

Summary
Hereditary coproporphyria (HCP) is an autosomal dominant acute hepatic porphyria due to the half-normal activity of the heme biosynthetic enzyme, coproporphyrinogen oxidase (CPOX). The enzyme catalyzes the step-wise oxidative decarboxylation of the heme precursor, coproporphyrinogen III to protoporphyrinogen IX via a tricarboxylic intermediate, harderoporphyrinogen. In autosomal dominant HCP, the deficient enzymatic activity results primarily in the accumulation of coproporphyrin III. To date, only a few homozygous HCP patients have been described, most having Harderoporphyria, a rare variant due to specific CPOX mutations that alter enzyme residues D400-K404, most patients described to date having at least one K404E allele. Here, we describe a Turkish male infant, the product of a consanguineous union, who presented with the Harderoporphyria phenotype including neonatal hyperbilirubinemia, hemolytic anemia, hepatosplenomegaly, and skin lesions when exposed to UV light. He was homoallelic for the CPOX missense mutation, c.980A>G (p.H327R), and had massively increased urinary uroporphyrins I and III (9250 and 2910 μM, respectively) and coproporphyrins I and III (895 and 19,400 μM, respectively). The patient expired at five months of age from an apparent acute neurologic porphyric attack. Structural studies predicted that p.H327R interacts with residue W399 in the CPOX active site, thereby accounting for the Harderoporphyria phenotype.
Introduction
Hereditary Coproporphyria (HCP; MIM 121300) is a rare autosomal dominant disorder of heme biosynthesis resulting from the half-normal activity of the mitochondrial enzyme, coproporphyrinogen oxidase (CPOX; EC 1.3.3.3) (Nordmann et al 1983; Anderson et al 2001). The homodimeric enzyme, encoded by the CPOX gene on chromosome 3q12.1, catalyzes the two-step oxidative decarboxylation of the heme precursor, coproporphyrinogen III (COPRO’gen III), to protoporphyrinogen IX (PROTO’gen IX) via the tricarboxylic intermediate, harderoporphyrinogen. Patients with HCP may experience acute, life-threatening neurovisceral attacks, and occasionally have skin lesions resembling those of patients with Porphyria Cutanea Tarda (PCT; MIM 176100) (Anderson et al 2001). HCP heterozygotes have markedly increased urinary and fecal porphyrins, predominantly coproporphyrin III (Anderson et al 2001). Urinary 5′-aminolevulinic acid (ALA) and porphobilinogen (PBG) are increased only during acute attacks, and typically revert to normal levels between acute attacks. The cloning of the CPOX gene (Martasek et al 1994; Taketani et al 1994), which contains seven exons and encodes the 454 amino acid residue subunit, permits mutation analyses to confirm the clinical and biochemical diagnoses. To date, over 50 CPOX mutations causing HCP have been described (The Human Gene Mutation Database, https://portal.biobase-international.com/hgmd/pro/start.php).
Patients with homozygous HCP can have early-onset of manifestations, including abdominal pain and skin manifestations (Table 1). These children, the offspring of heterozygous HCP parents, typically have less than 10% of normal CPOX enzymatic activity and have markedly elevated levels of urinary and fecal coproporphyrins (Table 2)during an acute attack. In 1955, Berger and Goldberg reported an 8 year old boy, the offspring of first cousins, who had milk intolerance, riboflavin deficiency and rickets and presented with short stature, diarrhea and failure to thrive. He had elevated urinary and fecal coproporphyrin (5720 μg/L and 2080 μg/g dry weight, respectively). Since both parents had elevated urinary and fecal coproporphyrins, homozygous HCP was suspected. However, the diagnosis was not confirmed by CPOX activity. Grandchamp et al (1977) described a four year old girl, the offspring of first cousins, who presented with skin pigmentation and hypertrichosis. At 10 years, she had persistent vomiting and abdominal pain, which resolved in a few weeks. After being in remission for 10 years she presented at age 20 years, with acute symptoms when she was pregnant. She was short (142 cm; 4′ 8″) and her urinary ALA and PBG were significantly elevated (34 mg/g creatinine and 39 mg/g creatinine, respectively), as were her urinary and fecal coproporphyrin levels (8.47 μg/g dry weight and 3,870 μg/g dry weight, respectively). She had a leukocyte coproporphyrin oxidase (CPOX) activity 2% of normal, while her parents activities were ~ 50% of mean normal. Sequencing of the family’s CPOX gene identified homozygosity for the missense mutation, p.R231W (renumbered p.R331W by Delfau-Larue et al 1994), predicting an arginine to tryptophan substitution (Martasek et al 1994). Doss et al (1999) described a 10 year old girl who had homozygous HCP. She presented with skin fragility, erythema and scarring of hands and feet, and erythrodontia. She was initially suspected to have Congenital Erythropoetic Porphyria based on her erythrodontia; however, biochemical studies revealed slightly elevated urinary ALA, normal urinary PBG, and markedly elevated urinary and fecal coproporphyrin III, consistent with an acute hepatic porphyria. Her leukocyte CPOX activity was markedly decreased, and was about half-normal in both parents. No acute attacks were described in this pre-pubertal patient. CPOX mutation analyses revealed compound heterozygosity for c.980A>G (p.H327R) and c.920A>G (p.H307R).
Table 1.
Clinical Findings in Previously Reported Homozygous HCP Patients
| Family/Patient | Age at Diagnosis/Report | Major features | Neonatal Jaundice | Hepato-splenomegaly | Hemolytic Anemia | Cutaneous Photo-sensitivity | Reddish Urine | CPOX Mutations | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Family 1 | |||||||||
| Patient 1 (M) | 8 y/11 y | Short Stature; Rickets; Riboflavin deficiency | − | − | − | − | − | Not done | Berger and Goldberg 1955 |
| Family 2 | |||||||||
| Patient 2 (F) | 4 y/20 y | Short Stature; Acute attacks | − | − | − | − | − | R331W/R331W |
Grandchamp et al 1977 Martasek et al 1994 |
| Family 3 | |||||||||
| Patient 3(F) | ?/10 y | Skin Fragility; Erythrodontia; Local erythema | − | − | − | + | − | H327R/H307R | Doss et al 1999 |
Abbreviations: CPOX, coproporphyrinogen oxidase.
Table 2.
Biochemical Findings in the Previously Reported Homozygous HCP Patients
| Family/Patient (Sex) | Urine | Feces | ||||||
|---|---|---|---|---|---|---|---|---|
| CPO Enzyme Activity a (% of Mean) | ALA | PBG | URO | COPRO | COPRO | URO | PROTO | |
| Family 1 | ||||||||
| Patient 1 (M) | NA | NA | Normal | Normal | 5,719 μg/L (57 X ULN) | 2,082 μg/gm dry wt (139 X ULN) | Normal | 56.8 μg/gm dry wt (2 X ULN) |
| Family 2 | ||||||||
| Patient 2 (F) | 2% | 34 mg/24 hr (19 X NM) | 39 mg/24 hr (39 X NM) | 2.75 μg/gm cr (~3 X NM) | 8.47 μg/gm cr (151 X NM) | 3,870 μg/gm dry wt Normal (250 X ULN) | 197 μg/gm dry wt (197 X ULN) | 108 μg/gm dry wt (4 X ULN) |
| Family 3 | ||||||||
| Patient 3 (F) | 2% | 60 μ/mol/24 hr (1.2 X ULN) | 4 μ/mol/24 hr (0.5 X ULN) | 1444 nmol/24 hr (50 X ULN) | 6,502 nmol/24 hr (55 X ULN) | 2835 nmol/gm dry wt (945 X ULN) | NA | 62 nmol/gm dry wt (2.4 X ULN) |
Abbreviations: CPOX, coproporphyrinogen oxidase; ALA, 5′-Aminolevulinic acid; PBG, porphobilinogen; URO, uroporphyrin; COPRO, coproporphyrin; HARDERO, harderoporphyrin; PROTO, protoporphyrinogen; Cr= creatinine; X ULN= times greater than the respective upper limits of normal; X NM= times greater than the respective normal mean; NA=not available
CPO Enzyme Activity: % Normal Mean [Picomoles protoporphyrin per hr per mg protein (Normal 483±95)]
A clinically distinct subtype of homozygous HCP, Harderoporphyria, has been identified based on its unique porphyrin excretion in which harderoporphyrin primarily accumulates in feces (Nordmann et al 1983). This form of homozygous HCP typically presents in infancy with neonatal jaundice and hemolytic anemia (Table 3). To date, only three families with five affected children have been described (Nordmann et al 1983; Lamoril et al 2001; Schmitt et al 2005). All patients either were homozygous for the CPOX p.K404E (c.1210A>G) mutation or had the p.K404E mutation and a splicing defect (IVS6+3) which deleted exon 6 (Lamoril et al 1995). These findings suggested the hypothesis that CPOX amino acids D400-K404 are in the active site and are required for the enzyme’s second step conversion of harderoporphyrin to protoporphyrinogen IX. Thus, it was postulated that mutations that are homozygous for these codons or are compound heterozygotes with a null mutation will result in the harderoporphyria phenotype (Schmitt et al 2005).
Table 3.
Clinical Findings in the Proband and Previously Reported Harderoporphyria Patients
| Family/Patient (Sex) | Age at Diagnosis/Report | Neonatal Jaundice | Hepato-splenomegaly | Hemolytic Anemia | Cutaneous Photosensitivity | Urine Reddish | CPOX Mutations | Reference |
|---|---|---|---|---|---|---|---|---|
| Family 1 | K404E/K404E | Nordmann et al 1983 | ||||||
| Patient 1 (M) | Birth/8 y | + | + | + | + | + | ||
| Patient 2 (M) | Birth/10 y | + | + | + | − | − | ||
| Patient 3 (F) | Birth/3 y | + | + | + | − | − | ||
| Family 2 | K404E/IVS6+3A>G | Lamoril et al 1998 | ||||||
| Patient 4 (M) | Birth/19 y | + | + | + | − | − | ||
| Family 3 | K404E/K404E | Schmitt et al 2005 | ||||||
| Patient 5 (M) | ~ 3 mo/adulthood | + | − | + | + | − | ||
| Family 4 | ||||||||
| Proband (M) | Birth/5 mo | + | + | + | + | + | H327R/H327R | Current Patient |
Here we describe a Turkish infant who presented with neonatal jaundice and hemolytic anemia and who was found to be homoallelic for CPOX mutation c.980A>G (p.H327R). This patient expands the clinical and mutational spectrum of Harderoporphyria to include a CPOX missense mutation in exon 5 that substitutes an arginine for the wild-type histidine at amino acid 327 (p.H327R), which is structurally involved in the enzyme’s conversion of harderoporphyrinogen to protoporphyrinogen IX.
Case Report
The proband was a four month old Turkish boy, the product of a first cousin union, who presented with neonatal jaundice and hemolytic anemia (Table 3). He was born full term to a healthy 24 year old mother after an uncomplicated pregnancy with a birth weight of 3600 g (50th percentile). Shortly after birth, he developed an indirect hyperbilirubinemia of 13 mg/dL, which required phototherapy. He experienced severe cutaneous photosensitivity following bili light exposure leading to a vesicular rash and blister formation. Other causes of neonatal jaundice were ruled out including galactosemia and tyrosinemia. The hyperbilirubinemia resolved and the patient was discharged. The skin lesions eventually healed with scarring leading to hypopigmentation on the exposed regions. At home, reddish urine was noted by the parents. At one and a half months of age, he developed signs and symptoms suggestive of an acute porphyric attack including elevated liver enzymes (aspartate transaminase (AST), 325 IU/L; alanine transaminase (ALT), 269 IU/L, and gamma glutamyl transpeptidase (GGT), 148 IU/L), dehydration, metabolic acidosis and vomiting. On admission, physical examination was significant for hepatosplenomegaly and failure to thrive. His severe hemolytic anemia (hemoglobin, 5.6 g/dL) required multiple packed erythrocyte transfusions. The direct Coombs test was negative and the hemolytic anemia was presumed to be secondary to the underlying metabolic disease. The patient ultimately succumbed to this condition at five months of age.
Biochemical studies at the DÜZEN Laboratory, Ankara, Turkey, demonstrated markedly elevated urinary ALA (2350 μMol/L, normal <534 μMol/L); PBG (2500 μMol/L, normal <88 μMol/L); and the urinary porphyrins were markedly elevated with predominantly uroporphyrin (6.9 μmol/L) and coproporphyrin III (3.36 μmol/L). Fecal porphyrins were not determined. Subsequent urinary porphyrin precursor and porphyrin analyses performed by the Porphyria Diagnostic Laboratory in the Department of Genetics and Genomic Sciences at the Mount Sinai School of Medicine confirmed the markedly increased urinary ALA (43 X normal mean) and PBG (138 X normal mean) and specifically determined the uroporphyrino I and III (299 and 208 X the upper limit of normal, respectively) and coproporphyrin I and III (31 and 236 X mean normal values, respectively) isomer levels, which were markedly elevated (Table 4). Urine porphyrin analyses were subsequently performed on the asymptomatic parents and older brother which showed slight elevations in coproporphyrin III.
Table 4.
Biochemical Findings in the Proband and Previously Reported Harderoporphyria Patients
| Family/Patient (Sex) | Urineb | Fecesb | |||||||
|---|---|---|---|---|---|---|---|---|---|
| CPO Enzyme Activitya (% of Mean) | ALA | PBG | URO | COPRO | Total Porphyrins | COPRO | HARDERO | PROTO | |
| Family 1 | |||||||||
| Patient 1 (M) | 10.1 | 57 | 20 | 150 | 2,144 | 342 | 26 | 66 | 8 |
| Patient 2 (M) | 9.3 | 50 | 11 | 35 | 1,980 | 656 | 29 | 66 | 4 |
| Patient 3 (F) | 9.5 | 40 | 11 | 320 | 2,560 | 272 | 21 | 65 | 14 |
| Family 2 | |||||||||
| Patient 4 (M) | 15.9 | 40 | 9 | 120 | 1,820 | 1,159 | 6 | 90 | 3 |
| Family 3 | |||||||||
| Patient 5 (M) | 3.7 | 17 | <5 | 150 | 1,118 | 672 | 19 | 72 | 9 |
| Family 4 | |||||||||
| Proband (M) | ND | 35 | 8.3 | 2,910 | 19,400 | ND | ND | ND | ND |
| Normal Levels for Proband (range) | 0.81 (0.14–2.54) μMol/Mol Cr | 0.06 (0.01–0.16) μMol/Mol Cr | 0 (0–30) nMol/L | <80 (50–100) nMol/L | − | − | − | − | |
Abbreviations: CPOX, coproporphyrinogen oxidase; ALA, 5′-Aminolevulinic acid; PBG, porphobilinogen; URO, uroporphyrin; COPRO, coproporphyrin; HARDERO, harderoporphyrin; PROTO, protoporphyrinogen
CPO Enzyme Activity: % Normal Mean [Picomoles protoporphyrin per hr per mg protein (Normal 483±95)]
Normal Values for Patients 1–5: Urinary -ALA <38 μMol/L; PBG <5 μMol/L; URO <50 nMol/L; COPRO <200 nMol/L; Fecal – Total Porphyrins <200 nMol/g min dry weight; COPRO 30% total; HARDERO <1% total, and PROTO 70% total.
Proband: URO I=9260 nmol/L; URO III=2910 nmol/L; COPRO I=895 nmol/L; COPRO III= 19,400 nmol/L
Based on the elevated urinary ALA, PBG, and porphyrin excretion pattern and infantile onset of symptoms, a homozygous acute porphyria was suspected. Complete gene sequencing of the probands hydroxymethylbilane synthase, CPOX, and protoporphyrinogen oxidasegenes revealed homozygosity for the CPOX missense mutation, c.980A>G (p.H327R), indicating the diagnosis of homozygous HCP. The parents and a four year old brother, were all heterozygous for the mutation and asymptomatic.
Discussion
Harderoporphyria is a rare autosomal recessive porphyria in which the unique accumulation of the tricarboxylic porphyrin, harderoporphyrin, results from specific missense mutations in the CPOX gene. The disease typically presents at birth with severe neonatal jaundice and hemolytic anemia. The clinical, biochemical, and molecular genetic features of the previous harderoporphyria cases from three affected families and the proband are summarized in Table 3. Of note, all cases shared the same clinical features, namely, neonatal jaundice, hemolytic anemia, and hepatosplenomegaly. The Turkish infant reported here presented shortly after birth with neonatal jaundice, hemolytic anemia, and photosensitivity. Subsequently, he had an acute episode of vomiting, metabolic acidosis, and elevated liver enzymes (AST, ALT, and GGT) at which time his urinary ALA and PBG levels were markedly elevated, consistent with an acute porphyric attack.
Based on the initial clinical presentation, a diagnosis of a cutaneous porphyria including Congenital Erythropoetic Porphyria (MIM 263700) and Hepatoerythropoetic Porphyria (MIM 176100) was considered. Since biochemical studies showed significant elevations of urinary ALA and PBG, a homozygous form of an acute porphyria was suspected, specifically Variegate Porphyria (MIM 176200), or HCP since both may have cutaneous photosensitivity. Although, a fecal porphyrin profile was not performed, the urine porphyrin analysis showed markedly elevated levels of coproporphyrin, in particular coproporphyrin III, suggestive of HCP.
Previous studies have shown slight elevations of urinary ALA and PBG (see Table 4) in the harderoporphyria patients (Schmitt et al 2005), However >40-fold elevations as seen in our patient have not been reported previously. This case expands the clinical spectrum of harderoporphyria to include symptoms suggestive of an acute hepatic porphyric attack characterized by vomiting and metabolic acidosis. Iron overload with high ferritin levels have been described in harderoporphyria (Schmitt et al 2005), however, this was not noted in our patient, although hisferritin (642 pmol/L, range: 13–400 pmol/L) was elevated during one of the acute attacks.
CPOX gene mutation analysis revealed that our patient was homozygous for the previously described missense mutation c.980A>G (Doss et al 1999) which predicts the substitution of a highly conserved histidine (Figure 1) by an arginine at amino acid 327 (p.H327R) in the CPOX enzyme subunit. Our proband’s asymptomatic parents and four year old healthy sibling were heterozygous for the mutation, consistent with their having autosomal dominant HCP (typically non-penetrant). Of the>50 CPOX mutations described to date in patients with HCP, the majority are missense mutations (The Human Gene Mutation Database, https://portal.biobase-international.com/hgmd/pro/start.php). Almost all of these are private mutations, which limits the ability to establish genotype/phenotype correlations. However, the K404E mutation, a lysine to glutamic acid substitution, first reported by Nordmann et al (1983), is unique. This mutation was homozygous in four affected children with harderoporphyria in two unrelated families (Table 3). These affected infants presented with neonatal jaundice and hemolytic anemia, and excreted high levels of coproporphyrins in their urines and feces, with the predominance of harderoporphyrin in the feces.
Figure 1. Amino acid Sequence Homology Between Human and Eukarayotic CPOX Sequences.
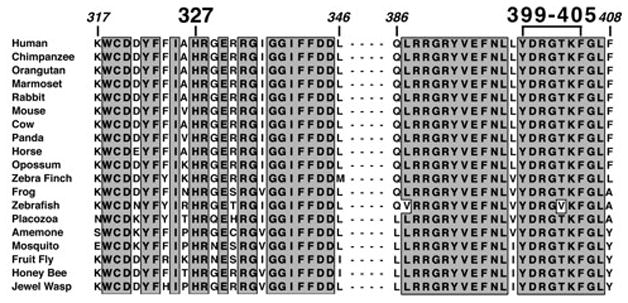
The human CPOX protein sequences in the regions encompassing residues 317 to 346 and 386 to 408 were compared to the sequences of 18 eurkaryotes throughout the animal phyla and extending to insects. The regions of sequence identity are boxed with a grey background. There was 100% identity for all of the sequences at residues H327 and Y399, predicted to be interacting in the structure (See Figure 2A and B).
To date, all reported cases of harderoporphyria have been homozygous for the highly conserved CPOX mutation c.1210A>G (p.K404E; Figure 1) or had the c.1210A>G mutation in combination with a splicing mutation (IVS6+3A>G) that caused skipping of exon 6 (Lamoril et al 1995). Another mutation, c.1201C>T (p.R401W), which was identified in a patient with HCP, was found to impair the in vitro decarboxylation of harderoporphyrinogen in a pattern similar to p.K404E (Lamoril et al 2001). These mutations lie in a highly conserved region of the CPOX protein (Figure 1) and presumably forms part of its catalytic site (Lamoril et al 1998; Lee et al 2005; Schmitt et al 2005). Subsequent in vitro mutagenesis of residues Y399 to K405 (individually mutated to alter polarity or charge) and mutant prokaryotic expression of each mutant allele revealed that the mutated enzymes had markedly reduced ability to convert harderoporphyrinogen to protoporphyrinogen IX (Schmitt et al 2005). Thus, it was shown that the amino acid residues Y399 to K405 are directly involved in the enzyme’s oxidative decarboxylation of harderoporphyrinogen.
Previously, analyses of the CPOX crystal structure predicted that the p.H327R amino acid substitution would alter the observed wild-type interaction between the highly conserved residue H327 in helix α7 and residue Y399 in helix α9 of the CPOX crystal structure (Lee et al 2005; Figure 10, supplemental data), thereby affecting the enzyme’s second step decarboxylation of harderoporphyrinogen to protoporphyrinogen IX. The finding that homozygosity for c.980A>G (p.H327R) in our patient resulted in the classic Harderoporphyria phenotype provided proof-of-concept for this structure-based prediction. Moreover, using the SWISS-MODEL server(Kiefer et al 2009)to derive the structure of the p.H327R mutant enzyme(Figure 2B), a similar interaction to that observed for the wild-type structure (Figure 2A) is predicted for the more bulky arginine residue, presumably accounting for the harderoporphyria phenotype. Since this region lies in the interface between the two subunits in the CPOX homodimer (Lee et al 2005), the arginine would not likely be free to move out into the bulk water to avoid clashing with p.Y399. Thus, the classic Harderoporphyria phenotype and homozygosity for c.980A>G (H327R) in the patient provides proof of this predicted interaction and supports the predicted structural requirements for the enzyme’s active site required for the second oxidative decarboxylation step.
Figure 2. Predicted CPOX Structures for the Wild-type and p.H327R Mutant Enzymes.
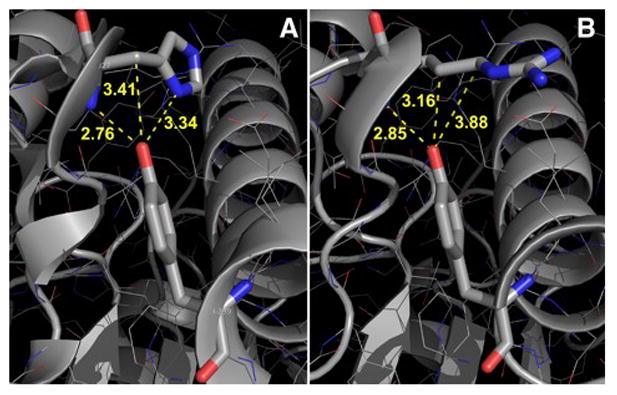
Panel A indicates the interacting region in the wild-type structure involved in the enzyme’s active site conversion of harderoporphyrinogen to protoporphyrinogen IX with the inter-atomic distances given between residues H327 and Y399. Panel B shows the modeled p.H327R substitution and close inter-atomic distances indicating that a clash would distort the region on helix α9 near residue R401, a mutation which previously was shown to result in harderoporphryin accumulation in vitro.
In conclusion, this report expands the clinical phenotype of harderoporphyria to include severe decompensations consistent with the acute attacks seen in the hepatic porphyrias. In addition, the enzymatic CPOX mechanism responsible for the accumulation of harderoporphyria was expanded to include a missense mutation c.980A>G (p.H327R) outside (but likely interacting with) the CPOX p.R401 to p.K404 region, which has been responsible for the unique phenotype in the previously reported patients. Thus, other CPOX mutations that interact in the active site’s conversion of harderoporphyrinogen, like the p.H327R mutant enzyme, can cause the unique phenotype and expand the genotype/phenotype correlations for this disease.
Acknowledgments
Details of funding for all research studies: This research was supported in part by grants from the National Institutes of Health (NIH) including a research grant (5 R01 DK026824) and a grant (1 U54 DK083909) for the Porphyria Consortium of the NIH Rare Diseases Clinical Research Network (RDCRN). Funding and/or programmatic support for this project has been provided by NIH Office of Rare Disease Clinical Research Network (ORDR). The views expressed in written materials or publications do not necessarily reflect the official policies of the Department of Health and Human Services.
Footnotes
Details of the contributions of individual authors: Making clear who has contributed pertinent aspects of the planning (RJD, MB, DFB, CSK, FSE), conduct (RJD, CSK, MB, AH, FSE, IO, LT, AC, IN, CY, SC, DFB), and reporting (RJD, MB, CSK, FSE) of the work described in the article.
Name of one author who serves as guarantor for the article: RJD accepts full responsibility for the work and/or the conduct of the study, had access to the data, and controlled the decision to publish.
A competing interest statement: All authors declare that the answer to all questions on the JIMD competing interest form are “no”, and therefore they have nothing to declare.
Details of ethics approval: Not Required.
A patient consent statement: The parents of the deceased child reported here have consented to these studies.
If vertebrate animals have been utilized: Not Applicable.
References
- Anderson KE, Sassa S, Bishop DF, Desnick RJ. Disorders of heme biosynthesis: X- linked sideroblastic anemia and the porphyrias. In: Scriver CR, Beaudet A, Sly WS, Valle D, editors. The metabolic and molecular basis of inherited diseases. McGraw-Hill; New York: 2001. pp. 2991–3062. [Google Scholar]
- Berger H, Goldberg A. Hereditary coproporphyria. Br Med J. 1955;2:85–88. doi: 10.1136/bmj.2.4931.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delfau-Larue MH, Martasek P, Grandchamp B. Coproporphyrinogen oxidase: gene organization and description of a mutation leading to exon 6 skipping. Hum Mol Genet. 1994;3:1325–1330. doi: 10.1093/hmg/3.8.1325. [DOI] [PubMed] [Google Scholar]
- Doss M, von Tiepermann R, Köpp W. Harderoporphyrin coproporphyria. Lancet. 1984;1:292. doi: 10.1016/s0140-6736(84)90169-7. [DOI] [PubMed] [Google Scholar]
- Doss MO, Gross U, Lamoril J, et al. Compound heterozygous hereditary coproporphyria with fluorescing teeth. Ann Clin Biochem. 1999;36:680–682. doi: 10.1177/000456329903600522. [DOI] [PubMed] [Google Scholar]
- Grandchamp B, Phung N, Nordmann Y. Homozygous case of hereditary coproporphyria. Lancet. 1977;2:1348–1349. doi: 10.1016/s0140-6736(77)90386-5. [DOI] [PubMed] [Google Scholar]
- Hindmarsch JT, Oliveras L, Greenway DC. Biochemical differentiation of the Porphyrias. Clin Biochem. 1999;32:609–619. doi: 10.1016/s0009-9120(99)00067-3. [DOI] [PubMed] [Google Scholar]
- Kiefer F, Arnold K, Künzli M, Bordoli L, Schwede T. The SWISS-MODEL Repository and associated resources. Nucl Acids Res. 2009;37:387–392. doi: 10.1093/nar/gkn750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamoril J, Martasek P, Deybach JC, Silva VD, Grandchamp B, Nordmann Y. A molecular defect in coproporphyrinogen oxidase gene causing harderoporphyria, a variant form of hereditary coproporphyria. Hum Mol Genet. 1995;4:275–278. doi: 10.1093/hmg/4.2.275. [DOI] [PubMed] [Google Scholar]
- Lamoril J, Puy H, Gouya L, et al. Neonatal hemolytic anemia due to inherited harderoporphyria: clinical characteristics and molecular basis. Blood. 1998;9:1453–1457. [PubMed] [Google Scholar]
- Lamoril J, Puy H, Whatley SD, et al. Characterization of mutations in the CPO gene in British patients demonstrates absence of genotype-phenotype correlation and identifies relationship between hereditary coproporphyria and harderoporphyria. Am J Hum Genet. 2001;68:1130–1138. doi: 10.1086/320118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D-S, Flachsová E, Bodnárová M, Demeler B, Mártasek P, Raman CS. Structural basis of hereditary coproporphyria. Proc Natl Acad Sci USA. 2005;102:14232–14237. doi: 10.1073/pnas.0506557102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martasek P, Nordmann Y, Grandchamp B. Homozygous hereditary coproporphyria caused by an arginine to tryptophane substitution in coprpporphyrinogen oxidase and common intragenic polymorphisms. Hum Mol Genet. 1994;3:477–480. doi: 10.1093/hmg/3.3.477. [DOI] [PubMed] [Google Scholar]
- Nordmann Y, Grandchamp B, de Verneuil H, Phung L, Cartigny B, Fontaine G. Haredroporphyria: a variant hereditary coproporphyria. J Clin Invest. 1983;72:1139–49. doi: 10.1172/JCI111039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt C, Gouya L, Malonova E, et al. Mutations in human CPO gene predict clinical expression of either hepatic hereditary coproporphyria or erythropoietic porphyria harderoporphyria. Hum Mol Genet. 2005;14:3089–3098. doi: 10.1093/hmg/ddi342. [DOI] [PubMed] [Google Scholar]
- Taketani S, Kohno H, Furukawa T, Yoshinaga T, Tokunaga R. Molecular cloning, sequencing and expression of cDNA encoding human coproporphyrinogen oxidase. Biochim Biophys Acta. 1994;1183:547–9. doi: 10.1016/0005-2728(94)90083-3. [DOI] [PubMed] [Google Scholar]