

Abstract
Unlike direct ESR, spin trap methodology depends on the absolute fidelity of the spin trap reaction. Two alternative reactions of 5,5-dimethyl-1-pyrroline N-oxide (DMPO) leading to radical adduct artifacts have been discovered and investigated: inverted spin trapping and the Forrester-Hepburn nucleophilic mechanisms. These two alternate pathways to radical adducts are a combination of one-electron oxidation and nucleophilic addition, in either order. In biological systems, serious artifacts have been reported due to the Forrester-Hepburn mechanism, which is initiated by the addition of a nucleophile to DMPO. It has recently been demonstrated that (bi)sulfite (hydrated sulfur dioxide) can react with DMPO via a nonradical, nucleophilic reaction, and it has been further proposed that DMPO/•SO3− formation in biological systems is an artifact and not the result of spin trapping of sulfur trioxide anion radical (•SO3−). The one-electron oxidation of (bi)sulfite catalyzed by horseradish peroxidase (HRP)/hydrogen peroxide (H2O2) has been reinvestigated by ESR spin trapping with DMPO and oxygen uptake studies to obtain further evidence for the radical reaction mechanism. In the absence of DMPO, the initial rate of (bi)sulfite-dependent oxygen and H2O2 consumption was determined to be half of the initial rate of DMPO/•SO3− radical adduct formation as determined by ESR, demonstrating that, under our experimental conditions, DMPO exclusively forms the radical adduct by trapping the •SO3−.
Keywords: ESR, spin trap, DMPO, sulfite radicals
In principle, the best method for characterizing free radicals in biological systems is direct detection by electron spin resonance [ESR; also known as electron paramagnetic resonance (EPR)] because this technique is based on fundamental physics and makes no assumptions. All other approaches of free radical detection are based on assumptions of radical chemistry. Many assays for superoxide have been developed based on the disproportionation of superoxide by superoxide dismutase.[1,2] The specificity of this enzyme has insured that these assays are robust and reliable. Unfortunately, enzymes have not evolved to disproportionate the multiplicity of radicals of interest in biology and medicine.
The major limitation of ESR is that its sensitivity, which is comparable to that of UV-visible spectroscopy of a strong chromophore, is poor relative to the instability of free radicals in biological solution. As a result, in the majority of cases in enzymatic systems, cells, and especially in vivo, the steady-state radical concentration is less than 1 nM, which is below the detection limit of ESR. In addition, for quantum mechanical reasons, diatomic radicals (e.g., hydroxyl and superoxide) are undetectable by direct ESR in solution.
For these reasons, a less direct, but highly successful ESR technique called spin trapping has been used (Eqn 1a).[3] The spin trapping depends on the specific reaction of free radicals with the double bond of the spin traps to form a stable nitroxide species:
| (1a) |
As a result, a nanomolar steady-state radical concentration can accumulate to high micromolar concentrations of spin adducts, also called radical adducts. Indeed, a number of free radical metabolites of xenobiotics, biochemicals, and proteins have been detected by ESR spin trapping both in vitro and in vivo.[3–5]
The most popular spin trap is 5,5-dimethyl-1-pyrroline N-oxide (DMPO), which has been cited in Medline more than 1,000 times (Eqn 1b). DMPO has significant advantages over other nitrone spin traps. First, it is the most redox inactive. Second, common nitrone spin traps other than DMPO such as α-phenyl-N-tert-butylnitrone (PBN) and α-(4-pyridyl-1-oxide)-N-tert-butylnitrone (POBN) have ESR spectra of their radical adducts which show relatively little dependence on the structure of the trapped radical, whereas the assignment of DMPO radical adducts of small free radicals can often be made from knowledge of the literature.[3] Although misassignments have occurred, many of these mistakes have been corrected, and a substantial knowledge base now exists for DMPO radical adducts.[6,7]
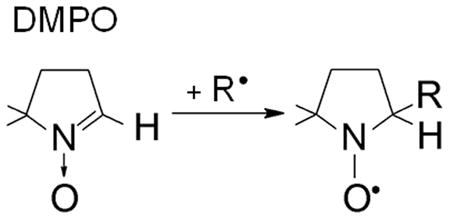 |
(1b) |
Unlike direct ESR, the spin trap methodology depends on the absolute fidelity of the spin-trapping reaction (Eqn 1b). Two alternate reactions of DMPO leading to radical adduct artifacts have been discovered and investigated – inverted spin trapping[8–10] and the Forrester-Hepburn mechanism.[11] These two alternate pathways to radical adducts are a combination of one-electron oxidation of the spin trap and nucleophilic addition, in either order (Scheme 1). The first of these reactions, inverted spin trapping, is of limited importance for DMPO because of its very high oxidation potential (EDMPO</sup>•+</sup>/DMPO = 1.63 V). DMPO is also very resistant to the two electron reduction to its hydroxylamine with its very low potential, −1.92 V. In fact, DMPO exhibits the widest potential window of any spin trap investigated.[12]
Scheme 1.

If DMPO is oxidized to its cation radical, water, as the most abundant nucleophile in biological systems, would be expected to react with it, to form DMPO/•OH. In fact, the DMPO cation radical is formed by ionizing radiation in dry Freon matrices at 77°K.[13] On melting in the absence of water, the DMPO•+ was very stable, but in very low concentrations in the presence of water, the cation radicals were converted into the DMPO/•OH radical adduct. This work went on to speculate that DMPO/•OH formation may occur in biological Fenton systems. Inverted spin trapping has also been described in photochemical oxidations or with strong oxidants such as tris(4-bromophenyl) aminium ion followed by further reaction of the cation radical with nucleophiles.[14]
To our knowledge, only two species capable of oxidizing DMPO to its cation radical have been reported in biological systems: the hydroxyl radical (EOH•/OH− = 2.8 V)[15] and sulfate anion radical, •OSO3− (ESO4•−/SO42− = 2.43 V).[16] In a biological Fenton system consisting of xanthine, xanthine oxidase, Fe(II), and [17O] oxygen (64 atom %), the molecular oxygen is enzymatically reduced to hydrogen peroxide (H2O2).[17] Fe(II) reduces this H2O2 to the hydroxyl radical. The vast majority of the DMPO/•OH formed in this system originated from the [17O] oxygen as indicated by the intensity of the 17O hyperfine coupling being close to the expected isotope ratio. If DMPO had been oxidized by the hydroxyl radical to its cation, then all of the oxygen would have originated from the [16O] water.
If DMPO were to be oxidized to its cation radical by •OSO3− as PBN (EPBN/PBN•+ = 1.47 V) is known to do, [18] it would react with water to form DMPO/•OH, not DMPO/•OSO3−. In fact, the sulfate anion is trapped by DMPO in a sulfite/HRP/H2O2 system (Fig. 1D).[19] Interestingly, the trapping of •OSO3− occurs only at the relatively low DMPO concentration of 3 mM. At the higher concentration of 100 mM DMPO, only the primary radical •SO3− is trapped (Fig. 1E). If oxidation of DMPO to its cation had any role in DMPO/•OSO3− formation, it should increase with DMPO. A surprising finding is that hydrolysis of spin adducts with strongly electronegative leaving groups such as SO42− is relatively fast, forming DMPO/•OH (Fig. 1C).[20] In summary, inverted spin trapping with DMPO in biological systems is not a significant problem because the oxidation potential of DMPO (EDMPO•+/DMPO = 1.63 V) is generally much higher than that generated in biological systems, and if inverted spin trapping did occur, only DMPO/•OH would form because of the very high concentrations of water in biological systems.
Fig. 1.

(Spectrum A) Formation of radical adducts in the reaction between Na2SO3 (10 mM), DMPO (3 mM), HRP (0.25 mg/mL) and H2O2 (10 μM) in 100 mM phosphate buffer, pH 7.9. (Spectrum B) Computer simulation of the DMPO/•OSO3− (aN = 13.8 G, , aHγ1 = 1.38 G, and aHγ2= 0.68 G), DMPO/•SO3− (aN = 14.7 G, ), and DMPO/•OH (aN = 15.0 G, ) radical adducts. (Spectrum C) Simulation of DMPO/•OH radical adduct. (Spectrum D) Simulation of DMPO/•OSO3− radical adduct. (Spectrum E) Simulation of DMPO/•SO3− radical adduct. This figure is adapted from ref. [19].
In biological systems, the most serious potential artifact is likely to be due to the Forrester-Hepburn mechanism, which is initiated by addition of a nucleophile to the spin trap (Scheme 1). Once formed, the hydroxylamine product is easily oxidized to give the radical adduct. Because water is the most abundant nucleophile in biological systems, it is not surprising that this reaction has been reported to form DMPO/•OH. Traces of DMPO/•OH are always detectable in water containing DMPO, and this has long been assumed to be due to slow hydrolysis of DMPO.[21] A much higher concentration of DMPO/•OH adduct has been reported to occur through nucleophilic addition of water in the presence of the Lewis acid Fe(III).[22] Due to the serious implications of these findings with respect to many spin-trapping studies, the suitability of DMPO as a hydroxyl radical spin trap was studied in typical Fenton systems. Using 17O-enriched water, this laboratory showed conclusively that nucleophilic addition of water occurs at the nitrone carbon (or C-2 position) of DMPO in the presence of either Fe(III) or Cu(II) ions.[23] Furthermore, these results demonstrate that this nucleophilic reaction is a major pathway to the DMPO/•OH adduct, even during the reaction of Fe(II) or Cu(I) with H2O2. When the reaction is carried out in various buffers, however, or in the presence of metal chelators, nucleophilic addition to DMPO from Fe(III) is effectively suppressed. [23] Chelators are necessary to suppress the reaction with Cu(II). Because chelators should always be present in biological systems to suppress trace transition metal-catalyzed reactions, nucleophilic addition of water to DMPO is not a major concern, even though in pure water DMPO is a ligand of Cu(II) and Fe(III).
In any case, to establish the existence of free hydroxyl radical in spin-trapping experiments, it is necessary to perform kinetic-based competition experiments with hydroxyl radical scavengers. For example, ethanol, formate, and dimethyl sulfoxide can be used in these competition experiments because, upon hydroxyl radical attack, they form carbon-centered radicals that can subsequently be trapped by DMPO.[24] Most artifacts leading to DMPO/•OH radical adduct formation will be excluded by the use of hydroxyl radical scavengers if the scavenger-derived radical adduct is detected and a corresponding decrease in the DMPO/•OH radical adduct concentration is found. Even quantitative kinetic criteria can be used. [25]
It has recently been reported that DMPO underwent nucleophilic addition of sulfite with the formation of the corresponding hydroxylamine adduct, which, upon mild one-electron oxidation, could form the radical adduct (Scheme 1).[26] Sulfur dioxide, one of the major atmospheric pollutants, is water soluble and, in aqueous solution at neutral pH, exists primarily as sulfite (SO32−) and (bi)sulfite (HSO3−) (pKa = 7.2).[27,28] Owing to its antioxidant and antimicrobial properties, (bi)sulfite is used extensively as a preservative in beverages and foods.[29] One-electron oxidation of (bi)sulfite to sulfate is catalyzed enzymatically by prostaglandin H synthase[30] and horseradish peroxidase (HRP).[31,32] The proposed mechanism of enzymatic oxidation of (bi)sulfite to the •SO3− radical by the HRP/H2O2 system occurs by two sequential, one-electron reduction reactions of compound I and compound II by (bi)sulfite:[19,30,32,33]
| (2) |
| (3) |
| (4) |
The net equation with trapping by DMPO is:
| (5) |
In the absence of DMPO, •SO3− reacts with molecular oxygen to form a peroxymonosulfate anion radical (−O3SOO•) which, upon reaction with excess (bi)sulfite, forms the sulfate anion radical (SO4•−). Both •SO3− and SO4•− were trapped by DMPO when sulfite was oxidized by the HRP/H2O2 system, where it was thought that DMPO/•SO3− formed through the radical’s addition across the double bond of the spin trap. However, Potapenko et al.[26] caution that the nonradical Forrester-Hepburn nucleophilic addition of sulfite may be the actual origin of the paramagnetic adduct and suggest that more careful analysis and additional experiments are necessary to prove the radical mechanism. Their primary evidence is that after one hour the NMR spectra of DMPO in the presence of sulfite showed significant concentrations of a new species identified as the hydroxylamine adduct. There is no question that that HRP forms •SO3− from sulfite because it has been detected directly with ESR and has a distinctive g value of 2.0031.[32] Nonetheless, the need to do additional experiments to test the origin of DMPO/•SO3− in this system was clear, because the mere formation of •SO3− does not prove that the radical adduct forms by trapping •SO3−.[34]
In order to distinguish between true spin trapping and nucleophilic addition, we first measured the predicted 2:1 stoichiometry of the DMPO/•SO3− radical adduct and H2O2 (Eqn. 5). Samples containing (bi)sulfite were incubated with 100 mM DMPO and 10 μM H2O2, and then the reaction was initiated with 10 μM HRP. The double-integrated intensity of each ESR spectrum (Fig. 2A) was plotted on a standard curve for the nitroxide tempol to calculate the concentration of DMPO/•SO3−. We obtained a maximum DMPO/•SO3− adduct yield of 19.5 ± 0.3 μM (Fig. 2B). This 2:1 stoichiometry (2DMPO/•SO3−: 1H2O2, 10 μM) is in excellent quantitative agreement with the calculated stoichiometry in Eqn (5). In the case of the Forrester-Hepburn nucleophilic mechanism, (bi)sulfite reacts with DMPO to form the corresponding hydroxylamine, which, in time, does happen.[26] The HRP/H2O2 system would then oxidize this hydroxylamine to DMPO/•SO3−, which would have exactly the same stoichiometry as the spin-trapping reaction, 2DMPO/•SO3−: 1H2O2. So, product yield cannot distinguish between these two mechanisms.
Fig. 2.

(A) Formation of DMPO/•SO3− radical adduct in the reaction between sodium sulfite (Na2SO3) and H2O2/HRP in the presence of DMPO. (Spectrum a) Reaction mixture containing Na2SO3 (50 mM), DMPO (100 mM) and H2O2 (10 μM) in 100 mM phosphate buffer, pH 7.4. After initiation with HRP (10 μM), the mixture was placed immediately into the flat cell. The double integral of the spectrum is proportional to the concentration of DMPO/•SO3− radical adduct. (Spectrum b) Same as in (spectrum a) without HRP. (Spectrum c) Same as in (spectrum a) without H2O2. (Spectrum d) Same as in (spectrum c) with 30,000 units of catalase/mL of incubation. (Spectrum e) Same as in (spectrum a), except the Na2SO3 was not added. (Spectrum f) Same as in (spectrum a) without the spin trap. (Spectrum g) Same as in (spectrum b) without H2O2. The double integral values are an average of 64 scans. (B) Dependence of the concentration of DMPO/•SO3− radical adduct on the (bi)sulfite concentrations in the presence of 10 μM H2O2. Reaction mixtures containing various concentrations of sodium sulfite (0.075, 0.150, 0.200, 0.300, 0.400, 0.500, 1, 5, 25 and 50 mM), DMPO (100 mM) and 10 μM H2O2 were initiated with 10 μM HRP in 100 mM phosphate buffer, pH 7.4. The concentration of the paramagnetic species was calculated from the double integrals of the ESR spectra. The estimated maximum concentration of DMPO/•SO3− radical adduct obtained from the extrapolated nonlinear fit is 19.5 ± 0.3 μM. Error bars represent the standard deviation of three independent experiments. This figure is adapted from ref. [34].
To provide a more complete understanding of the radical mechanism of sulfur-derived radicals in the HRP/H2O2 system, we extended the oxygen uptake experiments previously reported.[30,32] Molecular oxygen is mother nature’s spin trap and is similar to DMPO in this sense. In agreement with these reports, when millimolar (bi)sulfite (3–30 mM range) was incubated with 10 μM H2O2 and initiated with 10 μM HRP, very fast oxygen consumption was observed; control experiments confirmed that all the components had to be present in order to observe oxygen consumption. The oxygen consumption is a result of the formation of peroxymonosulfate radical (−O3SOO•) in the following free radical chain mechanism:[19,27,35]
| (6) |
| (7) |
| (8) |
| (9) |
| (10) |
This mechanism has been accepted and supported by various experiments.[19,36,37] To confirm the radical mechanism between the DMPO and •SO3− discussed above, we measured the stoichiometric molar ratio between the oxygen and (bi)sulfite from Eqn (10) and calculated the initial rate of oxygen consumption.
Figure 3A shows the simulation[38] of the kinetic system (bi)sulfite/HRP/H2O2 based on Eqns (7)–(9) and previously published rate constants.[16] The initial concentration of oxygen determined in phosphate buffer, pH 7.4, was 245 μM, which was used in the simulation program. The concentration of (bi)sulfite was 300 μM; the initiating concentration of the •SO3− anion radical was chosen to match the experimental curve and was determined to be only 1.4 × 10−13 M. This result supports the literature conclusions that •SO3− formation is dominated by the radical chain reaction. As expected, a complete conversion of (bi)sulfite to sulfate was calculated, and one-half as much oxygen (~ 147 μM) was predicted to be consumed (Fig. 3A), which is in agreement with the mechanism discussed above.
Fig. 3.

(A) Computer simulation of the concentration changes of reactants as a function of time. The initial concentrations were: [O2]0 = 245 μM, [SO32−]0 = 300 μM, [•SO3−]0 = 1.4 × 10−7 μM, and [SO42−]0 = 0 μM. (B) Oxygen uptake curves as a function of (bi)sulfite concentration. Sodium sulfite (Na2SO3) was placed in a chamber with 10 μM H2O2 in 100 mM phosphate buffer, pH 7.4, and the reaction was initiated with 10 μM HRP at the times indicated. The concentration of (bi)sulfite for each curve was as follows: (a) 0 μM, (b) 150 μM, (c) 225 μM, (d) 300 μM and (e) 500 μM. This figure is adapted from ref. [34].
Based on this simulation, the next set of oxygen uptake experiments used 0–500 μM (bi)sulfite mixed with 10 μM H2O2, and initiated with 10 μM HRP. Figure 3B demonstrates the dependence of oxygen consumption on (bi)sulfite concentration. A very good agreement between the calculated and experimental oxygen consumption was observed for spectrum d in Fig. 3B, where the (bi)sulfite concentration (300 μM) was the same as in the simulated data. The experimental oxygen consumption was 148.0 μM ± 2.6, in good agreement with the theoretical stoichiometric value of 150 μM and the theoretical oxygen consumption (Fig. 3A).
Next we evaluated the effect of DMPO concentration on oxygen uptake using 300 μM (bi)sulfite, 10 μM H2O2, and 10 μM HRP (Fig. 4). In the presence of only 100 μM DMPO (spectrum d), oxygen uptake was inhibited by more than 50%. Incubation of (bi)sulfite with higher DMPO concentrations (1–100 mM) showed that the inhibition of oxygen consumption positively correlated with spin trap concentration. Interestingly, at the concentrations of DMPO usually used in ESR experiments (100 mM), oxygen consumption was inhibited almost completely (Fig. 4, spectrum a). This result demonstrates again the very high efficiency of DMPO trapping of the •SO3− anion radical formed either during the HRP-catalyzed oxidation of (bi)sulfite or the subsequent chain reaction. This effect of DMPO was simulated for spectrum d using the known rate constant for the reaction between •SO3− and the spin trap, k = 1.2 × 107 M−1 s−1.[39]
Fig. 4.

Oxygen uptake curves as a function of DMPO concentration. DMPO was placed in a chamber with sodium sulfite (300 μM) and H2O2 (10 μM) in 100 mM phosphate buffer, pH 7.4, and the reaction was initiated with 10 μM HRP at the times indicated. The concentration of DMPO for each curve was as follows: (a) 100 mM, (b) 10 mM, (c) 1 mM, (d) 0.1 mM and (e) 0 mM. This figure is adapted from ref. [34].
To provide further insight into the radical mechanism of (bi)sulfite oxidation, kinetic ESR spin-trapping experiments were performed. The reaction rate of oxygen consumption in Eqn (10) can be represented by:
| (11) |
According to Eqn (5), the initial rate of H2O2 consumption was also predicted to be one half the rate at which (bi)sulfite disappears and one half the rate at which the radical adduct DMPO/•SO3− appears. Therefore, from Eqn (11) we obtain the initial rate stoichiometry:
| (12) |
Concentrations of the reactants were adjusted to give a reaction rate slow enough for conventional ESR instruments. Figure 5A shows a time course plot of the first peak of the DMPO/•SO3− signal. Micromolar concentrations of (bi)sulfite (75–500 μM) were mixed with 100 mM DMPO and 0.5 μM H2O2. The reaction was initiated with 0.5 μM HRP and immediately aspirated into the flat cell. In this incubation, the radical adduct formation increased linearly for the first 25–30 sec. At higher initial concentrations of (bi)sulfite (400 μM and 500 μM), the maximum accumulation of DMPO/•SO3− adduct was observed at ~100–150 sec after HRP initiation, followed by decay. This pattern could be explained by the absence of sufficient H2O2 to continue •SO3− generation in the presence of 100 mM DMPO, where the chain reaction is blocked.
Fig. 5.

(A) Time-sweep ESR kinetics formation of DMPO/•SO3− radical adduct. Reaction mixtures containing sodium sulfite as indicated, DMPO (100 mM) and H2O2 (0.5 μM) were initiated with 0.5 μM HRP in 100 mM phosphate buffer, pH 7.4. The instrument parameters were as follows: microwave frequency, 9.78 GHz; microwave power, 20 mW; modulation amplitude, 2.0 G; field position, 3468 G; receiver gain, 5 × 105; time constant, 328 ms; and sweep time, 336 s. (B) Oxygen uptake curves using identical conditions as in Fig. 4A, except without spin trap DMPO. The concentration of sodium sulfite for each curve was as follows: (a) 0 μM, (b) 75 μM, (c) 150 μM, (d) 300 μM, (e) 400 μM and (f) 500 μM. This figure is adapted from ref. [34].
Oxygen uptake experiments were performed using the same conditions (in the absence of DMPO) as those for the kinetic ESR spin-trapping experiments described above. When 75–500 μM (bi)sulfite was mixed with 0.5 μM H2O2 and 0.5 μM HRP as initiator, the dependence of oxygen consumption on (bi)sulfite concentration was measured (Fig. 5B). In order to test whether the formation of the sulfur trioxide anion radical is a result of the one-electron oxidation step of (bi)sulfite rather than the nucleophilic addition of (bi)sulfite to the spin trap DMPO followed by the oxidation of the resulting hydroxylamine adduct, we compared the initial rate of DMPO/•SO3− radical adduct formation for the ESR spin trapping with the initial rate of oxygen consumption under the same conditions. Figure 6A represents the time course of both techniques using 300 μM (bi)sulfite and 0.5 μM H2O2/0.5 μM HRP. The first 30 sec after reaction initiation were fitted using linear regression (the dotted lines), and the values obtained for the initial rates were 1.49 ± 0.02 and 2.83 ± 0.05 nM sec−1 for oxygen uptake and the ESR time course, respectively. These values are in very good agreement with Eqn (12), where the rate of DMPO/•SO3− radical adduct formation is twice that of the oxygen consumption. This result suggests that •SO3− radical formation is entirely responsible for the oxygen consumption and that DMPO/•SO3− radical adduct is a true product of •SO3−.
Fig. 6.

(A) ESR time course of DMPO/•SO3− adduct formation in comparison with oxygen uptake curve. The solutions for both instruments contained 300 μM Na2SO3, 0.5 μM H2O2, 0.5 μM HRP as initiator and 100 mM DMPO (only for ESR spin trapping of •SO3− radical). The instrumental conditions were the same as those described in the legend of Figure 5A for ESR. (B) Hydrogen peroxide consumption by HRP in the presence of sodium sulfite as a function of time. Na2SO3 was placed in a chamber with 0.5 μM H2O2 in 100 mM phosphate buffer, pH 7.4, and the reaction was initiated with 0.5 μM HRP at the times indicated. (a) H2O2 alone. The concentration of sodium sulfite for each curve was as follows: (b) 0 μM, (c) 100 μM, (d) 300 μM, (e) 400 μM and (f) 500 μM. This figure is adapted from ref. [34].
For comparison, the above experiments were repeated (in the absence of DMPO) using an H2O2-selective electrode to determine the initial rate of H2O2 consumption. As a control, HRP (0.5 μM) was added to a continuously stirred H2O2 (0.5 μM) buffer solution, where it catalyzed a very slow consumption of peroxide due to catalase activity (Fig. 6B, trace b). This background reaction was not taken into account for the calculation of the initial rate (see below). When the same experiments were repeated by adding increasing amounts of (bi)sulfite, the rate of H2O2 consumption was significantly increased (traces c–f), indicating that (bi)sulfite has a role in accelerating this process. The time course corresponding to the experiments shown in Fig. 6A is presented as trace d in Fig. 6B. The initial rate of H2O2 consumption was calculated by fitting the first 30 sec to obtain the value of 1.38 ± 0.09 nM sec−1, which is almost equal to the oxygen consumption rate and approximately 50% of the initial rates of DMPO/•SO3− radical adduct formation calculated above. From the rate equation (14), the ratio between the rates of H2O2 consumption and sulfur trioxide anion radical formation should be 1:2 and, in fact, this was observed. The present data demonstrate that the enzymatic oxidation of (bi)sulfite by HRP and H2O2 proceeds via a radical mechanism as demonstrated using quantitative kinetic oxygen uptake, H2O2 consumption, and ESR spin-trapping experiments.
Our ESR spin-trapping data (Fig. 5A) show that mixing DMPO with micromolar (bi)sulfite followed by immediate oxidation by HRP/H2O2 does form the DMPO/•SO3− radical adduct. Previous work from Potapenko and co-workers on the reaction mechanism of (bi)sulfite oxidation in the presence of DMPO or DEPMPO suggested that a non-radical addition of (bi)sulfite with DMPO was responsible for the ESR detection of the radical adduct.[26,40] A reversible nucleophilic addition of (bi)sulfite anion to the double bond of the spin traps was proposed, resulting in the formation of the hydroxylamine which, upon oxidation by ferricyanide or sodium dichromate, formed an ESR-detected DMPO/•SO3− complex. According to the authors, this radical adduct formation is an example of a detectable paramagnetic species that did not occur from radical chemistry.
In the current work, kinetic ESR and oxygen consumption experiments directly tested this hypothesis. The reported DMPO-sulfite hydroxylamine adduct was detected after pre-incubation of 100 mM (bi)sulfite with DMPO.[26] In short, high concentrations of both (bi)sulfite and DMPO were used for longer incubation times. These experimental conditions facilitated the relatively slow nucleophilic addition of (bi)sulfite to the double bond of the nitrone spin trap, resulting in the formation of NMR-detected hydroxylamine with a calculated equilibrium constant Keq ≈ 18 lmol−1. It is very unlikely that the immediate recording of the ESR spectra (within 3 min) allowed the corresponding DMPO-sulfite hydroxylamine to accumulate and be further oxidized as is described by Potapenko et al.[26] Furthermore, these prior studies, in which the radical adduct formation was reported to form by oxidation of the hydroxylamine by millimolar concentrations of ferricyanide or sodium dichromate, [26,40] neglected the fact that Fe(III) and Cr(VI) are known to catalyze the autoxidation of (bi)sulfite via
| (13) |
where M = Fe(III) or CrO42−.
We have demonstrated that, under our experimental conditions, the ESR-detected DMPO/•SO3− radical adduct is formed upon addition of the •SO3− radical across the double bond of DMPO, and not indirectly by nucleophilic addition of (bi)sulfite to the spin trap. The calculations from quantitative ESR experiments show that there is the predicted ratio between the rate of formation of DMPO/•SO3− radical adduct and the rates of oxygen and H2O2 uptake. It has been shown that the consumption of oxygen can proceed via radical chain chemistry of •SO3− and oxygen. Therefore, it would appear that DMPO is not susceptible to artifacts arising from nonradical chemistry (nucleophilic addition) under the experimental conditions we have used and that the Forrester-Hepburn reaction affected neither the quantitative analysis of the data nor the conclusion about the biochemical origin of the DMPO/•SO3− adduct.
The question of nucleophilic addition of (bi)sulfite leading to radical adduct formation under other experimental conditions must also be considered. It is not practical to investigate quantitatively and kinetically a wide range of concentrations of H2O2, HRP, sulfite, and DMPO, but another approach has been proposed to test any contribution of the Forrester-Hepburn nucleophilic mechanism to the formation of DMPO/•SO3− or any other radical adduct possible. Timmins et al.[48] proposed the isotopic labeling of the presumably slower nucleophilic addition of a spin trap such as 15N-DMPO during a pre-incubation period (Scheme 2). This pre-incubation could be as long as it takes to obtain an ESR spectrum, followed by rapid addition of an equal concentration of 14N-DMPO and the initiators of radical generation - in the case of sulfite, H2O2 and HRP. This would result in a differential isotopic labeling of the radical adduct from the Forrester-Hepburn and the spin-trapping reactions. If the experiment is repeated, reversing the order of isotope addition, a lack of non-radical addition will result in identical spectra. The occurrence of the Forrester-Hepburn addition will manifest itself in favoring the product of the initially added isotope. At high enough concentrations of sulfite and DMPO, the nucleophilic reaction could be fast enough to affect the results. In the unlikely case that the nucleophilic reaction was much faster than the radical reaction, the two spectra would also be the same regardless of the order of isotopic DMPO addition. If a free radical reaction did not, in fact, exist, then the nucleophilic reaction would dominate. It does not appear possible to prove that the Forrester-Hepburn nucleophilic mechanism cannot occur under any conditions as is generally the case when trying to prove a negative.
Scheme 2.

(adapted from ref [48])
In conclusion, the lower the concentration of DMPO and substrate the less likely is nucleophilic addition. Nevertheless, although to our knowledge neither inverted spin trapping nor nucleophilic addition has, in fact, led to misinterpretations in any biological systems, it is prudent to carry out experiments supporting free radical formation such as oxygen consumption, the inhibition of oxygen consumption by the spin trap, or other auxiliary experiments.
Acknowledgments
We would like to acknowledge Mrs. Mary Mason, Dr. Ann Motten, and Ms. Jean Corbett for their editing of the manuscript. This work was supported by the Intramural Research Program of the National Institutes of Health and the National Institute of Environmental Health Sciences.
References
- 1.Beauchamp C, Fridovich I. Anal Biochem. 1971;44:276. doi: 10.1016/0003-2697(71)90370-8. [DOI] [PubMed] [Google Scholar]
- 2.McCord JM, Fridovich I. J Biol Chem. 1968;243:5753. [PubMed] [Google Scholar]
- 3.Janzen EG, Haire DL. Adv Free Radic Chem. 1990;1:253. [Google Scholar]
- 4.Knecht KT, Mason RP. Arch Biochem Biophys. 1993;303:185. doi: 10.1006/abbi.1993.1272. [DOI] [PubMed] [Google Scholar]
- 5.Davies MJ, Hawkins CL. Free Radic Biol Med. 2004;36:1072. doi: 10.1016/j.freeradbiomed.2003.12.013. [DOI] [PubMed] [Google Scholar]
- 6.Buettner GR. Free Radic Biol Med. 1987;3:259. doi: 10.1016/s0891-5849(87)80033-3. [DOI] [PubMed] [Google Scholar]
- 7.Spin Trap Database. http://tools.niehs.nih.gov/stdb/index.cfm.
- 8.Chandra H, Symons MCR. J Chem Soc, Chem Commun. 1986:1301. [Google Scholar]
- 9.Cerri V, Frejaville C, Vila F, Allouche A, Gronchi G, Tordo P. J Org Chem. 1989;54:1447. [Google Scholar]
- 10.Eberson L. J Chem Soc Perkin Trans. 1994;2:171. [Google Scholar]
- 11.Forrester AR, Hepburn SP. J Chem Soc (C) 1971;4:701. [Google Scholar]
- 12.McIntire GL, Blount HN, Stronks HJ, Shetty RV, Janzen EG. J Phys Chem. 1980;84:916. [Google Scholar]
- 13.Bhattacharjee S, Khan MN, Chandra H, Symons MCR. J Chem Soc, Perkin Trans. 1996;2:2631. [Google Scholar]
- 14.Eberson L. J Chem Soc, Perkin Trans. 1992;2:1807. [Google Scholar]
- 15.Kim SM, Vogelpohl A. Chem Eng Technol. 1998;21:187. [Google Scholar]
- 16.Neta P, Huie RE, Ross AB. J Phys Chem Ref Data. 1988;17:1027. [Google Scholar]
- 17.Mottley C, Connor HD, Mason RP. Biochem Biophys Res Commun. 1986;141:622. doi: 10.1016/s0006-291x(86)80218-2. [DOI] [PubMed] [Google Scholar]
- 18.Zubarev V, Brede O. J Chem Soc Perkin Trans. 1994;2:1821. [Google Scholar]
- 19.Mottley C, Mason RP. Arch Biochem Biophys. 1988;267:681. doi: 10.1016/0003-9861(88)90077-x. [DOI] [PubMed] [Google Scholar]
- 20.Davies MJ, Gilbert BC, Stell JK, Whitwood AC. J Chem Soc, Perkin Trans. 1992;2:333. [Google Scholar]
- 21.Janzen EG, Jandrisits LT, Shetty RV, Haire DL, Hilborn JW. Chem Biol Interact. 1989;70:167. doi: 10.1016/0009-2797(89)90071-9. [DOI] [PubMed] [Google Scholar]
- 22.Makino K, Hagiwara T, Hagi A, Nishi M, Murakami A. Biochem Biophys Res Commun. 1990;172:1073. doi: 10.1016/0006-291x(90)91556-8. [DOI] [PubMed] [Google Scholar]
- 23.Hanna PM, Chamulitrat W, Mason RP. Arch Biochem Biophys. 1992;296:640. doi: 10.1016/0003-9861(92)90620-c. [DOI] [PubMed] [Google Scholar]
- 24.Buettner GR, Mason RP. Meth Enzymol. 1990;186:127. doi: 10.1016/0076-6879(90)86101-z. [DOI] [PubMed] [Google Scholar]
- 25.Castelhano AL, Perkins MJ, Griller D. Can J Chem. 1983;61:298. [Google Scholar]
- 26.Potapenko DI, Bagryanskaya EG, Reznikov VV, Clanton TL, Khramtsov VV. Magn Reson Chem. 2003;41:603. doi: 10.1002/mrc.1652. [DOI] [PubMed] [Google Scholar]
- 27.Hayon E, Treinin A, Wilf J. J Amer Chem Soc. 1972;94:47. [Google Scholar]
- 28.Neta P, Huie RE. Environ Health Perspect. 1985;64:209. doi: 10.1289/ehp.8564209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gunnison AF. Food Cosmet Toxicol. 1981;19:667. doi: 10.1016/0015-6264(81)90519-8. [DOI] [PubMed] [Google Scholar]
- 30.Mottley C, Mason RP, Chignell CF, Sivarajah K, Eling TE. J Biol Chem. 1982;257:5050. [PubMed] [Google Scholar]
- 31.Araiso T, Miyoshi K, Yamazaki I. Biochemistry. 1976;15:3059. doi: 10.1021/bi00659a019. [DOI] [PubMed] [Google Scholar]
- 32.Mottley C, Trice TB, Mason RP. Mol Pharmacol. 1982;22:732. [PubMed] [Google Scholar]
- 33.Roman R, Dunford HB. Can J Chem. 1973;51:588. [Google Scholar]
- 34.Ranguelova K, Mason RP. Free Radic Biol Med. 2009;47:128. doi: 10.1016/j.freeradbiomed.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reed GA, Curtis JF, Mottley C, Eling TE, Mason RP. Proc Natl Acad Sci USA. 1986;83:7499. doi: 10.1073/pnas.83.19.7499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McElroy WJ. Atmos Environ. 1986;20:323. [Google Scholar]
- 37.Erben-Russ M, Michel C, Bors W, Saran M. Radiat Environ Biophys. 1987;26:289. doi: 10.1007/BF01221974. [DOI] [PubMed] [Google Scholar]
- 38.Mendes P, Kell DB. Bioinformatics. 1998;14:869. doi: 10.1093/bioinformatics/14.10.869. [DOI] [PubMed] [Google Scholar]
- 39.Taniguchi H, Madden KP. J Am Chem Soc. 1999;121:11875. [Google Scholar]
- 40.Potapenko DI, Clanton TL, Bagryanskaya EG, Gritsan NP, Reznikov VA, Khramtsov VV. Free Radic Biol Med. 2003;34:196. doi: 10.1016/s0891-5849(02)01194-2. [DOI] [PubMed] [Google Scholar]
- 41.Bal Reddy K, Coichev N, van Eldik R. J Chem Soc, Chem Commun. 1991:481. [Google Scholar]
- 42.Berglund J, Fronaeus S, Elding LI. Inorg Chem. 1993;32:4527. doi: 10.1021/ic980225z. [DOI] [PubMed] [Google Scholar]
- 43.Shi X, Mao Y. Biochem Biophys Res Commun. 1994;205:141. doi: 10.1006/bbrc.1994.2641. [DOI] [PubMed] [Google Scholar]
- 44.Shi X. J Inorg Biochem. 1994;56:155. doi: 10.1016/0162-0134(94)85002-x. [DOI] [PubMed] [Google Scholar]
- 45.Brandt C, Fabian I, van Eldik R. Inorg Chem. 1994;33:687. [Google Scholar]
- 46.Brandt C, Elding LI. Atmos Environ. 1998;32:797. [Google Scholar]
- 47.Lima S, Bonifacio RL, Azzellini GC, Coichev N. Talanta. 2002;56:547. doi: 10.1016/s0039-9140(01)00584-7. [DOI] [PubMed] [Google Scholar]
- 48.Timmins GS, Barlow GK, Silvester JA, Wei X, Whitwood AC. Redox Report. 1997;3:125. doi: 10.1080/13510002.1997.11747099. [DOI] [PubMed] [Google Scholar]