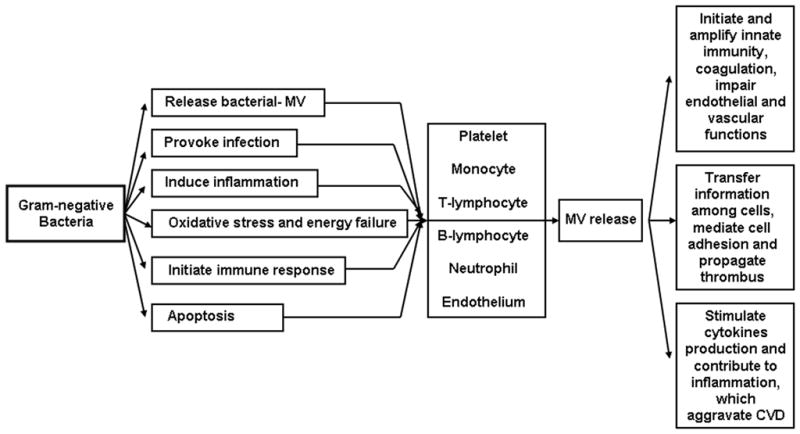
Schematic of how MV of gram-negative bacteria may interface with host cells to increase progression of cardiovascular disease.
MV in the initiation and amplification of coagulation and thrombosis
During formation of MV, the normal phospholipid asymmetry of the membrane is changed, exposing negatively charged phospholipids such as PS from the inner membrane leaflet to the outer membrane leaflet. Thrombin generation capacity of MV is proportional to the amount of PS bearing on the surface (105) or the number of MV bearing PS (106). Surface PS offers binding sites for coagulant factors II, Va, and Xa and provides a platform for the assembly of the prothrombinase complex, accelerating the conversion of prothrombin into thrombin and supporting a procoagulant phenotype (107, 108).
MV also represent a reservoir of blood borne tissue factor (TF) activity (109, 110). Monocytes and polymorphonuclear leukocytes are one source of TF-bearing MV, which when transferred or in association with platelets, trigger and propagate thrombosis (76). Whether platelets produce TF is controversial (109). There may be three distinct sources of platelet TF: 1) TF taken up from circulating monocyte-derived MV; 2) TF stored in the α-granules of platelets from precursor bone megakaryocytes or taken up from other sources; 3) TF that is synthesized and expressed on the plasma membrane of mature platelets (111, 112).
The rate of accumulation of TF in platelet-rich thrombi cannot be explained by recruitment of leukocytes onto adherent platelets (113, 114). One mechanism to explain this discrepancy is that TF-bearing MV adhere to platelets through binding of P-selectin glycoprotein ligand-1 (PSGL-1) on the MV and P-selectin on the platelets (113, 115, 116). Tissue factor exists in the blood in an inactive (latent) and active configuration (117) as TF-bearing MV circulate in healthy persons while TF activity was not detected (118). In addition to contributing to thrombin formation, fusion between TF-bearing MV and activated platelets facilitates transfer of both proteins and lipids to the platelet membrane (116). Therefore, MV modulate coagulation by directly initiating coagulation cascade or indirectly via activating platelets. It is proposed that TF bearing MV, mainly of monocytic and lymphocytic origin, contribute to thrombus formation following rupture of atherosclerotic plaque by binding to activated platelets at the site of plaque rupture (14, 113, 119).
MV and interactions with cells of the vascular wall
Using rat aortic rings as a bioassay, MV from patients with acute myocardial infarction initiated reduced endothelium-dependent relaxations and release of nitric oxide compared to MV from patients without ischemia (120). These reductions in endothelium-dependent relaxations are consistent with general vasomotor dysfunction observed after myocardial infarction, and potentially would contribute to increased formation of thrombus and decreased tissue perfusion.
Endothelium-dependent vasodilatation induced by acetylcholine was significantly reduced by circulating MV from patients with metabolic syndrome (121). MV derived from diabetic patients and HIV-infected individuals also reduce acetylcholine-induced and attenuate shear stress-induced vasodilatation through stimulating expression of caveolin-1 and down-regulation of endothelial nitric oxide synthase (eNOS) in cultured human endothelial cells (77). T-lymphocyte-derived MV may increase inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX2) through NF-κB - dependent transcription (122).
MV derived from monocytes and lymphocytes are found in atherosclerotic plaques in greater concentrations than those derived from platelets (14). This condition is opposite to the various numbers of MV found in blood and may reflect the activation of these cells as they migrate into vascular lesions or that the leukocyte-derive MV may be associated with platelets in the blood.
MV and intercellular communication
Regardless of their cellular origin (bacterial or host), MV can amplify progression of CVD through the transfer of biological information between cells. Transfer of integrins and selectins to target cells through MV facilitate leukocyte adhesion and interaction with vascular endothelium. In addition, platelet-derived MV enhance binding of neutrophils to other neutrophils under flow conditions via P-selectin on MV and PSGL-1 on neutrophils (123). Monocyte-endothelial interactions stimulated by platelet-derived MV up-regulated expression of cellular adhesion molecules (ICAM-1) on endothelium and CD11b/CD18 on monocytes, a process related to metabolism of arachidonic acid (124). Adhesion of monocytes and neutrophils to the endothelium is considered a crucial step in the early processes of atherosclerotic lesions (125).
It is likely that platelet-derived MV, rather than platelets deliver long-range signals to other cells throughout the circulation since the aggregation and binding properties of activated platelets make it difficult for them to travel through the circulatory system (126). Consequently, MV, instead of intact platelets, may be critical in enhancing thrombosis and modulating both innate and adaptive immune responses.
Processes of how MV attach to target cells is not completely understood. In addition to fusion of the cell membranes, receptor binding, as described in other sections, is important, and the receptors involved may differ depending upon the stimulus for MV generation. MV bearing ICAM-1 resulting from TNF-α stimulation of endothelial cells bind to β2 integrin on monocytes increasing TF gene expression in monocytes and induction of TF-dependent procoagulant activity (127). Alternatively, Fas/Fas ligand receptor interaction might be important in binding of apoptotic MV to target cells (122).
MV and amplification of infection-induced immune responses
As discussed above, LPS on the outer membrane of gram-negative bacteria or associated with bacterial derived MV activate host cells in part through TLR4 signaling mechanisms. Mutational deletion of TLR4 decreased risk of development of atherosclerosis but increased susceptibility for infection (128, 129). Other genetic polymorphisms of monocyte chemo-attractant protein (MCP-1) and its receptor CCR2 (chemokine {C-C motif} receptor 2) may increase susceptibility for chronic stable angina pectoris and myocardial infarction (129, 130). While pro-inflammatory cytokines stimulate MV production, MV in turn stimulate production of these same cytokines thus amplifying the signal through a type of positive feedback. For example, stimulation of leukocytes with endotoxin (a complex, not purified LPS) induced shedding of MV containing PAF, an agonist which stimulates innate immunity (72). Furthermore, after priming THP-1 monocytes with LPS, stimulation of those cells with ATP generated MV which were both PS positive and contained bioactive IL-1β (131). This convergent response initiated by two different agonists suggests that generation and release of MV could be a general pathway for secretion of some cytoplasmic proteins which modulate the immune system. Stimulation of dendritic cells by IL-1β initiates acute inflammatory response (132).
In addition to initiating immune response, MV contribute to maturation of immune cells. MV generated from thrombin-activated platelets carry CD40L and initiate maturation of monocyte-derived dendritic cells which in turn activate naïve T-cells (133). CD40L in platelet MV can be transferred to the adaptive immune B-cell compartments that may be distant from the site of platelet activation. This “long-range” signaling results in production of antigen-specific IgG (134). MV generated from endothelial cells in response to bacterial infection induce maturation of plasmacytoid dendritic cells, secretion of inflammatory cytokine (IL-6 and IL-8, but not IFN-α) and proliferation of allogenic naïve CD4+ T-cell. These responses are not activated by either platelet or T-cell derived MV (75). Collectively these responses demonstrate a coordinated interaction among processes responding to infection, vascular cell activation and cytokines implicated in progression of cardiovascular disease.
Summary and future directions
MV derived from invading pathogens and host cells can be considered as vectors of trans-cellular exchange of signals common to infection, general inflammation, and procoagulant properties of the blood contributing to progression of CVD. The virulent potential of MV derived from pathogens may explain in part the consistent finding that the burden of infection increases development of CVD in the absence of consistent identification of specific pathogens within atherosclerotic lesions (9, 135–137). While the literature abounds in studies of generation of cell-specific MV and their characterization in various diseases, general methodological challenges remain in the development of MV as a diagnostic or prognostic tool. These challenges include the need for standardized methods of blood collection and processing including issues related to anticoagulants and cell-free plasma preparation after blood collection which may themselves affect production, adhesion or secretion of MV in the collected sample (31, 36). A second challenge is to better understand how different stimuli affect production and content of MV or to differentiate a non-specific “molecular switch” common to generation of MV from various cell types.
A third challenge is to define how sex and age modulate formation of MV in various cell types which might influence thrombosis, immunity and vascular reactivity. For example, estrogen receptor beta knockout female mice show higher PS-expressing platelet-derived MV in the plasma and thrombin-generating capacity compared with wild type mice (91). In addition, total numbers of MV, those derived from platelets, monocytes and vascular endothelium, and those positive for phosphatidylserine and tissue factor were significantly greater (P<0.05) in newly menopausal women (circulating estrogen < 20pg/ml) compared to age-matched women who had estrogen >40pg/mL (138). These results infer hormonal regulation on MV generation and thrombotic risk. Since incidence of immunologic disorders and cardiovascular risk diverge with age and sex (139, 140), understanding the contribution of MV in association with these important variables will facilitate development of more effective preventive strategies for individuals.
Acknowledgments
Salary support for the authors is from grants from the American Heart Association, AHA30503Z; National Institutes of Health, HL90639; Kronos Longevity Research Institute, the Mayo Foundation and the China Scholarship Council.
Footnotes
Conflict of Interest: None
References
- 1.Kalvegren H, Majeed M, Bengtsson T. Chlamydia pneumoniae binds to platelets and triggers P-selectin expression and aggregation: a causal role in cardiovascular disease? Arterioscler Thromb Vasc Biol. 2003;23:1677–1683. doi: 10.1161/01.ATV.0000084810.52464.D5. [DOI] [PubMed] [Google Scholar]
- 2.Lee N, Hui D, Wu A, Chan P, Cameron P, Joynt GM, Ahuja A, Yung MY, Leung CB, To KF, Lui SF, Szeto CC, Chung S, Sung JJ. A major outbreak of severe acute respiratory syndrome in Hong Kong. N Engl J Med. 2003;348:1986–1994. doi: 10.1056/NEJMoa030685. [DOI] [PubMed] [Google Scholar]
- 3.Pasceri V, Patti G, Cammarota G, Pristipino C, Richichi G, Di Sciascio G. Virulent strains of Helicobacter pylori and vascular diseases: a meta-analysis. Am Heart J. 2006;151:1215–1222. doi: 10.1016/j.ahj.2005.06.041. [DOI] [PubMed] [Google Scholar]
- 4.Willershausen B, Kasaj A, Willershausen I, Zahorka D, Briseno B, Blettner M, Genth-Zotz S, Munzel T. Association between chronic dental infection and acute myocardial infarction. J Endod. 2009;35:626–630. doi: 10.1016/j.joen.2009.01.012. [DOI] [PubMed] [Google Scholar]
- 5.Smeeth L, Thomas SL, Hall AJ, Hubbard R, Farrington P, Vallance P. Risk of myocardial infarction and stroke after acute infection or vaccination. N Engl J Med. 2004;351:2611–2618. doi: 10.1056/NEJMoa041747. [DOI] [PubMed] [Google Scholar]
- 6.Casscells SW, Granger E, Kress AM, Linton A, Madjid M, Cottrell L. Use of oseltamivir after influenza infection is associated with reduced incidence of recurrent adverse cardiovascular outcomes among military health system beneficiaries with prior cardiovascular diseases. Circulation. 2009;2:108–115. doi: 10.1161/CIRCOUTCOMES.108.820357. [DOI] [PubMed] [Google Scholar]
- 7.Patel KN, Soubra SH, Lam FW, Rodriguez MA, Rumbaut RE. Polymicrobial sepsis and endotoxemia promote microvascular thrombosis via distinct mechanisms. J Thromb Haemost. 2010;8:1403–1409. doi: 10.1111/j.1538-7836.2010.03853.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heit JA, Melton LJI, Lohse CM, Petterson TM, Silverstein MD, Mohr DN, O’Fallon WM. Incidence of venous thromboembolism in hospitalized patients vs community residents. Mayo Clin Proc. 2001;76:1102–1110. doi: 10.4065/76.11.1102. [DOI] [PubMed] [Google Scholar]
- 9.Watson C, Alp NJ. Role of Chlamydia pneumoniae in atherosclerosis. Clin Sci (Lond) 2008;114:509–531. doi: 10.1042/CS20070298. [DOI] [PubMed] [Google Scholar]
- 10.Schwartz MK, Hunter LW, Huebner M, Lieske JC, Miller VM. Characterization of biofilm formed by human-derived nanoparticles. Nanomedicine. 2009;4:931–941. doi: 10.2217/nnm.09.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nieuwland R, Berckmans RJ, McGregor S, Boing AN, Romijn FP, Westendorp RG, Hack CE, Sturk A. Cellular origin and procoagulant properties of microparticles in meningococcal sepsis. Blood. 2000;95:930–935. [PubMed] [Google Scholar]
- 12.Ogura H, Tanaka H, Koh T, Fujita K, Fujimi S, Nakamori Y, Hosotsubo HYK, Shimazu T, Sugimoto H. Enhanced production of endothelial microparticles with increased binding to leukocytes in patients with severe systemic inflammatory response syndrome. J Trauma. 2004;56:823–830. doi: 10.1097/01.ta.0000084517.39244.46. Discussion 830–831. [DOI] [PubMed] [Google Scholar]
- 13.Larsson A, Nilsson B, Eriksson M. Thrombocytopenia and platelet microvesicle formation caused by Legionella pneumophila infection. Thromb Res. 1999;96:391–397. doi: 10.1016/s0049-3848(99)00119-x. [DOI] [PubMed] [Google Scholar]
- 14.Mallat Z, Hugel B, Ohan J, Leseche G, Freyssinet JM, Tedgui A. Shed membrane microparticles with procoagulant potential in human atherosclerotir plaques: a role for apoptosis in plaque thrombogenicity. Circulation. 1999;99:348–353. doi: 10.1161/01.cir.99.3.348. [DOI] [PubMed] [Google Scholar]
- 15.Mallat Z, Benamer H, Hugel B, Benessiano J, Steg PG, Freyssinet JM, Tedgui A. Elevated levels of shed membrane microparticles with procoagulant potential in the peripheral circulating blood of patients with acute coronary syndromes. Circulation. 2000;101:841–843. doi: 10.1161/01.cir.101.8.841. [DOI] [PubMed] [Google Scholar]
- 16.Piccin A, Murphy WG, Smith OP. Circulating microparticles: pathophysiology and clinical implications. Blood Rev. 2007;21:157–171. doi: 10.1016/j.blre.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 17.Cocucci E, Racchetti G, Meldolesi J. Shedding microvesicles: artefacts no more. Trends Cell Biol. 2009;19:43–51. doi: 10.1016/j.tcb.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 18.Morel O, Toti F, Hugel B, Freyssinet JM. Cellular microparticles: a disseminated storage pool of bioactive vascular effectors. Curr Opin Hematol. 2004;11:156–164. doi: 10.1097/01.moh.0000131441.10020.87. [DOI] [PubMed] [Google Scholar]
- 19.Jimenez JJ, Jy W, Mauro LM, Soderland C, Horstman LL, Ahn YS. Endothelial cells release phenotypically and quantitatively distinct microparticles in activation and apoptosis. Thromb Res. 2003;109:175–180. doi: 10.1016/s0049-3848(03)00064-1. [DOI] [PubMed] [Google Scholar]
- 20.Hashimoto K, Jayachandran M, Owen WG, Miller VM. Aggregation and microparticle production through toll-like receptor 4 activation in platelets from recently menopausal women. J Cardiovasc Pharmacol. 2009;54:57–62. doi: 10.1097/FJC.0b013e3181ab373d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu C-Y, Martel J, Young D, Young JD. Fetuin-A/Albumin-Mineral Complexes Resembling Serum Calcium Granules and Putative Nanobacteria: Demonstration of a Dual Inhibition-Seeding Concept. PLoS ONE. 2009;4:e8058. doi: 10.1371/journal.pone.0008058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Young JD, Martel J, Young D, Young A, Hung C-M, Young L, Chao Y-J, Young J, Wu C-Y. Characterization of Granulations of Calcium and Apatite in Serum as Pleomorphic Mineralo-Protein Complexes and as Precursors of Putative Nanobacteria. PLoS ONE. 2009;4:e5421. doi: 10.1371/journal.pone.0005421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schwartz MK, Lieske JC, Miller VM. Contribution of biologically derived nanoparticles to disease. Surgery. 2010;147:181–184. doi: 10.1016/j.surg.2009.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hunter LW, Shiekh FA, Pisimisis GT, Kim SH, Edeh SN, Miller VM, Lieske JC. Key role of alkaline phosphatase in the development of human-derived nanoparticles in vitro. Acta Biomaterialia. 2011;7:1319–1345. doi: 10.1016/j.actbio.2010.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sadallah S, Eken C, Schifferli JA. Ectosomes as modulators of inflammation and immunity. Clin Exp Immunol. 2011;163:26–32. doi: 10.1111/j.1365-2249.2010.04271.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Silverman JM, Reiner NE. Exosomes and other microvesicles in infection biology: organelles with unanticipated phenotypes. Cellular Microbiology. 2011;13:1–9. doi: 10.1111/j.1462-5822.2010.01537.x. [DOI] [PubMed] [Google Scholar]
- 27.Martinez MC, Tesse A, Zobairi F, Andriantsitohaina R. Shed membrane microparticles from circulating and vascular cells in regulating vascular function. Am J Physiol. 2005;288:H1004–1009. doi: 10.1152/ajpheart.00842.2004. [DOI] [PubMed] [Google Scholar]
- 28.Joop K, Berckmans RJ, Nieuwland R, Berkhout J, Romijn FP, Hack CE, Sturk A. Microparticles from patients with multiple organ dysfunction syndrome and sepsis support coagulation through multiple mechanisms. Thromb Haemost. 2001;85:810–820. [PubMed] [Google Scholar]
- 29.Brogan PA, Shah V, Brachet C, Harnden A, Mant D, Klein N, Dillon MJ. Endothelial and platelet microparticles in vasculitis of the young. Arthritis Rheum. 2004;50:927–936. doi: 10.1002/art.20199. [DOI] [PubMed] [Google Scholar]
- 30.Bernal-Mizrachi L, Jy W, Fierro C, Macdonough R, Velazques HA, Purow J, Jimenez JJ, Horstman LL, Ferreira A, de Marchena E, Ahn YS. Endothelial microparticles correlate with high-risk angiographic lesions in acute coronary syndromes. Int J Cardiol. 2004;97:439–446. doi: 10.1016/j.ijcard.2003.10.029. [DOI] [PubMed] [Google Scholar]
- 31.Simak J, Gelderman MP, Yu H, Wright V, Baird AE. Circulating endothelial microparticles in acute ischemic stroke: a link to severity, lesion volume and outcome. J Thromb Haemost. 2006;4:1296–1302. doi: 10.1111/j.1538-7836.2006.01911.x. [DOI] [PubMed] [Google Scholar]
- 32.Amabile N, Guerin AP, Leroyer A, Mallat Z, Nguyen C, Boddaert J, London GM, Tedgui A, Boulanger CM. Circulating endothelial microparticles are associated with vascular dysfunction in patients with end-stage renal failure. J Am Soc Nephrol. 2005;16:3381–3388. doi: 10.1681/ASN.2005050535. [DOI] [PubMed] [Google Scholar]
- 33.Simak J, Gelderman MP. Cell membrane microparticles in blood and blood products: potentially pathogenic agents and diagnostic markers. Transfus Med Rev. 2006;20:1–26. doi: 10.1016/j.tmrv.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 34.Horstman LL, Ahn YS. Platelet microparticles: a wide-angle perspective. Crit Rev Oncol Hematol. 1999;30:111–142. doi: 10.1016/s1040-8428(98)00044-4. [DOI] [PubMed] [Google Scholar]
- 35.Gilbert GE, Sims PJ, Wiedmer T, Furie B, Furie BC, Shattil SJ. Platelet-derived microparticles express high affinity receptors for factor VIII. J Biol Chem. 1991;266:17261–17268. [PubMed] [Google Scholar]
- 36.Gemmell CH, Sefton MV, Yeo EL. Platelet-derived microparticle formation involves glycoprotein IIb-IIIa. Inhibition by RGDS and a Glanzmann’s thrombasthenia defect. J Biol Chem. 1993;268:14586–14589. [PubMed] [Google Scholar]
- 37.Wiedmer T, Sims PJ. Participation of protein kinases in complement C5b-9-induced shedding of platelet plasma membrane vesicles. Blood. 1991;78:2880–2886. [PubMed] [Google Scholar]
- 38.Sapet C, Simoncini S, Loriod B, Puthier D, Sampol J, Nguyen C, Dignat-George F, Anfosso F. Thrombin-induced endothelial microparticle generation: identification of a novel pathway involving ROCK-II activation by caspase-2. Blood. 2006;108:1868–1876. doi: 10.1182/blood-2006-04-014175. [DOI] [PubMed] [Google Scholar]
- 39.Miyoshi H, Umeshita K, Sakon M, Imajoh-Ohmi S, Fujitani K, Gotoh M, Oiki E, Kambayashi J, Monden M. Calpain activation in plasma membrane bleb formation during tert-butyl hydroperoxide-induced rat hepatocyte injury. Gastroenterology. 1996;110:1897–1904. doi: 10.1053/gast.1996.v110.pm8964416. [DOI] [PubMed] [Google Scholar]
- 40.Azevedo LC, Pedro M, Laurindo FR. Circulating microparticles as therapeutic targets in cardiovascular diseases. Recent patents on cardiovascular drug discovery. 2007;2:41–51. doi: 10.2174/157489007779606121. [DOI] [PubMed] [Google Scholar]
- 41.Peterson DB, Sander T, Kaul S, Wakim BT, Halligan B, Twigger S, Pritchard KA, Jr, Oldham KT, Ou JS. Comparative proteomic analysis of PAI-1 and TNF-alpha-derived endothelial microparticles. Proteomics. 2008;8:2430–2446. doi: 10.1002/pmic.200701029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Satta N, Toti F, Feugeas O, Bohbot A, Dachary-Prigent J, Eschwege V, Hedman H, Freyssinet JM. Monocyte vesiculation is a possible mechanism for dissemination of membrane-associated procoagulant activities and adhesion molecules after stimulation by lipopolysaccharide. J Immunol. 1994;153:3245–3255. [PubMed] [Google Scholar]
- 43.Miguet L, Pacaud K, Felden C, Hugel B, Martinez MC, Freyssinet JM, Herbrecht R, Potier N, van Dorsselaer A, Mauvieux L. Proteomic analysis of malignant lymphocyte membrane microparticles using double ionization coverage optimization. Proteomics. 2006;6:153–171. doi: 10.1002/pmic.200500133. [DOI] [PubMed] [Google Scholar]
- 44.Moskovich O, Fishelson Z. Live cell imaging of outward and inward vesiculation induced by the complement c5b-9 complex. J Biol Chem. 2007;282:29977–29986. doi: 10.1074/jbc.M703742200. [DOI] [PubMed] [Google Scholar]
- 45.Pula G, Perera S, Prokopi M, Sidibe A, Boulanger CM, Mayr M. Proteomic analysis of secretory proteins and vesicles in vascular research. Proteomics Clin Appl. 2008;2:882–891. doi: 10.1002/prca.200800040. [DOI] [PubMed] [Google Scholar]
- 46.Beveridge TJ, Kadurugamuwa JL. Periplasm, periplasmic spaces, and their relation to bacterial wall structure: novel secretion of selected periplasmic proteins from Pseudomonas aeruginosa. Microb Drug Resist. 1996;2:1–8. doi: 10.1089/mdr.1996.2.1. [DOI] [PubMed] [Google Scholar]
- 47.Yaron S, Kolling GL, Simon L, Matthews KR. Vesicle-mediated transfer of virulence genes from Escherichia coli O157:H7 to other enteric bacteria. Appl Environ Microbiol. 2000;66:4414–4420. doi: 10.1128/aem.66.10.4414-4420.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kadurugamuwa JL, Beveridge TJ. Membrane vesicles derived from Pseudomonas aeruginosa and Shigella flexneri can be integrated into the surfaces of other gram-negative bacteria. Microbiology. 1999;145 (Pt 8):2051–2060. doi: 10.1099/13500872-145-8-2051. [DOI] [PubMed] [Google Scholar]
- 49.Spiewak R, Dutkiewicz J. In vitro study of pro-inflammatory and anti-tumour properties of microvesicles from bacterial cell wall of Pantoea agglomerans. Ann Agric Environ Med. 2008;15:153–161. [PubMed] [Google Scholar]
- 50.Kolling GL, Matthews KR. Export of virulence genes and Shiga toxin by membrane vesicles of Escherichia coli O157:H7. Appl Environ Microbiol. 1999;65:1843–1848. doi: 10.1128/aem.65.5.1843-1848.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meyer DH, Fives-Taylor PM. Evidence that extracellular components function in adherence of Actinobacillus actinomycetemcomitans to epithelial cells. Infect Immun. 1993;61:4933–4936. doi: 10.1128/iai.61.11.4933-4936.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Renelli M, Matias V, Lo RY, Beveridge TJ. DNA-containing membrane vesicles of Pseudomonas aeruginosa PAO1 and their genetic transformation potential. Microbiology (Reading, England) 2004;150:2161–2169. doi: 10.1099/mic.0.26841-0. [DOI] [PubMed] [Google Scholar]
- 53.Vidakovics ML, Jendholm J, Morgelin M, Mansson A, Larsson C, Cardell LO, Riesbeck K. B cell activation by outer membrane vesicles--a novel virulence mechanism. PLoS pathogens. 2010;6:e1000724. doi: 10.1371/journal.ppat.1000724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pham K, Feik D, Hammond BF, Rams TE, Whitaker EJ. Aggregation of human platelets by gingipain-R from Porphyromonas gingivalis cells and membrane vesicles. Platelets. 2002;13:21–30. doi: 10.1080/09537100120104863. [DOI] [PubMed] [Google Scholar]
- 55.Sharma A, Novak EK, Sojar HT, Swank RT, Kuramitsu HK, Genco RJ. Porphyromonas gingivalis platelet aggregation activity: outer membrane vesicles are potent activators of murine platelets. Oral Microbiol Immunol. 2000;15:393–396. doi: 10.1034/j.1399-302x.2000.150610.x. [DOI] [PubMed] [Google Scholar]
- 56.Ellis TN, Kuehn MJ. Virulence and immunomodulatory roles of bacterial outer membrane vesicles. Microbiol Mol Biol Rev. 2010;74:81–94. doi: 10.1128/MMBR.00031-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dutkiewicz J, Skorska C, Burrell R, Szuster-Ciesielska A, Sitkowska J. Immunostimulative effects of repeated inhalation exposure to microvesicle-bound endotoxin of Pantoea agglomerans. Ann Agric Environ Med. 2005;12:289–294. [PubMed] [Google Scholar]
- 58.Nunez-del AA, Salas-Tellez E, de la Garza M, Diaz-Aparicio E, Tenorio-Gutierrez V. Identification of an immunogenic protein of Actinobacillus seminis that is present in microvesicles. Can J Vet Res. 2006:70. [PMC free article] [PubMed] [Google Scholar]
- 59.Tashiro Y, Ichikawa S, Shimizu M, Toyofuku M, Takaya N, Nakajima-Kambe T, Uchiyama H, Nomura N. Variation of physiochemical properties and cell association activity of membrane vesicles with growth phase in Pseudomonas aeruginosa. Appl Environ Microbiol. 2010;76:3732–3739. doi: 10.1128/AEM.02794-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cognasse F, Hamzeh-Cognasse H, Lafarge S, Delezay O, Pozzetto B, McNicol A, Garraud O. Toll-like receptor 4 ligand can differentially modulate the release of cytokines by human platelets. Br J Haematol. 2008;141:84–91. doi: 10.1111/j.1365-2141.2008.06999.x. [DOI] [PubMed] [Google Scholar]
- 61.Kuroki Y, Tsuchida K, Go I, Aoyama M, Naganuma T, Takemoto Y, Nakatani T. A study of innate immunity in patients with end-stage renal disease: special reference to toll-like receptor-2 and -4 expression in peripheral blood monocytes of hemodialysis patients. Int J Mol Med. 2007;19:783–790. [PubMed] [Google Scholar]
- 62.Faure E, Equils O, Sieling PA, Thomas L, Zhang FX, Kirschning CJ, Polentarutti N, Muzio M, Arditi M. Bacterial lipopolysaccharide activates NF-kappaB through toll-like receptor 4 (TLR-4) in cultured human dermal endothelial cells. Differential expression of TLR-4 and TLR-2 in endothelial cells. J Biol Chem. 2000;275:11058–11063. doi: 10.1074/jbc.275.15.11058. [DOI] [PubMed] [Google Scholar]
- 63.Wang JG, Manly D, Kirchhofer D, Pawlinski R, Mackman N. Levels of microparticle tissue factor activity correlate with coagulation activation in endotoxemic mice. J Thromb Haemost. 2009;7:1092–1098. doi: 10.1111/j.1538-7836.2009.03448.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Eriksson M, Nelson D, Nordgren A, Larsson A. Increased platelet microvesicle formation is associated with mortality in a porcine model of endotoxemia. Acta Anaesthesiol Scand. 1998;42:551–557. doi: 10.1111/j.1399-6576.1998.tb05165.x. [DOI] [PubMed] [Google Scholar]
- 65.Larsson A, Eriksson M, Lundahl T, Lundkvist K, Lindahl T. Impaired platelet function in endotoxemic pigs analyzed by flow cytometry. Platelets. 1998;9:115–119. doi: 10.1080/09537109876898. [DOI] [PubMed] [Google Scholar]
- 66.Lipcsey M, Larsson A, Olovsson M, Sjolin J, Eriksson MB. Early endotoxin-mediated haemostatic and inflammatory responses in the clopidogrel-treated pig. Platelets. 2005;16:408–414. doi: 10.1080/09537100500163168. [DOI] [PubMed] [Google Scholar]
- 67.de Vos AF, Pater JM, van den Pangaart PS, de Kruif MD, van ’t Veer C, van der Poll T. In vivo lipopolysaccharide exposure of human blood leukocytes induces cross-tolerance to multiple TLR ligands. J Immunol. 2009;183:533–542. doi: 10.4049/jimmunol.0802189. [DOI] [PubMed] [Google Scholar]
- 68.Jayachandran M, Miller VM, Brunn GJ, Owen WG. Platelet response as a sentinel marker of toll-like receptor 4 activation in mice. Thromb Res. 2009 doi: 10.1016/j.thromres.2009.05.005. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jayachandran M, Brunn GJ, Karnicki K, RSM, Owen WG, Miller VM. In vivo effects of lipopolysaccharide and TLR4 on platelet production and activity: Implications for thrombotic risk. J Appl Physiol. 2007;102:429–433. doi: 10.1152/japplphysiol.01576.2005. [DOI] [PubMed] [Google Scholar]
- 70.Stahl AL, Sartz L, Nelsson A, Bekassy ZD, Karpman D. Shiga toxin and lipopolysaccharide induce platelet-leukocyte aggregates and tissue factor release, a thrombotic mechanism in hemolytic uremic syndrome. PLoS One. 2009;4:e6990. doi: 10.1371/journal.pone.0006990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liu ML, Reilly MP, Casasanto P, McKenzie SE, Williams KJ. Cholesterol enrichment of human monocyte/macrophages induces surface exposure of phosphatidylserine and the release of biologically-active tissue factor-positive microvesicles. Arterioscler Thromb Vasc Biol. 2007;27:430–435. doi: 10.1161/01.ATV.0000254674.47693.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Watanabe J, Marathe GK, Neilsen PO, Weyrich AS, Harrison KA, Murphy RC, Zimmerman G, McIntyre TM. Endotoxins stimulate neutrophil adhesion followed by synthesis and release of platelet-activating factor in microparticles. J Biol Chem. 2003;278:33161–33168. doi: 10.1074/jbc.M305321200. [DOI] [PubMed] [Google Scholar]
- 73.Gauley J, Pisetsky DS. The release of microparticles by RAW 264.7 macrophage cells stimulated with TLR ligands. J Leukoc Biol. 2010;87:1115–1123. doi: 10.1189/jlb.0709465. [DOI] [PubMed] [Google Scholar]
- 74.Kim SK, Kim YM, Yeum CE, Jin SH, Chae GT, Lee SB. Rifampicin Inhibits the LPS-induced Expression of Toll-like Receptor 2 via the Suppression of NF-kappaB DNA-binding Activity in RAW 264.7 Cells Korean. J Physiol Pharmacol. 2009;13:475–482. doi: 10.4196/kjpp.2009.13.6.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Angelot F, Seilles E, Biichle S, Berda Y, Gaugler B, Plumas J, Chaperot L, Dignat-George F, Tiberghien P, Saas P, Garnache-Ottou F. Endothelial cell-derived microparticles induce plasmacytoid dendritic cell maturation: potential implications in inflammatory diseases. Haematologica. 2009;94:1502–1512. doi: 10.3324/haematol.2009.010934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rauch U, Bonderman D, Bohrmann B, Badimon JJ, Himber J, Riederer MA, Nemerson Y. Transfer of tissue factor from leukocytes to platelets is mediated by CD15 and tissue factor. Blood. 2000;96:170–175. [PubMed] [Google Scholar]
- 77.Martin S, Tesse A, Hugel B, Martinez MC, Morel O, Freyssinet JM, Andriantsitohaina R. Shed membrane particles from T lymphocytes impair endothelial function and regulate endothelial protein expression. Circulation. 2004;109:1653–1659. doi: 10.1161/01.CIR.0000124065.31211.6E. [DOI] [PubMed] [Google Scholar]
- 78.Distler JH, Pisetsky DS, Huber LC, Kalden JR, Gay S, Distler O. Microparticles as regulators of inflammation: novel players of cellular crosstalk in the rheumatic diseases. Arthritis Rheum. 2005;52:3337–3348. doi: 10.1002/art.21350. [DOI] [PubMed] [Google Scholar]
- 79.Mesri M, Altieri DC. Endothelial cell activation by leukocyte microparticles. J Immunol. 1998;161:4382–4387. [PubMed] [Google Scholar]
- 80.Miller VM, Redfield MM, McConnell JP. Use of BNP and CRP as Biomarkers in Assessing Cardiovascular disease: Diagnosis Versus Risk. Current Vascular Pharmacology. 2007;5:15–25. doi: 10.2174/157016107779317251. [DOI] [PubMed] [Google Scholar]
- 81.Ridker PM, Silvertown JD. Inflammation, C-reactive protein, and atherothrombosis. J Periodontol. 2008;79:1544–1551. doi: 10.1902/jop.2008.080249. [DOI] [PubMed] [Google Scholar]
- 82.Barichello T, Savi GD, Silva GZ, Generoso JS, Bellettini G, Vuolo F, Petronilho F, Feier G, Comim CM, Quevedo J, Dal-Pizzol F. Antibiotic therapy prevents, in part, the oxidative stress in the rat brain after meningitis induced by Streptococcus pneumoniae. Neurosci Lett. 2010;478:93–96. doi: 10.1016/j.neulet.2010.04.072. [DOI] [PubMed] [Google Scholar]
- 83.Calvino FM, Parra CT. H. pylori and mitochondrial changes in epithelial cells. The role of oxidative stress. Rev Esp Enferm Dig. 2010;102:41–50. doi: 10.4321/s1130-01082010000100006. [DOI] [PubMed] [Google Scholar]
- 84.Patel KD, Zimmerman GA, Prescott SM, McIntyre TM. Novel leukocyte agonists are released by endothelial cells exposed to peroxide. J Biol Chem. 1992;267:15168–15175. [PubMed] [Google Scholar]
- 85.Kamat JP, Devasagayam TP, Priyadarsini KI, Mohan H. Reactive oxygen species mediated membrane damage induced by fullerene derivatives and its possible biological implications. Toxicology. 2000;155:55–61. doi: 10.1016/s0300-483x(00)00277-8. [DOI] [PubMed] [Google Scholar]
- 86.Misra MK, Sarwat M, Bhakuni P, Tuteja R, Tuteja N. Oxidative stress and ischemic myocardial syndromes. Med Sci Monit. 2009;15:RA209–219. [PubMed] [Google Scholar]
- 87.Forstermann U. Nitric oxide and oxidative stress in vascular disease. Pflugers Arch. 2010;459:923–939. doi: 10.1007/s00424-010-0808-2. [DOI] [PubMed] [Google Scholar]
- 88.Ceriello A, Motz E. Is oxidative stress the pathogenic mechanism underlying insulin resistance, diabetes, and cardiovascular disease? The common soil hypothesis revisited. Arterioscler Thromb Vasc Biol. 2004;24:816–823. doi: 10.1161/01.ATV.0000122852.22604.78. [DOI] [PubMed] [Google Scholar]
- 89.Shargorodsky M, Debby O, Matas Z, Zimlichman R. Effect of long-term treatment with antioxidants (vitamin C, vitamin E, coenzyme Q10 and selenium) on arterial compliance, humoral factors and inflammatory markers in patients with multiple cardiovascular risk factors. Nutr Metab. 2010;7:55. doi: 10.1186/1743-7075-7-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Helal O, Defoort C, Robert S, Marin C, Lesavre N, Lopez-Miranda J, Riserus U, Basu S, Lovegrove J, McMonagle J, Roche HM, Dignat-George F, Lairon D. Increased levels of microparticles originating from endothelial cells, platelets and erythrocytes in subjects with metabolic syndrome: Relationship with oxidative stress. Nutr Metab Cardiovasc Dis. 2010 doi: 10.1016/j.numecd.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 91.Jayachandran M, Preston CC, Hunter LW, Jahangir A, Owen WG, Korach KS, Miller VM. Loss of Estrogen Receptor β Decreases Mitochondrial Energetic Potential and Increases Thrombogenicity of Platelets in Aged Female Mice. Age. 2010;32:109–121. doi: 10.1007/s11357-009-9119-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Freikman I, Amer J, Cohen JS, Ringel I, Fibach E. Oxidative stress causes membrane phospholipid rearrangement and shedding from RBC membranes--an NMR study. Biochim Biophys Acta. 2008;1778:2388–2394. doi: 10.1016/j.bbamem.2008.06.008. [DOI] [PubMed] [Google Scholar]
- 93.Vince RV, Chrismas B, Midgley AW, McNaughton LR, Madden LA. Hypoxia mediated release of endothelial microparticles and increased association of S100A12 with circulating neutrophils. Oxidative medicine and cellular longevity. 2009;2:2–6. doi: 10.4161/oxim.2.1.7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rossig L, Haendeler J, Mallat Z, Hugel B, Freyssinet JM, Tedgui A, Dimmeler S, Zeiher AM. Congestive heart failure induces endothelial cell apoptosis: protective role of carvedilol. J Am Coll Cardiol. 2000;36:2081–2089. doi: 10.1016/s0735-1097(00)01002-0. [DOI] [PubMed] [Google Scholar]
- 95.Rossig L, Hoffmann J, Hugel B, Mallat Z, Haase A, Freyssinet JM, Tedgui A, Aicher A, Zeiher AM, Dimmeler S. Vitamin C inhibits endothelial cell apoptosis in congestive heart failure. Circulation. 2001;104:2182–2187. doi: 10.1161/hc4301.098284. [DOI] [PubMed] [Google Scholar]
- 96.Essayagh S, Xuereb JM, Terrisse AD, Tellier-Cirioni L, Pipy B, Sie P. Microparticles from apoptotic monocytes induce transient platelet recruitment and tissue factor expression by cultured human vascular endothelial cells via a redox-sensitive mechanism. Thromb Haemost. 2007;98:831–837. [PubMed] [Google Scholar]
- 97.Herkert O, Djordjevic T, BelAiba RS, Gorlach A. Insights into the redox control of blood coagulation: role of vascular NADPH oxidase-derived reactive oxygen species in the thrombogenic cycle. Antioxidants & redox signaling. 2004;6:765–776. doi: 10.1089/1523086041361695. [DOI] [PubMed] [Google Scholar]
- 98.Wesche-Soldato DE, Swan RZ, Chung CS, Ayala A. The apoptotic pathway as a therapeutic target in sepsis. Current drug targets. 2007;8:493–500. doi: 10.2174/138945007780362764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Abid Hussein MN, Boing AN, Sturk A, Hau CM, Nieuwland R. Inhibition of microparticle release triggers endothelial cell apoptosis and detachment. Thromb Haemost. 2007;98:1096–1107. doi: 10.1160/th05-04-0231. [DOI] [PubMed] [Google Scholar]
- 100.Yu JL, May L, Lhotak V, Shahrzad S, Shirasawa S, Weitz JI, Coomber BL, Mackman N, Rak JW. Oncogenic events regulate tissue factor expression in colorectal cancer cells: implications for tumor progression and angiogenesis. Blood. 2005;105:1734–1741. doi: 10.1182/blood-2004-05-2042. [DOI] [PubMed] [Google Scholar]
- 101.van Doormaal FF, Kleinjan A, Di Nisio M, Buller HR, Nieuwland R. Cell-derived microvesicles and cancer. Neth J Med. 2009;67:266–273. [PubMed] [Google Scholar]
- 102.Boing AN, Hau CM, Sturk A, Nieuwland R. Platelet microparticles contain active caspase 3. Platelets. 2008;19:96–103. doi: 10.1080/09537100701777295. [DOI] [PubMed] [Google Scholar]
- 103.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 104.Kiechl S, Egger G, Mayr M, Wiedermann CJ, Bonora E, Oberhollenzer F, Muggeo M, Xu Q, Wick G, Poewe W, Willeit J. Chronic infections and the risk of carotid atherosclerosis. Prospective results from a large population study. Circulation. 2001;103:1064–1070. doi: 10.1161/01.cir.103.8.1064. [DOI] [PubMed] [Google Scholar]
- 105.Weerheim AM, Kolb AM, Sturk A, Nieuwland R. Phospholipid composition of cell-derived microparticles determined by one-dimensional high-performance thin-layer chromatography. Anal Biochem. 2002;302:191–198. doi: 10.1006/abio.2001.5552. [DOI] [PubMed] [Google Scholar]
- 106.Jayachandran M, Litwiller RD, Owen WG, Heit JA, Behrenbeck TR, Mulvagh SL, Araoz PA, Budoff MJ, Harman SM, Miller VM. Characterization of Blood Borne Microparticles as Markers of Premature Coronary Calcification in Newly Menopausal Women. Am J Physiol Heart Circ Physiol. 2008;295:931–938. doi: 10.1152/ajpheart.00193.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Miller VM, Jayachandran M, Hashimoto K, Heit JA, Owen WG. Estrogen, Inflammation, and Platelet Phenotype. Gender Medicine. 2008;5:S91–S102. doi: 10.1016/j.genm.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 108.Morel O, Toti F, Hugel B, Camoin-Jau L, Dignat-George F, Freyssinet JM. Procoagulant Microparticles: Disrupting the Vascular Homeostasis Equation? Arterioscler Thromb Vasc Biol. 2006;26:2594–2604. doi: 10.1161/01.ATV.0000246775.14471.26. [DOI] [PubMed] [Google Scholar]
- 109.Bouchard BA, Mann KG, Butenas S. No evidence for tissue factor on platelets. Blood. 2010;166:854–855. doi: 10.1182/blood-2010-05-285627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Muller I, Klocke A, Alex M, Kotzsch M, Luther T, Morgenstern E, Zieseniss S, Zahler S, Preissner K, Engelmann B. Intravascular tissue factor initiates coagulation via circulating microvesicles and platelets. FASEB J. 2003;17:476–478. doi: 10.1096/fj.02-0574fje. [DOI] [PubMed] [Google Scholar]
- 111.Panes O, Matus V, Saez CG, Quiroga T, Pereira J, Mezzano D. Human platelets synthesize and express functional tissue factor. Blood. 2007;109:5242–5250. doi: 10.1182/blood-2006-06-030619. [DOI] [PubMed] [Google Scholar]
- 112.Key NS. Platelet tissue factor: how did it get there and is it important? Semin Hematol. 2008;45(2 Suppl 1):S16–20. doi: 10.1053/j.seminhematol.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 113.Falati S, Liu Q, Gross P, Merrill-Skoloff G, Chou J, Vandendries E, Celi A, Croce K, Furie BC, Furie B. Accumulation of tissue factor into developing thrombi in vivo is dependent upon microparticle P-selectin glycoprotein ligand 1 and platelet P-selectin. J Exp Med. 2003;197:1585–1598. doi: 10.1084/jem.20021868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Collen D, Hoylaerts MF. Relationship betwen inflammation and venous thromboembolism as studied by microparticle assessment in plasma. J Am Coll Cardiol. 2005;45:1472–1473. doi: 10.1016/j.jacc.2005.02.029. [DOI] [PubMed] [Google Scholar]
- 115.Furie B, Furie BC. Role of platelet P-selectin and microparticle PSGL-1 in thrombus formation. Trends in molecular medicine. 2004;10:171–178. doi: 10.1016/j.molmed.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 116.Del Conde I, Shrimpton CN, Thiagarajan P, Lopez JA. Tissue-factor-bearing microvesicles arise from lipid rafts and fuse with activated platelets to initiate coagulation. Blood. 2005;106:1604–1611. doi: 10.1182/blood-2004-03-1095. [DOI] [PubMed] [Google Scholar]
- 117.Bach R, Rifkin DB. Expression of tissue factor procoagulant activity: regulation by cytosolic calcium. Proc Natl Acad Sci U S A. 1990;87:6995–6999. doi: 10.1073/pnas.87.18.6995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Butenas S, Bouchard BA, Brummel-Ziedins KE, Parhami-Seren B, Mann KG. Tissue factor activity in whole blood. Blood. 2005;105:2764–2770. doi: 10.1182/blood-2004-09-3567. [DOI] [PubMed] [Google Scholar]
- 119.Giesen PLA, Rauch U, Bohrmann B, Kling D, Roque M, Fallon JT, Badimon JJ, Himber J, Riederer MA, Nemerson Y. Blood-borne tissue factor: Another view of thrombosis. Proc Natl Acad Sci USA. 1999;96:2311–2315. doi: 10.1073/pnas.96.5.2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Boulanger CM, Scoazec A, Ebrahimian T, Henry P, Mathieu E, Tedgui A, Mallat Z. Circulating microparticles from patients with myocardial infarction cause endothelial dysfunction. Circulation. 2001;104:2649–2652. doi: 10.1161/hc4701.100516. [DOI] [PubMed] [Google Scholar]
- 121.Agouni A, Lagrue-Lak-Hal AH, Ducluzeau PH, Mostefai HA, Draunet-Busson C, Leftheriotis G, Heymes C, Martinez MC, Andriantsitohaina R. Endothelial dysfunction caused by circulating microparticles from patients with metabolic syndrome. Am J Pathol. 2008;173:1210–1219. doi: 10.2353/ajpath.2008.080228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tesse A, Martinez MC, Hugel B, Chalupsky K, Muller CD, Meziani F, Mitolo-Chieppa D, Freyssinet JM, Andriantsitohaina R. Upregulation of proinflammatory proteins through NF-kappaB pathway by shed membrane microparticles results in vascular hyporeactivity. Arterioscler Thromb Vasc Biol. 2005;25:2522–2527. doi: 10.1161/01.ATV.0000189298.62240.5d. [DOI] [PubMed] [Google Scholar]
- 123.Forlow SB, McEver RP, Nollert MU. Leukocyte-leukocyte interactions mediated by platelet microparticles under flow. Blood. 2000;95:1317–1323. [PubMed] [Google Scholar]
- 124.Barry OP, Pratico D, Savani RC, FitzGerald GA. Modulation of monocyte-endothelial cell interactions by platelet microparticles. J Clin Invest. 1998;102:136–144. doi: 10.1172/JCI2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Libby P, Theroux P. Pathophysiology of coronary artery disease. Circulation. 2005;111:3481–3488. doi: 10.1161/CIRCULATIONAHA.105.537878. [DOI] [PubMed] [Google Scholar]
- 126.Ludwig RJ, Schultz JE, Boehncke WH, Podda M, Tandi C, Krombach F, Baatz H, Kaufmann R, von Andrian UH, Zollner TM. Activated, not resting, platelets increase leukocyte rolling in murine skin utilizing a distinct set of adhesion molecules. J Invest Dermatol. 2004;122:830–836. doi: 10.1111/j.0022-202X.2004.22318.x. [DOI] [PubMed] [Google Scholar]
- 127.Sabatier F, Roux V, Anfosso F, Camoin L, Sampol J, Dignat-George F. Interaction of endothelial microparticles with monocytic cells in vitro induces tissue factor-dependent procoagulant activity. Blood. 2002;99:3962–3970. doi: 10.1182/blood.v99.11.3962. [DOI] [PubMed] [Google Scholar]
- 128.Kiechl S, Lorenz E, Reindl M, Wiedermann CJ, Oberhollenzer F, Bonora E, Willeit J, Schwartz DA. Toll-like receptor 4 polymorphisms and atherogenesis. N Engl J Med. 2002;347:185–192. doi: 10.1056/NEJMoa012673. [DOI] [PubMed] [Google Scholar]
- 129.de Kleijn D, Pasterkamp G. Toll-like receptors in cardiovascular diseases. Cardiovasc Res. 2003;60:58–67. doi: 10.1016/s0008-6363(03)00348-1. [DOI] [PubMed] [Google Scholar]
- 130.Bucova M, Bernadic M, Buckingham T. C-reactive protein, cytokines and inflammation in cardiovascular diseases. Bratisl Lek Listy. 2008;109:333–340. [PubMed] [Google Scholar]
- 131.MacKenzie A, Wilson HL, Kiss-Toth E, Dower SK, North RA, Surprenant A. Rapid secretion of interleukin-1beta by microvesicle shedding. Immunity. 2001;15:825–835. doi: 10.1016/s1074-7613(01)00229-1. [DOI] [PubMed] [Google Scholar]
- 132.Hart DN. Dendritic cells: unique leukocyte populations which control the primary immune response. Blood. 1997;90:3245–3287. [PubMed] [Google Scholar]
- 133.Kaneider NC, Kaser A, Tilg H, Ricevuti G, Wiedermann CJ. CD40 ligand-dependent maturation of human monocyte-derived dendritic cells by activated platelets. International journal of immunopathology and pharmacology. 2003;16:225–231. doi: 10.1177/039463200301600307. [DOI] [PubMed] [Google Scholar]
- 134.Sprague DL, Elzey BD, Crist SA, Waldschmidt TJ, JJR, Ratliff TL. Platelet-mediated modulation of adaptive immunity: unique delivery of CD154 signal by platelet-derived membrane vesicles. Blood. 2008;111:5028–5036. doi: 10.1182/blood-2007-06-097410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Epstein SE, Zhu J, Burnett MS, Zhou YF, Vercellotti GM, Hajjar D. Infection and atherosclerosis: Potential roles of pathogen burden and molecular mimicry. Arterioscler Thromb Vasc Biol. 2000;20:1417–1420. doi: 10.1161/01.atv.20.6.1417. [DOI] [PubMed] [Google Scholar]
- 136.Zhu J, Nieto FJ, Horne BD, Anderson JL, Muhlestein JB, Epstein SE. Prospective study of pathogen burden and risk of myocardial infarction or death. Circulation. 2001;103:45–51. doi: 10.1161/01.cir.103.1.45. [DOI] [PubMed] [Google Scholar]
- 137.Espinola-Klein C, Rupprecht HJ, Blankenberg S, Bickel C, Kopp H, Victor A, Hafner G, Prellwitz W, Schlumberger W, Meyer J. Impact of infectious burden on progression of carotid atherosclerosis. Stroke. 2002;33:2581–2586. doi: 10.1161/01.str.0000034789.82859.a4. [DOI] [PubMed] [Google Scholar]
- 138.Jayachandran M, Litwiller RD, Owen WG, Miller VM. Circulating Microparticles and Endogenous Estrogen in Newly Menopausal Women. Climacteric. 2009;12:177–184. doi: 10.1080/13697130802488607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Denti L. The hormone replacement therapy (HRT) of menopause: focus on cardiovascular implications. Acta Biomed. 2010;81 (Suppl 1):73–76. [PubMed] [Google Scholar]
- 140.Bittner V. Menopause, age, and cardiovascular risk: a complex relationship. J Am Coll Cardiol. 2009;54:2374–2375. doi: 10.1016/j.jacc.2009.10.008. [DOI] [PubMed] [Google Scholar]