

Abstract
The first Suzuki–Miyaura cross-coupling reactions of the synthetically versatile O-aryl carbamate and O-sulfamate groups is described. The transformations utilize the inexpensive, bench-stable catalyst NiCl2(PCy3)2 to furnish biaryls in good to excellent yields. A broad scope for this methodology has been demonstrated. Substrates with electron-donating and electron-withdrawing groups (EDGs, EWGs) are tolerated, in addition to those that possess ortho substitutents. Furthermore, heteroaryl substrates may be employed as coupling partners. A computational study providing the full catalytic cycles for these cross-coupling reactions is described. The oxidative additions with carbamates and sulfamates occur via a five-centered transition state, resulting in the exclusive cleavage of the Ar–O bond. Water is found to stabilize the Ni–carbamate catalyst resting state, and thus provides rationalization of the relative decreased rate of coupling of carbamates. Several synthetic applications are presented to showcase the utility of the methodology in the synthesis of polysubstituted aromatic compounds of natural product and bioactive molecule interest.
Introduction
Transition metal-catalyzed cross-coupling reactions provide a powerful means to assemble carbon–carbon (C–C) and carbon–heteroatom (C–X) bonds.1 Although halides are most commonly employed as the electrophilic partner,1,2 phenolic derivatives (Figure 1), or ‘pseudohalides’, offer a valuable alternative given that phenols are typically inexpensive and readily available materials.3 Cross-couplings of aryl sulfonates have been most widely studied and a range of C–C and C–X bond forming reactions are now established.1,4,5 Recent studies have focused on the development of less common phenol-based electrophiles,6 such as ethers,7 esters,8 carbamates,9 and sulfamates9b,10 since they are commonly more robust, typically unreactive toward Pd-catalysis, and show synthetic advantage for the regioselective construction of aromatics by C–H activation and directed ortho metalation (DoM) chemistry.11,12,13,14
Figure 1.

Phenol-Based Cross-Coupling Partners.
Inspired by the reasons outlined above, our laboratories have pursued the development of cross-coupling reactions involving phenol-derived carbamates and sulfamates (Scheme 1). Previous studies have demonstrated the utility of these reaction partners in nickel-catalyzed Kumada couplings.9d,9e,10 However, the corresponding Suzuki–Miyaura couplings of these substrates have remained elusive, despite the numerous benefits of organoboronate coupling methodologies. Such advantages include the low toxicity, wide availability, and pronounced stability of organoboronates, in addition to their broad functional group tolerance.1,15 In this article, we report a) the development of the Ni-catalyzed Suzuki–Miyaura cross-coupling reactions of aryl O-carbamates and O-sulfamates, b) the broad scope of these transformations, which includes the cross-coupling of heterocyclic substrates, c) computational studies that elucidate the complete catalytic cycle of these couplings, and d) a variety of synthetic applications, including DoM – linked tactics and a concise synthesis of the anti-inflammatory drug flurbiprofen.16
Scheme 1.

Results and Discussion
Suzuki–Miyaura Cross-coupling Reactions of Aryl O-Carbamates
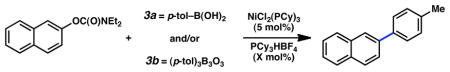
A key challenge in achieving the Suzuki–Miyaura cross-coupling of aryl carbamates lies in activating the fairly inert aryl carbon–oxygen bond of these substrates. A similar obstacle had been overcome in our previously reported Suzuki–Miyaura coupling of aryl pivalates.8a Encouraged by our prior success, we explored NiCl2(PCy3)2-promoted conditions to effect the desired Suzuki–Miyaura coupling of aryl carbamates (Table 1). Of note, NiCl2(PCy3)2 is readily available, considerably stable to air and water, and can be used on the bench-top rather than in a glovebox.17,18,19 Initial studies were directed toward the coupling of fused-aromatic systems, which are typically superior substrates in Ni-catalyzed couplings of phenolic derivatives.7,8,9 Unfortunately, applying our optimal conditions for pivalate coupling (i.e., NiCl2(PCy3)2 (5 mol%) and K3PO4 (4.5 equiv) in toluene at 80 °C) to a 1-naphthyl carbamate substrate led only to trace amounts of cross-coupled product. By raising the temperature to 110 °C, however, the desired biaryl was obtained in 51% yield (entry 1). Further optimization ultimately established more forcing conditions that delivered the targeted product in 86% yield (entry 2).
Table 1.
Cross-coupling of aryl carbamates with arylboronic acidsa
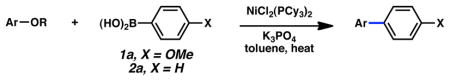
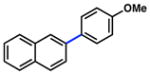
Conditions: NiCl2(PCy3)2 (10 mol%), ArB(OH)2 (4 equiv), K3PO4 (7.2 equiv), toluene (0.3 M), 130 °C for 24 h.
Conditions: NiCl2(PCy3)2 (5 mol%), ArB(OH)2 (2.5 equiv), K3PO4 (4.5 equiv), toluene (0.3 M), 110 °C, 24 h.
Yields of isolated products.
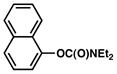
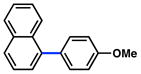
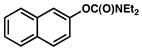
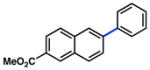
Additional carbamate substrates were examined under our Ni-catalyzed reaction conditions (Table 1).20 2-Naphthyl carbamates gave products in lower yields (entries 3 and 4). The reaction proved tolerant of an EWG (–CO2Me, entry 4) and an EDG (–OMe, entry 5) on the naphthyl ring. The corresponding reactions of aryl carbamates proved more challenging. Nonetheless, carbamates derived from phenol and p-methoxyphenol were converted to the corresponding cross-coupled products in 52% and 41% yield, respectively (entries 6 and 7).
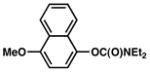
Further studies were undertaken to uncover higher yielding and more generally useful reaction conditions. The N,N-diethyl carbamate of 2-naphthol was subjected to NiCl2(PCy3)2 with variations in temperature, solvent, ligand additive, and organoboron species (Table 2). When employing o-xylenes at 150 °C, cross-coupling with boroxine 3b proceeded sluggishly but nevertheless furnished the desired biaryl in 61% yield (entry 1). Mixtures of boronic acids and boroxines were also examined. Using a 1:1 mixture of 3a:3b, the desired biaryl was obtained in only 26% yield (entry 2). These results, coupled with the observation that 3a liberates excessive water in organic solvents, led us to hypothesize that, although some water is necessary to generate the catalytically active boronate species, excessive water can be detrimental to the carbamate cross-coupling reaction.21 Furthermore, Shi had previously reported the critical role of water in the Suzuki–Miyaura coupling of aryl pivalate esters.8b By using a 1:10 ratio of 3a:3b,22 and thereby minimizing the water content, a quantitative yield of cross-coupled product was obtained (entry 3). Conducting the reaction at 120 °C, with toluene as solvent and 10 mol% Ni catalyst, gave a lower yield of the biaryl adduct (entry 4).23
Table 2.
Optimization studies for naphthyl 2-O-carbamate coupling
 | ||||||
|---|---|---|---|---|---|---|
| entryb | solvent | temp | PCy3HBF4 | K3PO4 | ArB(OR)2 | yielda |
| 1 | o-xylene | 150 °C | 10 mol% | 5 equiv | 3b (2.5 equiv) | 61% |
| 2 | o-xylene | 150 °C | 10 mol% | 5 equiv | 3a:3b (1:1, 2.5 equiv) | 26% |
| 3 | o-xylene | 150 °C | 10 mol% | 5 equiv | 3a:3b (1:10, 2 equiv) | 100% (84%) |
| 4c | toluene | 120 °C | - | 7.2 equiv | 3a:3b (1:10, 2.5 equiv) | 62% |
Yield by GC/MS analysis (yield of isolated product).
All reactions were run for 20 h with the exception of entry 4, which was run for 5 h.
10 mol% NiCl2(PCy3)2.
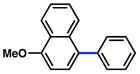
Having found optimal and reproducible conditions, we turned our attention to defining the scope and functional group tolerance of the carbamate cross-coupling reaction (Table 3).20 Substrates derived from 2-naphthol, 1-naphthol, and phenol underwent smooth coupling (entries 1–3). Furthermore, although a substrate bearing the electron-withdrawing fluoro substituent was tolerated (entry 4), coupling in the presence of a cyano derivative was less fruitful (entry 5). The latter result can be explained by competitive cross-coupling at the cyano group, a transformation reported recently by Shi.24 Finally, an electron-rich substrate gave only low yields of product (entry 6).
Table 3.
Cross-coupling of aryl carbamates under improved conditionsa
 | ||||
|---|---|---|---|---|
| entry | Ar–OC(O)NEt2 | (RO)2B–Ar | product | yieldb |
| 1 |
|
3 |
|
100% (84%) |
| 2 |
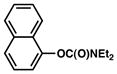
|
2 |
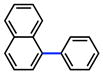
|
100% (82%) |
| 3 |
|
1 |
|
64% (58%) |
| 4 |
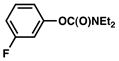
|
1 |
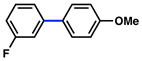
|
80% (69%) |
| 5 |
|
2 |
|
36% (28%) |
| 6 |
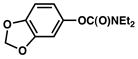
|
2 |
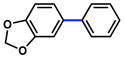
|
23% (31%) |
Conditions: NiCl2(PCy3)2 (5 mol%), PCy3HBF4 (10 mol%), ArB(OR)2 (2.5 equiv), ratio of Ar3B3O3:ArB(OH)2 = 10:1 (4 equiv), K3PO4 (5 equiv).
Yield by GC/MS analysis (yield of isolated product).
Several ortho-substituted aryl carbamates were tested in the Suzuki–Miyaura coupling (Table 4). Substrates of this type can be readily synthesized by DoM12 or transition metal-catalyzed C–H functionalization.14b–e Substrates with o-benzyl, -alkenyl, and -phenyl groups were all tolerated (entries 1–3). Coupling of the o-methoxy substrate proceeded in modest yield (entry 4), whereas coupling of a 2,4-dimethylated substrate was unsuccessful (entry 5). In view of the coupling of other ortho-substituted systems to afford good to excellent yields of products (entries 1 – 3), rationalization of these results based on steric effects is premature.
Table 4.
Cross-coupling of ortho-substituted aryl O-carbamatesa
 | |||
|---|---|---|---|
| entry | Ar–OC(O)NEt2 | product | yieldb |
| 1 |
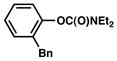
|
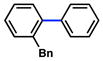
|
60% (70%) |
| 2 |
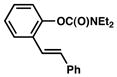
|
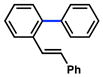
|
99% (93%) |
| 3 |
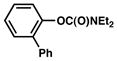
|
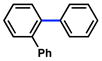
|
69% (50%) |
| 4 |
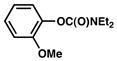
|
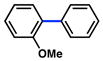
|
40% (36%) |
| 5 |
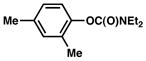
|
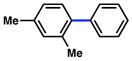
|
8% |
Conditions: NiCl2(PCy3)2 (5 mol%), PCy3HBF4 (10 mol%), 2 (2.5 equiv), ratio of Ph3B3O3:PhB(OH)2 = 10:1 (4 equiv), K3PO4 (5 equiv).
Yield by GC/MS analysis (yield of isolated product).
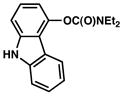
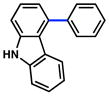
As shown in Table 5, the carbamate cross-coupling methodology is also applicable to heterocyclic substrates. Thus, the 3-pyridyl-carbamate was efficiently cross-coupled with a variety of organoboron species (entries 1–5). In addition, a quinoline-derived substrate was tolerated (entry 6) and a carbazole-containing substrate underwent conversion to the desired biaryl under our optimal reaction conditions albeit in modest yield (entry 7).
Table 5.
Cross-coupling of heterocyclic aryl O-carbamates and scope of aryl boronatesa
 | ||||
|---|---|---|---|---|
| entry | HetAr–OC(O)NEt2 | (RO)2B–Ar | product | yieldb |
| 1 |
|
1 |
|
(84%) |
| 2 |
|
2 |
|
100% (85%) |
| 3 |
|
3 |
|
(87%) |
| 4 |
|
4 |
|
(70%) |
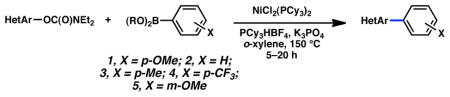
| 5 |
|
5 |
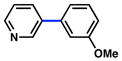
|
(65%) |
| 6 |
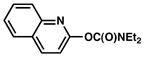
|
2 |
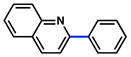
|
100% (51%) |
| 7 |
|
2 |
|
45% (36%) |
Conditions: NiCl2(PCy3)2 (5 mol%), PCy3HBF4 (10 mol%), ArB(OR)2 (2.5 equiv), ratio of Ar3B3O3:ArB(OH)2 = 10:1 (4 equiv), K3PO4 (5 equiv).
Yield by GC/MS analysis (yield of isolated product).
Suzuki–Miyaura Cross-coupling Reactions of Aryl Sulfamates
Concurrent with the above studies on aryl carbamates, aryl sulfamates were targeted as substrates for the Ni-catalyzed Suzuki–Miyaura cross-coupling reactions. Although initial efforts to effect this transformation with dppp as ligand gave initial encouragement,10a employing the tricyclohexylphosphine ligand led to improved results, ultimately rendering aryl sulfamates superior Suzuki–Miyaura coupling partners to the corresponding carbamates (Table 6).20 Naphthyl substrates were smoothly converted to biaryl products,25 even in the presence of an EWG or EDG (entries 1–3). Most strikingly, the reaction proceeded comparably well when operating on aryl derivatives (entries 4–9). Methyl and the electron-withdrawing CF3 substituents are tolerated (entries 5–7). Substrates bearing electron-rich methoxy or amino substituents also afforded very good yields of coupled products (entries 8 and 9). Moreover, an enone sulfamate participated in the Suzuki–Miyaura cross-coupling reaction (entry 10).
Table 6.
Cross-coupling of aryl sulfamatesa
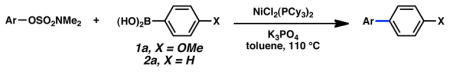 | ||||
|---|---|---|---|---|
| entry | Ar–OSO2NMe2 | (HO)2B–Ar | product | yieldb |
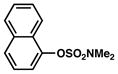
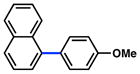
| 1 |
|
1a |
|
95% |
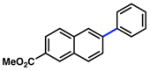
| 2 |
|
2a |
|
72% |
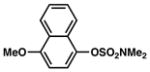
| 3 |
|
2a |
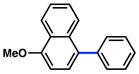
|
92% |
| 4 |
|
1a |
|
87% |
| 5 |
|
1a |
|
89% |
| 6 |
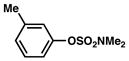
|
1a |
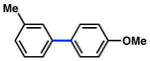
|
91% |
| 7 |
|
2a |
|
81% |
| 8 |
|
2a |
|
80% |
| 9 |
|
2a |
|
76% |
| 10 |
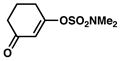
|
2a |
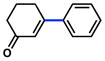
|
75% |
Conditions: NiCl2(PCy3)2 (5 mol%), ArB(OH)2 (2.5 equiv), K3PO4 (4.5 equiv), toluene (0.3 M), 110 °C for 24 h.
Yields of isolated products.
In view of the availability of many ortho-substituted aryl sulfamates by DoM chemistry, such derivatives were also evaluated in the Suzuki–Miyaura cross-coupling reaction (Table 7).20,26 The transformation was found to be tolerant of an ortho-cresol derived substrate, in addition to the sterically burdened sulfamate prepared from 2,6-dimethylphenol (entries 1 and 2). Furthermore, substrates bearing ortho-trimethylsilyl, -phenyl, and -methoxy substituents underwent cross-coupling to give the corresponding products in excellent yields (entries 3–5). Interestingly, a substrate possessing a bulky ortho-t-butylketone substitutent could also be utilized in this methodology (entry 6).
Table 7.
Cross-coupling of o-substituted aryl sulfamatesa
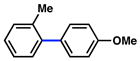
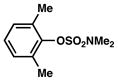
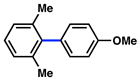
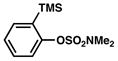
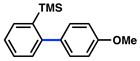
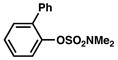
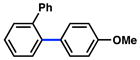
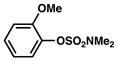
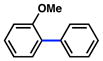
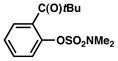
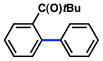
Conditions: NiCl2(PCy3)2 (5 mol%), ArB(OH)2 (2.5 equiv), K3PO4 (4.5 equiv), toluene (0.3 M), 110 °C for 24 h.
Yields of isolated products.
Conditions: NiCl2(PCy3)2 (10 mol%), ArB(OH)2 (4 equiv), K3PO4 (7.2 equiv), toluene (0.3 M), 130 °C for 24 h.
Although the DoM chemistry of aryl sulfamates was initially reported using N,N-diethyl substrates,10a the corresponding N,N-dimethyl aryl sulfamates were found to undergo metalation under the identical reported reaction conditions. Scheme 2 highlights syntheses of substrates 8–11 beginning from phenyl sulfamate 6, which, in turn, is easily prepared from phenol and commercially available dimethylsulfamoyl chloride27 in quantitative yield. Compounds 9 and 10 were obtained by lithiation of phenyl sulfamate 6, followed by quenching with TMSCl and PivCl, respectively. Similarly, the boronate 7 was derived by quenching the intermediate lithio species with B(OMe)3, followed by treatment with pinacol.10a Boronate 7 served as the common precursor to substituted sulfamates 8 and 11. Whereas methoxysulfamate 11 was prepared by a straightforward oxidation28/methylation sequence, ortho-phenyl sulfamate 8 was accessed by a Pd-catalyzed Suzuki–Miyaura cross-coupling. It is notable that the sulfamate remains undisturbed under the Pd-mediated reaction conditions.
Scheme 2.

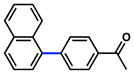
The scope of the sulfamate cross-coupling reaction was also found to be broad with respect to the boronic acid component (Table 8). A methyl substituent was tolerated at the para, meta, and ortho positions (entries 1–3), as was a 4-methoxymethyl group (entry 4). Cross-coupling of a boronic acid bearing the electron-donating methoxy group proceeded in 95% yield (entry 5). Finally, electron-withdrawing trifluoromethyl, fluoro, and acetyl substituents were compatible with the sulfamate coupling methodology (entries 6–8).
Table 8.
Scope of aryl boronic acid in the Suzuki–Miyaura cross-coupling of aryl O-sulfamatesa
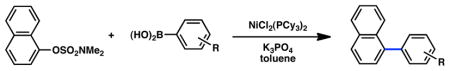
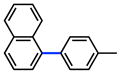
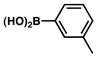
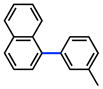
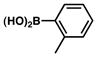
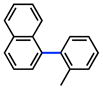
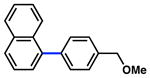
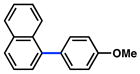
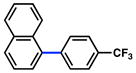
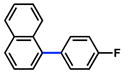
Conditions: NiCl2(PCy3)2 (5 mol%), ArB(OH)2 (2.5 equiv), K3PO4 (4.5 equiv), toluene (0.3 M), 110 °C for 24 h.
Yields of isolated products.
Conditions: NiCl2(PCy3)2 (10 mol%), ArB(OH)2 (4 equiv), K3PO4 (7.2 equiv), toluene (0.3 M), 130 °C for 24 h.
Conditions: NiCl2(PCy3)2 (5 mol%), ArB(OH)2 (2.5 equiv), K3PO4 (4.5 equiv), toluene (0.3 M), 120 °C for 24 h.
Suzuki–Miyaura Cross-coupling Reactions of Heterocyclic O-Sulfamates
Given the importance of heterocycles in medicinal agents, we probed the use of heterocyclic partners in the sulfamate Suzuki–Miyaura cross-coupling process.29 As shown in Table 9, a variety of heterocyclic aryl sulfamates were suitable for this methodology, although more forcing reaction conditions were often required to achieve synthetically useful yields. Coupling of a dihydrobenzofuran-derived substrate afforded the desired biaryl in 88% yield (entry 1). Similar success was observed in the coupling of nitrogen-containing heteroaryl sulfamates (entries 2–6). In addition to indole and carbazole (entries 2 and 3), the pyridine and quinoline heterocycles, each possessing basic amine functionality, were also tolerated (entries 4–6).
Table 9.
Cross-coupling of heterocyclic aryl O-sulfamates with phenyl boronic acida
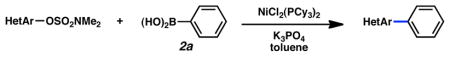
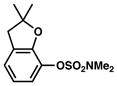
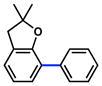
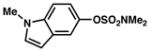
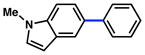
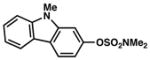
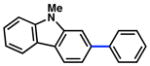
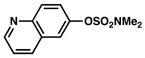
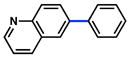
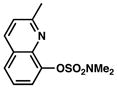
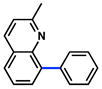
Conditions: NiCl2(PCy3)2 (5 mol%), 2a (2.5 equiv), K3PO4 (4.5 equiv), toluene (0.3 M), 110 °C, 24 h.
Yields of isolated products.
Conditions: NiCl2(PCy3)2 (10 mol%), 2a (4 equiv), K3PO4 (7.2 equiv), toluene (0.3 M), 130 °C for 24 h.
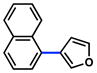
The scope of the sulfamate cross-coupling reaction with respect to heteroaryl boronic acids is summarized in Table 10. Benzofuran- and furan-containing substrates underwent smooth cross-coupling under our standard reaction conditions (entries 1 and 2). Furthermore, a sulfur-containing heterocyclic boronic acid could be employed (entry 3). A pyridine 3-boronic acid derivative was also tolerated in our Suzuki–Miyaura coupling methodology (entry 4).
Table 10.
Cross-coupling of naphthyl 1-O-sulfamates with heterocyclic aryl boronic acidsa
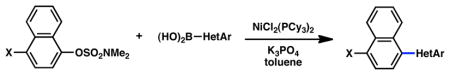 | |||
|---|---|---|---|
| entry | (HO)2B–HetAr | product | yieldb |
| 1 |
|
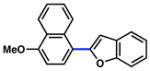
|
67% |
| 2 |
|
|
79% |
| 3 |
|
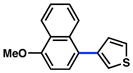
|
80% |
| 4 |
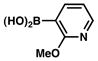
|
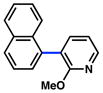
|
80% |
Conditions: NiCl2(PCy3)2 (5 mol%), HetArB(OH)2 (2.5 equiv), K3PO4 (4.5 equiv), toluene (0.3 M), 110 °C, 24 h.
Yields of isolated products.
As an additional important test of the sulfamate coupling methodology, we attempted a Suzuki–Miyaura reaction wherein both coupling partners were heterocyclic substrates (Scheme 3).29 We were delighted to find that the desired cross-coupling between quinoline-derived sulfamate 12 and pyridinyl boronic acid 13 proceeded smoothly to furnish biaryl 14 in 97% yield. This result underscores the critical tolerance of the sulfamate cross-coupling process to basic nitrogen substituents.
Scheme 3.

Mechanistic Studies
Pd-catalyzed Suzuki-Miyaura cross-couplings have been studied computationally by various groups.30,31 The three key steps in the catalytic cycle, oxidative addition,32 transmetallation,31 and reductive elimination,33 have been studied carefully for reactions involving a variety of substrates. The mechanism of Ni-catalyzed Suzuki-Miyaura cross-coupling with aryl acetates has been recently investigated theoretically by Li et al.34 Here we report the first theoretical study of the catalytic cycles of the Ni-catalyzed Suzuki–Miyaura cross-coupling with O-carbamates and O-sulfamates using density functional theory (DFT). The selectivities between couplings with the Ar–O bond and the O–carbonyl/sulfonyl bond and the effects of water on the reactivities are also described.
Figures 3 and 4 depict the catalytic cycles for the Suzuki–Miyaura cross-coupling of aryl carbamates and sulfamates, respectively, as determined by DFT calculations. The geometries of important transition structures are shown in Figure 5 for the coupling of N,N-dimethyl phenyl carbamate and N,N-dimethyl phenyl sulfamate with phenyl boronic acid. The PCy3 ligand used in the experiments was also used in the calculations. Geometry optimizations and frequency calculations were performed using B3LYP35 and a mixed basis set employing LANL2DZ for metal and 6–31G(d) for other atoms. Conformational searches of the PCy3 ligand were performed. The initial geometry of PCy3 was taken from the crystal structure of Ni(PCy3)(C2H4)2.36 Several rotamers of the PCy3 ligands in the Ni complexes were tested as the initial geometry in the optimizations. Energies reported are Gibbs free energies in solution, which involve zero-point vibrational energy corrections, thermal corrections to Gibbs free energy at 298 K, and solvation free energy corrections computed by singlet point CPCM37 calculations on gas-phase optimized geometries (toluene was used as solvent). The molecular cavities were built up using the United Atom Topological Model (UAHF). Vibrational frequencies were calculated for all optimized structures to confirm the nature of the stationary points. All calculations were performed using Gaussian 03.38
Figure 3.

Gibbs free energy profile of Ni-catalyzed Suzuki-Miyaura cross-coupling reaction of phenyl N,N-dimethyl O-carbamate 15 with phenylboronic acid. PCy3 was used as ligand in the calculations. For clarity, the cyclohexyl groups on the ligand are not shown.
Figure 4.

Gibbs free energy profile of the Ni-catalyzed Suzuki-Miyaura cross-coupling reaction of N,N-dimethyl phenyl O-sulfamate with phenylboronic acid. PCy3 was used as ligand in the calculations. For clarity, the cyclohexyl groups on the ligand are not shown.
Figure 5.

Transition state structures of Ni-catalyzed oxidative additions of (a) N,N-dimethyl phenyl O-carbamate and (b) N,N-dimethyl phenyl O-sulfamate. PCy3 was used as ligand in the calculations. For clarity, the cyclohexyl groups on the ligand are not shown.
The oxidative addition of aryl carbamates may occur via several different pathways: the Ni may be mono- or bis-ligated; the oxidative addition may occur at the Ph–O bond or the O–carbonyl bond of the carbamates. Previous theoretical studies suggested that the oxidative addition of aryl halides to Pd(0) catalysts involves formation of an η2 LPd(ArX) pre-reaction complex.32 The η2 LPd(ArX) complex may be generated through ligand dissociation from PdL2 followed by coordination with aryl halide or through a concerted or stepwise associative displacement pathway.39 Recent density functional calculations by Li et al. suggested that the oxidative addition of phenylacetates in Ni-catalyzed Suzuki–Miyaura couplings also involves an η2 LNi(ArX) pre-reaction complex.8c Upon dissociation of a PCy3 ligand, the Ni catalyst coordinates with the substrate to generate an η2 complex 16, which is slightly less stable than Ni(PCy3)2.40 Li et al. suggested that the oxidative addition of phenylacetates occurs via a three-centered transition state, and the weaker PhO–carbonyl bond is more reactive compared to the Ph–O bond.8c We investigated the possible pathways in the oxidative additions with phenyl carbamates (Figures 3 and 5) and found that the preferred pathway involves oxidative addition at the Ph–O bond via a five-centered transition state (TS17, Figures 3 and 5) in which Ni is mono-ligated and coordinated with the carbonyl oxygen.41,42 The corresponding three-centered transition state (TS30) that uses a single oxygen of the carboxylate to bridge requires 7.4 kcal/mol higher activation energy. In contrast to previous theoretical studies by Li et al., the Ph–O bond in carbamates is more reactive in oxidative addition than the PhO–carbonyl bond, although the former is a stronger bond in terms of bond dissociation energies.43 Oxidative addition at the O–carbonyl bond can only occur via a three-centered transition state (TS31) and requires 3.9 kcal/mol higher energy than the oxidative addition at the Ph–O bond (TS17). Thus, the oxidative addition occurs exclusively at the Ph–O bond due to a favorable five-centered transition state. The carbonyl group is acting as a directing group to activate the Ph–O bond in the oxidative addition. It is conceivable that such activating effects by adjacent oxygenation is also present in oxidative additions with carbonates, sulfonates, and sulfamates, etc.
A stable phenyl Ni(II)-carbamate complex 18 is formed after the oxidative addition (Figure 3). The carbamate is κ2-coordinated with Ni. Subsequent ligand exchange of the carbamate complex 18 with phenylboronate leads to phenyl Ni(II) boronate complex 20, which is 5.5 kcal/mol less stable than the Ni-carbamate complex. The detailed mechanism of this ligand exchange step has been suggested to be stepwise.15a,44 Previous theoretical studies suggested that the ligand exchange does not have large barrier.8c,31b,g Thus, we assume the transformation from 18 to 20 is facile.
The following transmetallation (TS21) is the rate-determining step of the catalytic cycle, and requires an activation energy of 30.2 kcal/mol from the catalyst resting complex 18. The transmetallation transition state (TS21) is consistant with the four-center transition state proposed in previous theoretical studies of Pd- and Ni-catalyzed Suzuki-Miyaura couplings.31, 8c The two aryl groups are cis to each other in the transmetallation transition state. The trans transition state is 3.5 kcal/mol less stable, presumably due to greater steric repulsions between the ligand and the aryl groups. Subsequent reductive elimination of the diphenyl Ni(II) complex (23 → TS24) is facile, requiring only a 2.9 kcal/mol activation energy.
Similarly, the oxidative addition of N,N-dimethyl phenyl sulfamate occurs via a mono-ligated five-membered transition state (TS27).41 Three-center transition states TS32 and TS33 are both much higher in energy (Figure 5). The activation barrier of oxidative additions of sulfamate 6 is 10.4 kcal/mol with respect to the Ni(PCy3)2 complex, which is 3.1 kcal/mol lower than that of the oxidative addition of N,N-dimethyl phenyl carbamate 15. The higher reactivity of the sulfamate in oxidative addition is due to the weaker Ph–O bond in sulfamate than the corresponding Ph–O bond in the carbamate group. Nonetheless, the oxidative addition with aryl carbamates and aryl sulfamates are both predicted to be very facile. The differences of their reactivities are attributed to the different activation barriers in the rate-determining transmetalation step. After the oxidative addition of the carbamate, a stable phenyl Ni(II)-carbamate complex 18 is formed. Subsequent ligand exchange with phenyl boronate requires 5.5 kcal/mol energy to form the Ni(II) boronate complex 20. In contrast, since sulfamate is a better leaving group, the Ni(II) boronate complex 20 is formed spontaneously from the Ni(II)-sulfamate complex 28. The activation barrier of the rate-determining transmetalation step for the cross-coupling with the sulfamate (ΔG‡ = 24.7 kcal/mol) is much lower than that for the corresponding carbamate (ΔG‡ = 30.2 kcal/mol). The subsequent steps after transmetalation are identical for the coupling reactions for carbamates and sulfamates.
To support the computational finding that transmetallation is the rate-determining step in the cross-coupling reactions described above, a series of experiments were carried out. Sulfamate 34 was independently subjected to reactions with boronic acids 1a, 2a, and 4a in the presence of NiCl2(PCy3)2 and K3PO4 in toluene at 80 °C (Figure 61H NMR analysis using hexamethylbenzene as internal standard. The relative rate of cross-coupling was found to be dependent on the individual boronic acid employed, with a direct correlation between electron-richness of the boronic acid and reaction rate (i.e., relative rate of conversion: 1a>2a>3a).45 These findings are consistent with a rate-determing transmetalation step for the sulfamate cross-coupling process.46
Figure 6.

Qualitative relative rates of cross-coupling depending on boronic acid
Similar to the reports by Shi in related Ni-catalyzed Suzuki–Miyaura cross-couplings,8b we have observed that water can play a critical role in the success or failure of a coupling reaction (vida supra). To better understand these experimental findings, we examined the role of water computationally. In the coupling with phenyl carbamate, water can coordinate with Ni and stabilize the catalyst resting state, the Ni-carbamate complex 18. A six-membered cyclic Ni(II)-water-carbamate complex 19 is formed and is 1.1 kcal/mol more stable than 18 (Figure 3).47 Coordination with water increases the barrier of transmetalation to 31.3 kcal/mol (19 → TS21), and thus decreases the reactivity of the coupling for the carbamate. In the coupling with phenyl sulfamate, the catalyst resting state is the Ni(II)-boronate complex 20. Upon coordination with a water molecule, a similar six-membered cyclic complex 29 is formed in equilibrium. However, 29 is 7.7 kcal/mol less stable than 20. This suggests that coordination with water does not affect the barrier of transmetalation in the coupling reaction of the sulfamate. This agrees with the experimental observation that Suzuki–Miyaura cross-couplings of aryl sulfamates are less sensitive to water than couplings of aryl carbamates.
In contrast to the high reactivity of the Ni catalyst in oxidative addition with carbamates and sulfamates, Pd catalysts are much less reactive in the oxidative addition step. Oxidative addition of phenyl N,N-dimethylcarbamate with Pd also prefers a five-membered mono-ligated transition state. The activation barrier is 42.2 kcal/mol with respect to the Pd(PCy3)2 complex, much higher than that of the corresponding Ni catalyst. Similarly, oxidative addition of phenyl N,N-dimethylsulfamate also requires a very high activation barrier of 39.7 kcal/mol. These observations are in agreement with previous mechanistic and theoretical studies that the oxidative addition step to Ni(0) is more facile than that to Pd(0).32,40 Therefore, due to the extremely high activation barriers for oxidative addition, Pd catalysts are not effective in couplings with carbamates and sulfamates.
Synthetic applications
The scope and limitations of our sulfamate and carbamate coupling methodologies were further examined by way of a variety of synthetic applications. In each of the studies undertaken, the synthetically useful capability of carbamates and sulfamates to function as directed metalation groups (DMGs) and Suzuki–Miyaura coupling partners was exploited. These studies showcase the utility of our methodology in the synthesis of polysubstituted aromatic compounds, with relevance to natural product and bioactive molecule synthesis.
Scheme 4 depicts a concise synthesis of 5-phenyl-2H-chromene 38 beginning from bis(carbamate) precursor 36. Thus, in a one-pot procedure involving sequential treatment with t-BuLi, 3-methylbut-2-enal, and AcOH, compound 36 was converted into 2H-chromene carbamate 37.48,49 Subsequent Suzuki–Miyaura cross-coupling provided biaryl 38 in 56% yield. Biaryl 38 possesses a heterocyclic framework of bioactivity50 and natural product interest.51
Scheme 4.

In another application, the unique heterotriaryl 43 was constructed using carbamate DoM and cross-coupling methodology (Scheme 5). ortho-Methoxy carbamate 39 was first transformed into boronic acid 40 in 88% yield using a standard lithiation/borylation protocol. Subsequent Pd-catalyzed Suzuki–Miyaura cross-coupling with iodobenzofuran 41 delivered arylated product 42 without disturbance of the aryl carbamate under these conditions.52 The subsequent Ni-catalyzed carbamate cross-coupling with phenyl boronic acid provided the targeted heterotriaryl 43.53
Scheme 5.

An illustration of the DoM/cross-coupling protocol beginning from a heteroaryl carbamate is presented in Scheme 6. Lithiation/borylation of 44 afforded the boropinacolate 45, which was subjected to standard Suzuki-Miyaura cross-coupling conditions with bromide 46 using Pd catalysis to furnish heterobiaryl 47.54 Heteroaryl carbamate 47 was found to be an excellent substrate for the Ni-catalyzed cross-coupling using our standard conditions, to afford the heterotriaryl product 48 in 91% yield. Compound 48 represents a class of pyridines with nonidentical diaryl substitution for which only two synthetic methods are available.55
Scheme 6.

As an application to bioactive molecule synthesis, the anti-inflammatory drug flurbiprofen56 was prepared using sulfamate methodology (Scheme 7). Boronic acid 49, derived from o-lithiation/borylation of N,N-dimethyl phenyl sulfamate, was fluorinated using the conditions described by Furuya and Ritter57 to provide fluorosulfamate 50. Alternative routes to generate 50 by direct lithiation/fluorination of N,N-dimethyl phenyl sulfamate were unsuccessful despite numerous attempts.58 Nonetheless, para-selective electrophilic iodination of 50 furnished 51 in 64% yield. With the aryl sulfamate being inert to Pd catalysis, we carried out a site-selective enolate coupling to install the necessary propionate side chain. Whereas enolate coupling of aryl iodide 51 under Buchwald’s Pd-based conditions was feasible,59 higher yields of 52 were obtained using a Ni-catalyzed variant.60 With the sulfamate remaining undisturbed, exposure of 52 to our Ni-catalyzed conditions facilitated the key sulfamate cross-coupling and delivered the biaryl 53. Acid-mediated hydrolysis furnished flurbiprofen (54) in 84% yield over the two steps. It should be emphasized that the aryl fluoride of 52 was chemically inert under our Ni-catalyzed cross-coupling conditions.61
Scheme 7.

We have observed that aryl carbamates amd sulfamates are unreactive toward Pd(0) catalysis (vida supra) and related processes.62 This feature allows the sequential cross-coupling of an aryl halide, followed by either an aryl sulfamate or carbamate coupling process (see Scheme 7). To further probe related issues of orthogonality, we questioned if it would be possible to couple aryl sulfamates in the presence of aryl carbamates. Although aryl sulfamates generally provide higher yields of cross-coupled products compared to aryl carbamates, the relative reactivity of these substrates had not been determined previously. As shown in Figure 7, an equimolar mixture of phenyl carbamate 55 and phenyl sulfamate 6 was treated with an excess of boronic acid 3a under Ni-catalyzed cross-coupling conditions. Although significant selectivity was observed at elevated temperatures, complete selectivity for sulfamate coupling was readily achieved at 50 °C, as determined by 1H NMR analysis with hexamethylbenzene as internal standard.45 The selectivity for sulfamate coupling is attributed to the lower oxidative addition barrier than that in the corresponding step for carbamates (see above computational studies). Analogous experiments were conducted on the naphthyl-based substrates, carbamate 56 and sulfamate 57, and a high selectivity for naphthyl sulfamate over carbamate cross-coupling was observed at 40 °C.45 We expect that these observations will be of synthetic value.
Figure 7.

Intermolecular competition experiments of aryl sulfamates and aryl carbamates.
Conclusion
In summary, we have discovered the first Suzuki–Miyaura cross-coupling reactions of the synthetically versatile O-aryl carbamate and O-sulfamate groups. The transformations utilize the inexpensive, bench-stable catalyst NiCl2(PCy3)2 to deliver biaryls in good to excellent yields. The methodology is tolerant of substrates bearing electron-donating and electron-withdrawing groups, in addition to those that possess ortho substitutents and heterocyclic frameworks. Furthermore, a computational study has revealed the full catalytic cycles for these cross-coupling reactions, thus shedding light on various mechanistic details, rationalizing sulfamate over carbamate higher reactivity, and indicating the role of water in the transition state. As demonstrated by the given synthetic applications, the methodology provides an efficient means to access polysubstituted aromatic compounds, with relevance to both natural product and bioactive molecule synthesis. The orthogonal use of the sulfamate or carbamate reactivities, in combination with directed ortho metalation (DoM) and other aryl O-based cross-coupling reactions in arene and heteroarene synthesis, may be anticipated.
Supplementary Material
Acknowledgments
The authors are grateful to NSERC Canada (Discovery Grant program) (V.S.), the NIH-NIGMS Grant R00-GM079922 (N.K.G.), Boehringer Ingelheim (N.K.G), DuPont (N.K.G), Eli Lilly (N.K.G), and the National Science Foundation Grant CHE-0548209 (K.N.H) for financial support. The Chemistry-Biology Interface training program (A.L.S., USPHS National Research Service Award GM08496), the Hungarian Scholarship Board Office (A. K.), the ACS Division of Organic Chemistry (K.W.Q.), and the Foote family (K.W.Q.), are acknowledged for graduate student fellowships. We thank the Garcia-Garibay laboratory (UCLA) for access to instrumentation, Michelle Riener and Krastina Petrova for experimental assistance, Dr. Françoise Sauriol (Queen’s) for NMR, and Dr. John Greaves (UC Irvine) and Dr. Lina Yuan (Queen’s) for mass spectral assistance. We express our gratitude to Frontier, Inc. for provision of samples of boronic acids. Calculations were performed on the National Science Foundation Terascale Computing System at the NCSA.
Footnotes
Supporting information available. Detailed experimental details, compound characterization data, optimized Cartesian coordinates and energies and complete authorship of reference 38 (Gaussian 03). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Diederich F, Meijere A, editors. Metal-Catalyzed Cross-Coupling Reactions. Wiley-VCH; Weinheim: 2004. [Google Scholar]; (b) Hassan J, Sévignon M, Gozzi C, Schulz E, Lemaire M. Chem Rev. 2002;102:1359–1469. doi: 10.1021/cr000664r. [DOI] [PubMed] [Google Scholar]; (c) Miyaura N, editor. Topics in Current Chemistry. Vol. 219. Springer-Verlag; New York: 2002. [Google Scholar]; (d) Corbet J, Mignani G. Chem Rev. 2006;106:2651–2710. doi: 10.1021/cr0505268. [DOI] [PubMed] [Google Scholar]; (e) Negishi E. Bull Chem Soc Jpn. 2007;80:233–257. [Google Scholar]
- 2.For a pertinent review, see: Littke AF, Fu GC. Angew Chem Int Ed. 2002;41:4176–4211. doi: 10.1002/1521-3773(20021115)41:22<4176::AID-ANIE4176>3.0.CO;2-U.
- 3.Rappoport Z. The Chemistry of Phenols. John Wiley & Sons Ltd; Chichester: 2003. [Google Scholar]
- 4.For recent examples of C–C bond formation involving aryl sulfonates, see: Naber JR, Fors BP, Wu X, Gunn JT, Buchwald SL. Heterocycles. 2010;80:1215–1226. doi: 10.3987/COM-09-S(S)105.Chow WK, So CM, Lau CP, Kwong FY. J Org Chem. 2010;75:5109–5112. doi: 10.1021/jo100846t.Bhayana B, Fors BP, Buchwald SL. Org Lett. 2009;11:3954–3957. doi: 10.1021/ol9015892.Munday RH, Martinelli JR, Buchwald SL. J Am Chem Soc. 2008;130:2754–2755. doi: 10.1021/ja711449e.So CM, Lau CP, Chan ASC, Kwong FY. J Org Chem. 2008;73:7731–7734. doi: 10.1021/jo8014819.Zhang L, Meng T, Wu J. J Org Chem. 2007;72:9346–9349. doi: 10.1021/jo7019064.Limmert ME, Roy AH, Hartwig JF. J Org Chem. 2005;70:9364–9370. doi: 10.1021/jo051394l.Tang ZY, Hu QS. J Am Chem Soc. 2004;126:3058–3059. doi: 10.1021/ja038752r.
- 5.For recent examples of C–X bond formation involving aryl sulfonates, see: Dooleweerdt K, Fors BP, Buchwald SL. Org Lett. 2010;12:2350–2353. doi: 10.1021/ol100720x.Vo GD, Hartwig JF. J Am Chem Soc. 2009;131:11049–11061. doi: 10.1021/ja903049z.So CM, Zhou Z, Lau CP, Kwong FY. Angew Chem Int Ed. 2008;47:6402–6406. doi: 10.1002/anie.200802157.Ogata T, Hartwig JF. J Am Chem Soc. 2008;130:13848–13849. doi: 10.1021/ja805810p.Fors BP, Watson DA, Biscoe MR, Buchwald SL. J Am Chem Soc. 2008;130:13552–13554. doi: 10.1021/ja8055358.Gao CY, Yang LM. J Org Chem. 2008;73:1624–1627. doi: 10.1021/jo7022558.
- 6.For pertinent reviews and highlights, see: Rosen BM, Quasdorf KW, Wilson DA, Zhang N, Resmerita A-M, Garg NK, Percec V. Chem Rev. doi: 10.1021/cr100259t.Yu DG, Li BJ, Shi ZJ. Acc Chem Res. 2010;43:1486–1495. doi: 10.1021/ar100082d.Knappke CEI, Jacobi von Wangelin A. Angew Chem Int Ed. 2010;49:3568–3570. doi: 10.1002/anie.201001028.Gooben LJ, Gooben K, Stanciu C. Angew Chem Int Ed. 2009;48:3569–3571. doi: 10.1002/anie.200900329.
- 7.(a) Tobisu M, Shimasaki T, Chatani N. Chem Lett. 2009;38:710–711. [Google Scholar]; (b) Tobisu M, Shimasaki T, Chatani N. Angew Chem Int Ed. 2008;47:4866–4869. doi: 10.1002/anie.200801447. [DOI] [PubMed] [Google Scholar]; (c) Guan BT, Xiang SK, Wu T, Sun ZP, Wang BQ, Zhao KQ, Shi ZJ. Chem Commun. 2008:1437–1439. doi: 10.1039/b718998b. [DOI] [PubMed] [Google Scholar]; (d) Dankwardt JW. Angew Chem Int Ed. 2004;43:2428–2432. doi: 10.1002/anie.200453765. [DOI] [PubMed] [Google Scholar]; (e) Wenkert E, Michelotti EL, Swindell CS. J Am Chem Soc. 1979;101:2246–2247. [Google Scholar]
- 8.(a) Quasdorf KW, Tian X, Garg NK. J Am Chem Soc. 2008;130:14422–14423. doi: 10.1021/ja806244b. [DOI] [PubMed] [Google Scholar]; (b) Guan BT, Wang Y, Li BJ, Yu DG, Shi ZJ. J Am Chem Soc. 2008;130:14468–14470. doi: 10.1021/ja8056503. [DOI] [PubMed] [Google Scholar]; (c) Li Z, Zhang SL, Fu Y, Guo QX, Liu L. J Am Chem Soc. 2009;131:8815–8823. doi: 10.1021/ja810157e. [DOI] [PubMed] [Google Scholar]; (d) Li BJ, Li YZ, Lu XY, Liu J, Guan BT, Shi ZJ. Angew Chem Int Ed. 2008;47:10124–10127. doi: 10.1002/anie.200803814. [DOI] [PubMed] [Google Scholar]; (e) Shimasaki T, Tobisu M, Chatani N. Angew Chem Int Ed. 2010;49:2929–2932. doi: 10.1002/anie.200907287. [DOI] [PubMed] [Google Scholar]; (f) Molander GA, Beaumard F. Org Lett. 2010;12:4022–4025. doi: 10.1021/ol101592r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Antoft-Finch A, Blackburn T, Snieckus V. J Am Chem Soc. 2009;131:17750–17752. doi: 10.1021/ja907700e. [DOI] [PubMed] [Google Scholar]; (b) Quasdorf KW, Riener M, Petrova KV, Garg NK. J Am Chem Soc. 2009;131:17748–17749. doi: 10.1021/ja906477r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Xi L, Li BJ, Wu ZH, Lu XY, Guan BT, Wang BQ, Zhao KQ, Shi ZJ. Org Lett. 2010;12:884–887. doi: 10.1021/ol9029534. [DOI] [PubMed] [Google Scholar]; (d) Dallaire C, Kolber I, Gingras M. Org Synth. 2002;78:42. [Google Scholar]; (e) Sengupta S, Leite M, Raslan DS, Quesnelle C, Snieckus V. J Org Chem. 1992;57:4066–4068. [Google Scholar]
- 10.(a) Macklin TK, Snieckus V. Org Lett. 2005;7:2519–2522. doi: 10.1021/ol050393c. [DOI] [PubMed] [Google Scholar]; (b) Wehn PM, Du Bois J. Org Lett. 2005;7:4685–4688. doi: 10.1021/ol051896l. [DOI] [PubMed] [Google Scholar]; (c) Ramgren SD, Silberstein AL, Yang Y, Garg NK. Angew Chem Int Ed. 2011;50:2171–2173. doi: 10.1002/anie.201007325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.For functionalization by electrophilic aromatic substitution, which favors para substitution, see: Smith MB, March J. March’s Advanced Organic Chemistry. 6. John Wiley & Sons, Inc; New Jersey: 2007. p. 670.
- 12.For functionalization by directed ortho metalation, see: Snieckus V. Chem Rev. 1990;90:879–933.Hartung CG, Snieckus V. In: Modern Arene Chemistry. Astruc D, editor. Wiley-VCH; New York: 2002. pp. 330–367.Macklin T, Snieckus V. In: Handbook of C-H Transformations. Dyker G, editor. Wiley-VCH; New York: 2005. pp. 106–119.
- 13.For functionalization using directed ortho metalation/ipso-desilylation, see: Zhao Z, Snieckus V. Org Lett. 2005;7:2523–2526. doi: 10.1021/ol0506563.Bracegirdle S, Anderson EA. Chem Commun. 2010;46:3454–3456. doi: 10.1039/b924135c.
- 14.For Pd-catalyzed ortho functionalization of aryl pivalates, see: Xiao B, Fu Y, Xu J, Gong TJ, Dai JJ, Yi J, Liu L. J Am Chem Soc. 2010;132:468–469. doi: 10.1021/ja909818n.For Pd-catalyzed ortho functionalization of aryl carbamates, see: Bedford RB, Webster RL, Mitchell CJ. Org Biomol Chem. 2009;7:4853–4857. doi: 10.1039/b916724m.Zhao X, Yeung CS, Dong VM. J Am Chem Soc. 2010;132:5837–5844. doi: 10.1021/ja100783c.Nishikata T, Abela AR, Huang S, Lipshutz BH. J Am Chem Soc. 2010;132:4978–4979. doi: 10.1021/ja910973a.For a recent Ir-catalyzed process, see: Yamazaki K, Kawamorita S, Ohmiya H, Sawamura M. Org Lett. 2010;12:3978–3981. doi: 10.1021/ol101493m.
- 15.(a) Miyaura N, Suzuki A. Chem Rev. 1995;95:2457–2483. [Google Scholar]; (b) Suzuki A. Chem Commun. 2005:4759–4763. doi: 10.1039/b507375h. [DOI] [PubMed] [Google Scholar]; (c) Doucet H. Eur J Org Chem. 2008:2013–2030. [Google Scholar]
- 16.(a) For preliminary communications involving this work, see references 9a and 9b. (b) Following these publications, Shi and coworkers reported an alternate protocol for the Suzuki–Miyaura coupling of aryl carbamates (see ref 9c). (c) For Suzuki–Miyaura couplings of aryl carbamates and sulfamates under microwave conditions, see: Baghbanzadeh M, Pilger C, Kappe CO. J Org Chem. 2011;76:1507–1510. doi: 10.1021/jo1024464.
- 17.NiCl2(PCy3)2 (CAS# 19999-87-2) is commercially available from Strem Chemicals, Inc. (catalog #28-0091) and Aldrich Chemical Co., Inc. (catalog #708526). Alternatively, it can be prepared in multigram quantities following a simple one-step protocol, see reference 8a and: Stone PJ, Dori Z. Inorg Chim Acta. 1970;5:434–438.Barnett KW. J Chem Educ. 1974;51:422–423.
- 18.Quasdorf KW, Garg NK. Encyclopedia of Reagents for Organic Synthesis. [DOI] [Google Scholar]
- 19.In the presence of excess arylboronic acid, NiCl2(PCy3)2 likely undergoes reduction to an active Ni(0) catalyst; see: Zim D, Monteiro AL. Org Lett. 2001;3:3049. doi: 10.1021/ol016526l.
- 20.The selection of boronic acid coupling partner was made in order to facilitate product purification.
- 21.Control experiments show that a large excess of water significantly reduces catalytic activity, likely by forming inactive nickel hydroxides/oxides, see: Inada K, Miyaura N. Tetrahedron. 2000;56:8657–8660.
- 22.(a) Since the addition of water to the boroxine is inaccurate on small-scale operation, the boronic acids were heated under vacuum for 1–12 h using Kugelrohr apparatus to achieve a 1:10 ratio of boronic acid:boroxine. The ratio was determined by 1H NMR analysis immediately prior to use. (b) For comment on similar observations, see: Storgaard M, Ellman JA. Org Synth. 2009;86:360–373.. (c) For the crucial role of water in aryl acetate cross-couplings, see ref 8b.
- 23.Comparable yields were obtained from reactions containing 10 or 20 mol% of the PCy3HBF4 additive.
- 24.Yu DG, Yu M, Guan BT, Li BJ, Zheng Y, Wu ZH, Shi ZJ. Org Lett. 2009;11:3374–3377. doi: 10.1021/ol901217m. [DOI] [PubMed] [Google Scholar]
- 25.Comparable results were obtained when N,N-diethyl sulfamates were used in place of N,N-dimethyl sulfamates. The N,N-dimethyl derivatives were pursued because the N,N-dimethyl sulfamoylating reagent is commercially available, see reference 27.
- 26.Aryl sulfamates have been shown to be less effective, but still synthetically useful, substrates in DoM reactions compared to aryl carbamates; see reference 10a.
- 27.N,N-Dimethylsulfamoyl chloride from Aldrich Chemical Co., Inc. costs approximately $0.80 per gram (CAS# 13360-57-1).
- 28.de Silva SO, Reed JN, Billedeau RJ, Wang X, Norris DJ, Snieckus V. Tetrahedron. 1992;48:4863–4878. [Google Scholar]
- 29.For examples of Pd-catalyzed Suzuki–Miyaura couplings that are tolerant of heterocycles, see: Billingsley K, Buchwald SL. J Am Chem Soc. 2007;129:3358–3366. doi: 10.1021/ja068577p.Guram AS, Wang X, Bunel E, Faul MM, Larsen RD, Martinelli MJ. J Org Chem. 2007;72:5104–5112. doi: 10.1021/jo070341w.For a pertinent review, see: Slagt VF, de Vries AHM, de Vries JG, Kellogg RM. Org Proc Res Dev. 2010;14:30–47.
- 30.For reviews of theoretical studies, see: Braga AAC, Ujaque G, Maseras F. In: Computational Modeling for Homogeneous and Enzymatic Catalysis A Knowledge-Base for Designing Efficient Catalysis. Morokuma K, Musaev DG, editors. WILEY-VCH GmbH & Co; KGaA, Weinheim: 2008. pp. 109–130.Xue LQ, Lin ZY. Chem Soc Rev. 2010;39:1692–1705. doi: 10.1039/b814973a.
- 31.For theoretical studies of Pd-catalyzed Suzuki–Miyaura couplings, see: Sumimoto M, Iwane N, Takahama T, Sakaki S. J Am Chem Soc. 2004;126:10457–10471. doi: 10.1021/ja040020r.Braga AAC, Morgon NH, Ujaque G, Maseras F. J Am Chem Soc. 2005;127:9298–9307. doi: 10.1021/ja050583i.Goossen LJ, Koley D, Hermann HL, Thiel W. J Am Chem Soc. 2005;127:11102–11114. doi: 10.1021/ja052435y.Goossen LJ, Koley D, Hermann HL, Thiel W. Organometallics. 2006;25:54–67.Braga AAC, Ujaque G, Maseras F. Organometallics. 2006;25:3647–3658.Braga AAC, Morgon NH, Ujaque G, Lledos A, Maseras F. J Organomet Chem. 2006;691:4459–4466.Sicre C, Braga AAC, Maseras F, Cid MM. Tetrahedron. 2008;64:7437–7443.Huang YL, Weng CM, Hong FE. Chem-Eur J. 2008;14:4426–4434. doi: 10.1002/chem.200800011.Gourlaouen C, Ujaque G, Lledós A, Medio-Simon M, Asensio G, Maseras F. J Org Chem. 2009;74:4049–4054. doi: 10.1021/jo900178c.Jover J, Fey N, Purdie M, Lloyd-Jones GC, Harvey JN. J Mol Catal A: Chem. 2010;324:39–47.
- 32.(a) Ahlquist M, Fristrup P, Tanner D, Norrby PO. Organometallics. 2006;25:2066–2073. [Google Scholar]; (b) Ahlquist M, Norrby PO. Organometallics. 2007;26:550–553. [Google Scholar]; (c) Li Z, Fu Y, Guo QX, Liu L. Organometallics. 2008;27:4043–4049. [Google Scholar]; (d) McMullin CL, Jover J, Harvey JN, Fey N. Dalton Trans. 2010;39:10833–10836. doi: 10.1039/c0dt00778a. [DOI] [PubMed] [Google Scholar]
- 33.(a) Ananikov VP, Musaev DG, Morokuma K. J Am Chem Soc. 2002;124:2839–2852. doi: 10.1021/ja017476i. [DOI] [PubMed] [Google Scholar]; (b) Ananikov VP, Musaev DG, Morokuma K. Organometallics. 2005;24:715–723. doi: 10.1021/om050255r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zuidema E, van Leeuwen PWNM, Bo C. Organometallics. 2005;24:3703–3710. [Google Scholar]; (d) Ananikov VP, Musaev DG, Morokuma K. Eur J Inorg Chem. 2007:5390–5399. [Google Scholar]; (e) Koizumi T, Yamazaki A, Yamamoto T. Dalton Trans. 2008:3949–3952. doi: 10.1039/b802408a. [DOI] [PubMed] [Google Scholar]; (f) Ariafard A, Yates BF. J Organomet Chem. 2009;694:2075–2084. [Google Scholar]
- 34.For a theoretical study of Ni-catalyzed Suzuki–Miyaura cross-couplings of aryl acetates, see ref 8c.
- 35.(a) Becke AD. J Chem Phys. 1993;98:5648–5652. [Google Scholar]; (b) Lee C, Yang W, Parr RG. Phys Rev B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 36.Kruger C, Tsay YH. J Organomet Chem. 1972;34:387–395. [Google Scholar]
- 37.(a) Barone V, Cossi M. J Phys Chem A. 1998;102:1995–2001. [Google Scholar]; (b) Cossi M, Rega N, Scalmani G, Barone V. J Comp Chem. 2003;24:669–681. doi: 10.1002/jcc.10189. [DOI] [PubMed] [Google Scholar]
- 38.Frisch MJ, et al. Gaussian 03, Revision D.01. Gaussian, Inc; Wallingford CT: 2004. [Google Scholar]
- 39.The exact mechanism of the formation of the η2 LPd(ArX) complex is controlled by the size of the ligand, see ref. 31j for a recent theoretical study on the details of this process.
- 40.For a mechanistic study on the formation of Ni η2 complexes with aryl halides, see: Yoshikai N, Matsuda H, Nakamura E. J Am Chem Soc. 2008;130:15258–15259. doi: 10.1021/ja807000a.
- 41.Oxidative addition of O-aryl thiocarbamates using Pd catalyst also occurs via a similar five-membered transition state: Harvey JN, Jover J, Lloyd-Jones GC, Moseley JD, Murray P, Renny JS. Angew Chem, Int Ed. 2009;48:7612–7615. doi: 10.1002/anie.200903908.
- 42.See Supporting Information for details of calculations of bis-ligated species.
- 43.The gas phase bond dissociation enthalpy of the Ph–O bond in phenyl N,N-dimethyl carbamate is 22.4 higher than that of the O–carbonyl bond according to B3LYP/6-31G(d) calculations. See the Supporting Information for details.
- 44.Miyaura N. J Organomet Chem. 2002;653:54–57. [Google Scholar]
- 45.See Supporting Information for details.
- 46.Reductive elimination to form the biaryl product is considered to be a facile step in the catalytic cycle, see ref 8c and references therein.
- 47.In gas phase calculations, 19 is 3.2 kcal/mol more stable than 18.
- 48.Chauder BA, Kalinin AV, Snickus V. Synthesis. 2001:140–144. [Google Scholar]
- 49.Chauder BA, Kalinin AV, Taylor NJ, Snickus V. Angew Chem Int Ed. 1999;38:1435–1438. doi: 10.1002/(SICI)1521-3773(19990517)38:10<1435::AID-ANIE1435>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 50.(a) Petasis NA, Butkevich AN. J Organomet Chem. 2009;694:1747–1753. doi: 10.1016/j.jorganchem.2008.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nicolaou KC, Pfefferkorn JA, Roecker AJ, Cao GQ, Barleunga S, Mitchell HJ. J Am Chem Soc. 2000;122:9939–9953. [Google Scholar]
- 51.Veitch NC, Grayer RJ. Nat Prod Rep. 2008;25:555–611. doi: 10.1039/b718040n. [DOI] [PubMed] [Google Scholar]
- 52.For the general DoM – directed remote metalation (DreM) – Suzuki cross-coupling approach to this and related systems, see: James CA, Coelho AL, Gevaert M, Forgione P, Snieckus V. J Org Chem. 2009;74:4094–4103. doi: 10.1021/jo900146d.
- 53.The low yield of 43 is a result of reductive cleavage and arylation of benzofuran, rather than low conversion of 42 owing to steric hindrance effects.
- 54.Alessi M, Larkin AL, Ogilvie KA, Green LA, Lai S, Lopez S, Snieckus V. J Org Chem. 2007;72:1588–1594. doi: 10.1021/jo0620359. [DOI] [PubMed] [Google Scholar]
- 55.(a) Chang MY, Lin CY, Hung CY. Tetrahedron. 2007;63:3312–3320. [Google Scholar]; (b) Karig G, Thasana N, Gallagher T. Synlett. 2002:808–810. [Google Scholar]
- 56.For pertinent reviews, see: Richy F, Rabehnda V, Mawet A, Reginster JY. Int J Clin Pract. 2007;61:1396–1406. doi: 10.1111/j.1742-1241.2007.01452.x.Kumar P, Pathak PK, Gupta VK, Srivastava BK, Kushwaha BS. Asian J Chem. 2004;16:558–562.
- 57.Furuya T, Ritter T. Org Lett. 2009;11:2860–2863. doi: 10.1021/ol901113t. [DOI] [PubMed] [Google Scholar]
- 58.For the DoM approach to ortho-fluorination, see: Snieckus V, Beaulieu F, Morhi K, Han W, Murphy CK, Davis FA. Tetrahedron Lett. 1994;35:3465–3468.
- 59.Moradi WA, Buchwald SL. J Am Chem Soc. 2001;123:7996–8002. doi: 10.1021/ja010797+. [DOI] [PubMed] [Google Scholar]
- 60.Durandetti M, Gosmini C, Périchon J. Tetrahedron. 2007;63:1146–1153. [Google Scholar]
- 61.For Ni-catalyzed Kumada and Suzuki–Miyaura cross-coupling reactions of aryl fluorides, see: Yoshikai N, Mashima H, Nakamura E. J Am Chem Soc. 2005;127:17978–17979. doi: 10.1021/ja056327n.Dankwardt JW. J Organomet Chem. 2005;690:932–938.Schaub T, Backes M, Radius U. J Am Chem Soc. 2006;128:15964–15965. doi: 10.1021/ja064068b.
- 62.The iodide of 51 could also be used in copper-catalyzed C–N bond forming reactions without disturbing the aryl sulfamate; Ramgren SD, Silberstein AL, Garg NK. Unpublished work. University of California; Los Angeles, CA: 2010.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.