

Abstract
Pancreatic acinar cells are secretory cells whose main function is to synthesize, store and finally release digestive enzymes into the duodenum. However, in response to noxious stimuli, acinar cells behave like real inflammatory cells because of their ability to activate signalling transduction pathways involved in the expression of inflammatory mediators. Mediated by the kinase cascade, activation of Nuclear factor-κB, Activating factor-1 and Signal transducers and activators of transcription transcription factors has been demonstrated in acinar cells, resulting in overexpression of inflammatory genes. In turn, kinase activity is down-regulated by protein phosphatases and the final balance between kinase and phosphatase activity will determine the capability of the acinar cells to produce inflammatory factors. The kinase/phosphatase pair is a redox-sensitive system in which kinase activation overwhelms phosphatase activity under oxidant conditions. Thus, the oxidative stress developed within acinar cells at early stages of acute pancreatitis triggers the activation of signalling pathways involved in the up-regulation of cytokines, chemokines and adhesion molecules. In this way, acinar cells trigger the release of the first inflammatory signals which can mediate the activation and recruitment of circulating inflammatory cells into the injured pancreas. Accordingly, the role of acinar cells as promoters of the inflammatory response in acute pancreatitis may be considered. This concept leads to amplifying the focus from leukocyte to acinar cells themselves, to explain the local inflammation in early pancreatitis.
Keywords: Acinar cells, Inflammatory mediators, Signal pathways, Acute pancreatitis
INTRODUCTION
Under physiological conditions the function of pancreatic acinar cells is to secrete hydrolytic enzymes into the duodenum that are used for the breakdown of large food molecules. Many of them are synthesized and stored as inactive pro-enzymes (zymogens) and they only become active when they reach the duodenum. However, in acute pancreatitis, activation of zymogens within acinar cells occurs which leads to the autodigestion of the pancreas. Classically, it has been accepted that the pancreatic injury would trigger a local and systemic inflammatory response in which the immune cells would play the central role. However, recently the concept has emerged that pancreatic acinar cells are responsible for releasing the first inflammatory signals in response to the injury initiated within them and, subsequently these signals activate the immune response. This notion is supported by different studies showing that acinar cells are able to activate signalling pathways involved in the expression of inflammatory mediators[1-3].
INFLAMMATORY MEDIATORS PRODUCED BY ACINAR CELLS
Acinar cells have demonstrated the ability to produce different types of inflammatory mediators in response to noxious stimuli.
Cytokines
Cytokines are a small group of low molecular weight soluble proteins which act as mediators of cell communication[4,5]. They are pleiotropic factors which after the interaction with highly specific cell surface receptors show the ability to enhance the production of themselves as well as other inflammatory mediators, thus contributing to the amplification of the inflammatory response in acute pancreatitis.
Overexpression of cytokines in pancreatic tissue during experimental acute pancreatitis has been reported in different studies, but the first evidence of cytokine production by a non-inflammatory cell in the pancreas was shown by Gukovskaya et al[6], who demonstrated that pancreatic acinar cells are able to produce, release and respond to TNF-α. Blinman et al[7] also reported expression of TNF-α and Il-6 in acinar cells as a result of the isolation procedure of acini. Increases in the production of TNF-α, Il-6 and Il-1β have been reported by Kim et al[8], who analyzed the response of pancreatic acinar cells cultured in the presence of neutrophils primed with 4β-phorbol 12 β-myristate 13 α-acetate (PMA). More recently, Ramudo et al[9] corroborated the acinar expression of TNF-α in in vitro studies, in which pancreatic acinar cells were cultured in the presence of pancreatitis-associated ascitic fluid (PAAF) as well as in in vivo studies carried out in acinar cells from rats with acute pancreatitis induced by bile-pancreatic duct obstruction (BPDO)[10]. In the BPDO model of AP, the acinar expression of the anti-inflammatory cytokine Il-10 has also been demonstrated at early stages of the disease[11].
Cytokines produced by acinar cells could play a detrimental role in the progression of acute pancreatitis through the induction of regulatory genes in the acinar cell, including those encoding inflammatory factors and their receptors[6] and cholecystokinin (CCK) receptors[12]. In addition, the acinar production of TNF-α may contribute to the severity of acute pancreatitis through the induction of apoptosis of acinar cells[6,13].
Chemokines
Chemokines are a family of small cytokines with activating and chemotactic effects on leukocytes, which provide a key stimulus for directing the migration of inflammatory cells into injured tissues[14]. On a structural basis, chemokines have been classified into four subfamilies: C, CC, CXC and CX3C, according to the position of the first two cysteine residues[15]. It is believed that each chemokine subfamily acts selectively upon different leukocyte subsets. CC chemokines, such as monocyte chemoattractant protein-1 (MCP-1), macrophage inflammatory protein (MIP)-1α, and regulated upon activation, normal T expressed and secreted (RANTES), would act mainly on monocytes. CXC chemokines, such as Il-8 or its analogue in the rat cytokine-induced neutrophil chemoattractant (CINC), Mob-1 (the homologue of human IP-10) and MIP-2, would mainly act on neutrophils. Experimental data support the notion that chemokines play a key role in the development and evolution of acute pancreatitis[16-19].
Pancreatic acinar cells have demonstrated the ability to produce chemokines as first inflammatory signals during acute pancreatitis. Acinar expression of MCP-1 has been reported following induction of acute pancreatitis with either supramaximal doses of caerulein or retrograde infusion of sodium taurocholate[17,20] and recently in the BPDO model[21]. Overexpression of CINC in acinar cells was also found in experimental models of acute pancreatitis different from hyperstimulation with caerulein[22], a finding that suggests different regulation for CC and CXC chemokine synthesis in acinar cells.
The capability of acinar cells to produce chemokines has also been shown in in vitro studies. Blinman et al[7] showed expression of KC, MCP-1 and MIP-2 in acini during the isolation procedure. MCP-1 was found overexpressed in acinar cells in response to caerulein[22,23], TNF-α[24] and PAAF[2]. Mob-1 and RANTES were reported to be synthesized in acinar cells stimulated with CCK or its analogue caerulein[22,23,25,26]. Xie et al[27] reported expression of Il-8 in the AR42J cells cultured in the presence of menadione, TNF-α and TGF-β.
Adhesion molecules
Three families of cell adhesion molecules play a central role in leukocyte-endothelial interactions: selectins, integrins and the immunoglobulin family.
Only ICAM-1 has been reported to be synthesized by pancreatic acinar cells. ICAM-1 is a cell surface glycoprotein belonging to the immunoglobulin family which binds to the β2 integrin counter-receptors LFA-1 and MAC-1 for the leukocyte transmigration into inflamed tissues[28].
ICAM-1 is constitutively expressed at very low levels but it is upregulated in response to inflammation[28,29]. Pancreatic acinar cells demonstrated the ability to upregulate ICAM-1 expression in response to supramaximal doses of caerulein and, as a result, the adhesion of neutrophils to acinar cells was found in in vitro studies[30]. Also, in vivo studies carried out in rats with AP induced by BPDO showed overexpression of ICAM-1 in acinar cells at early stages of the disease[31]. These data suggest that acinar cells themselves are able to release the first signals from the pancreas during AP for the recruitment of circulating inflammatory cells into the injured pancreas.
SIGNAL PATHWAYS INVOLVED IN THE EXPRESSION OF INFLAMMATORY FACTORS IN ACINAR CELLS
Nuclear factor-κB (NF-κB)
NF-κB is a transcription factor which forms homo- or heterodimers composed of members of the Rel subfamily, which bind to DNA and activate a wide range of genes, many of them associated with inflammation[32,33]. NF-κB is kept latent in the cytosol via interaction with inhibitory proteins of IκB family that prevent NF-κB translocation to the nucleus and its binding to DNA. NF-κB activation requires the phosphorylation of IκB proteins at specific regulatory amino acid residues and subsequently IκBs are targeted for degradation by the 26S proteasome, thus allowing NF-κB dimers to translocate to the nucleus where they bind to DNA and activate target genes[34].
The involvement of NF-κB transcription factor in the expression of inflammatory mediators in acinar cells has been demonstrated in rats with AP experimentally induced by caerulein[1,6,23], BPDO[10,21,35,36] and retrograde infusion of NaTc into the bile-pancreatic duct[21,35]. Overexpression of cytokines, chemokines and adhesion molecules has been associated to NF-κB activation in acinar cells cultured in the presence of CCK or its analogue caerulein[25,30,37], substance P[38], PAAF[9] and TNF-α[6].
Activating factor-1 (AP-1)
AP-1 is a collective term referring to dimeric transcription factors composed of Jun, Fos or ATF (activating transcription factor) subunits that bind a common DNA site, the AP-1 binding site. Most of the genes that encode AP-1 components behave as “immediate-early” genes, such as those related with the inflammatory response[39].
AP-1 activation has been demonstrated to be involved in the signal transduction mechanisms for the acinar expression of cytokines and chemokines during AP induced by NaTc[40], caerulein and ethanol[41]. In addition, AP-1 has also been found to be activated in AR42J cells in response to lysophosphatidylcholine[42] and in mouse pancreatic acinar cells stimulated with substance P for chemokine synthesis[3,43].
Signal transducers and activators of transcription (STAT)
The STAT family of transcription factors was initially identified as cytokine and growth factor-inducible DNA binding[44]. They are silent in the cytoplasm and in response to stimuli such as cytokines, growth factors and reactive oxygen species (ROS), become phosphorylated by members of the Janus kinase (JAK) family[45] and Src kinase family[46].
The STAT pathway has been shown to act as down-stream in the expression of inflammatory mediators in acinar cells of rats with AP induced by either BPDO or NaTc[21,35], as well as in isolated acinar cells cultured in the presence of either PAAF[2] or substance P[3]. Activation of STAT1 and STAT3 has also been reported to be involved in cytokine synthesis in AR42J cells stimulated either with cytokines[47,48] or cerulein[49].
Kinases
Different kinds of kinases have been associated with the phosphorylation required for the activation of transcription factors mediating the expression of inflammatory mediators in acinar cells.
The mitogen activated protein kinase (MAPK) family is composed of five major classes of MAPKs, which include p38MAPK, extracellular-regulated kinase (ERK)1/2 and Jun N-terminal kinase (JNK). Emerging evidence has implicated these three MAPKs in the pathophysiology of AP because of the pivotal role they play in the inflammatory response developed within different cell types including acinar cells. According to different reports[7,9,10,50], the overproduction of ROS generated in acinar cells in the course of AP might activate the MAPK cascade and, as a result, inflammatory genes are expressed. In this line, the use of N-Acetilcysteine (NAC) as an antioxidant treatment reduced the acinar expression of cytokines and chemokines mediated by MAPK activation in rats with AP induced either by BPDO or NaTc[10,21]. This notion is also supported by in vitro studies in which p38MAPK, ERK1/2 and JNK activation was found in acinar cells treated with H2O2[51]. MAPK inhibition or transfection with dominant mutant MAPK genes abolished stress-induced cytokine expression[7,52]. It has been shown that ERK and JNK act as upstreams of NF-κB and AP-1-mediated chemokine expression in acinar cells[3]. Accordingly, the evidence indicates that MAPKs would be involved in the control of the inflammatory response in acinar cells by directing cytokine expression.
Protein kinase C (PKC) is a family of serine threonine kinases with 10 isoforms. In pancreatic acinar cells, PKC activation is required to mediate NF-κB activation in response to CCK-8 and TNF-α[37]. The activity of PKCs is regulated by phosphorylation, Src kinases being important mediators in this regulation[53]. Recently, it has been reported that Src kinases are involved in the signalling cascade which triggers chemokine synthesis in acinar cells in response to substance P[3]. However, Src kinases do not play an essential role in the inflammatory response of acinar cells to either CCK-8 or TNF-α. Also, protein kinase D has been associated with NF-κB activation as early events during caerulein-induced pancreatitis[54].
Phosphatases
Protein phosphatases act on protein kinases decreasing their activity by dephosphorylation. Phosphatase activity exerts a negative regulation of MAPK action on the expression of inflammatory genes[55]. Phosphatases may regulate the duration and strength of MAPK activation, which are critical determinants of their biological effects.
There are two major groups of protein phosphatases, protein tyrosine phosphatases (PTPs) and serine/threonine phosphatases (PPPs). PTPs include cytosolic members such as MAPK phosphatases (MKP) (which down-regulates MAPK activity[56]), SHP-1 and SHP-2, and membrane phosphatases such as CD45.
A decrease in PP2A activity associated with up-regulation of pro-inflammatory genes early in the course of AP has recently been reported[57]. Pancreatic overexpression of MKPs[58] and SHPs[59] has been reported as an early response during AP induced by caerulein. Regarding membrane phosphatases, CD45, a tyrosine phosphatase traditionally considered to be expressed exclusively in nucleated hematopoietic cells[60], was unexpectedly found in the acinar cell membrane[61]. This result strongly supports the theory that the acinar cell may function like an immunocompetent cell. Decrease in CD45 expression associated to the inflammatory response triggered by acinar cells was found in response to PAAF[61]. Additionally, CD45 expression in acinar cells has been shown to be down-regulated in the course of BPDO-induced AP in parallel with an increase in TNF-α production and both effects were prevented by NAC treatment[62]. It is generally thought that CD45 couples Src kinases to maintain them in a dephosphorylated state[63]. Given that Src kinases are required to initiate the signal transduction cascade in the inflammatory gene expression, the role of CD45 in acinar cells might be to modulate the inflammatory response developed in acinar cells in response to a stressful environment. However, given that acinar cell phosphatases are down-regulated by redox-sensitive mechanisms[57,62], they would not be able to operate as negative regulators of the inflammatory response developed by acinar cells during AP when the overproduction of ROS overwhelms the natural antioxidant mechanisms.
From the data reviewed (Figure 1), it is evident that acinar cells are able to behave like an inflammatory cell. More basic research in this area is expected to advance in the knowledge on the contribution of acinar cells in the complex inflammatory cascade triggered in AP.
Figure 1.
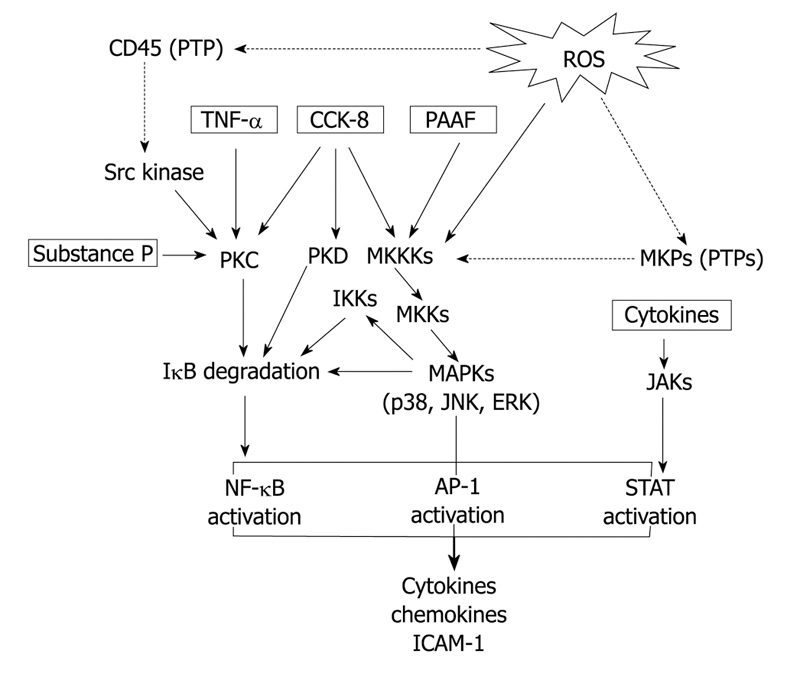
Signal transduction pathways involved in the expression of inflammatory mediators in pancreatic acinar cells. Abbreviations: ROS: Reactive oxygen species; PTPs: Proteine tyrosine phosphatases; TNF-α: Tumor necrosis factor-α; CCK-8: Cholecystokinin-8; PAAF: Pancreatitis-associated ascitic fluid; PKC: Protein kinase C; PKD: Protein kinase D; MKKKs: Mitogen activated protein kinase kinase kinases; MKKs: Mitogen activated protein kinase kinases; MAPKs: Mitogen activated protein kinases; MKPs: Mitogen kinase phosphatases; IKKs: ΙκB kinases; JNK: Jun N-terminal kinase; ERK: Extracellular-regulated kinase; JAKs: Janus kinases; NF-κB: Nuclear factor-κB; AP-1: Activating factor-1; STAT: Signal transducers and activators of transcription. Arrows: Activation; Dotted arrows: Inhibition.
Acknowledgments
The author thanks Ms. Elizabeth Nestor for her linguistic assistance
Footnotes
Supported by A grant from Insituto de Salud Carlos III, Spain, PI08/0035
Peer reviewer: Ilker Tasci, MD, Associate Professor, Department of Internal Medicine, Gulhane School of Medicine, Etlik 06018, Ankara, Turkey
S- Editor Li LF L- Editor Roemmele A E- Editor Yang C
References
- 1.Gukovsky I, Gukovskaya AS, Blinman TA, Zaninovic V, Pandol SJ. Early NF-kappaB activation is associated with hormone-induced pancreatitis. Am J Physiol. 1998;275:G1402–G1414. doi: 10.1152/ajpgi.1998.275.6.G1402. [DOI] [PubMed] [Google Scholar]
- 2.Ramudo L, Yubero S, Manso MA, Vicente S, De Dios I. Signal transduction of MCP-1 expression induced by pancreatitis-associated ascitic fluid in pancreatic acinar cells. J Cell Mol Med. 2009;13:1314–1320. doi: 10.1111/j.1582-4934.2008.00529.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ramnath RD, Sun J, Bhatia M. Involvement of SRC family kinases in substance P-induced chemokine production in mouse pancreatic acinar cells and its significance in acute pancreatitis. J Pharmacol Exp Ther. 2009;329:418–428. doi: 10.1124/jpet.108.148684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lowry SF. Cytokine mediators of immunity and inflammation. Arch Surg. 1993;128:1235–1241. doi: 10.1001/archsurg.1993.01420230063010. [DOI] [PubMed] [Google Scholar]
- 5.Aderem AA. How cytokines signal messages within cells. J Infect Dis. 1993;167 Suppl 1:S2–S7. doi: 10.1093/infdis/167.supplement_1.s2. [DOI] [PubMed] [Google Scholar]
- 6.Gukovskaya AS, Gukovsky I, Zaninovic V, Song M, Sandoval D, Gukovsky S, Pandol SJ. Pancreatic acinar cells produce, release, and respond to tumor necrosis factor-alpha. Role in regulating cell death and pancreatitis. J Clin Invest. 1997;100:1853–1862. doi: 10.1172/JCI119714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blinman TA, Gukovsky I, Mouria M, Zaninovic V, Livingston E, Pandol SJ, Gukovskaya AS. Activation of pancreatic acinar cells on isolation from tissue: cytokine upregulation via p38 MAP kinase. Am J Physiol Cell Physiol. 2000;279:C1993–C2003. doi: 10.1152/ajpcell.2000.279.6.C1993. [DOI] [PubMed] [Google Scholar]
- 8.Kim H, Seo JY, Roh KH, Lim JW, Kim KH. Suppression of NF-kappaB activation and cytokine production by N-acetylcysteine in pancreatic acinar cells. Free Radic Biol Med. 2000;29:674–683. doi: 10.1016/s0891-5849(00)00368-3. [DOI] [PubMed] [Google Scholar]
- 9.Ramudo L, Manso MA, De Dios I. Biliary pancreatitis-associated ascitic fluid activates the production of tumor necrosis factor-alpha in acinar cells. Crit Care Med. 2005;33:143–148; discussion 248. doi: 10.1097/01.ccm.0000150654.13653.5b. [DOI] [PubMed] [Google Scholar]
- 10.Ramudo L, Manso MA, Sevillano S, de Dios I. Kinetic study of TNF-alpha production and its regulatory mechanisms in acinar cells during acute pancreatitis induced by bile-pancreatic duct obstruction. J Pathol. 2005;206:9–16. doi: 10.1002/path.1747. [DOI] [PubMed] [Google Scholar]
- 11.Ramudo L, Manso MA, Vicente S, De Dios I. Pro- and anti-inflammatory response of acinar cells during acute pancreatitis. Effect of N-acetyl cysteine. Cytokine. 2005;32:125–131. doi: 10.1016/j.cyto.2005.07.017. [DOI] [PubMed] [Google Scholar]
- 12.Viguerie N, Bertrand V, Dufresne M, Davis E, Lefort S, Vita N, Vaysse N, Pradayrol L, Bastie MJ. Interleukin-6 regulation of CCK/gastrin receptors and amylase secretion in a rat pancreatic acinar cell line (AR4-2J) Eur Cytokine Netw. 1994;5:433–440. [PubMed] [Google Scholar]
- 13.Yasuda H, Kataoka K, Ichimura H, Mitsuyoshi M, Iida T, Kita M, Imanishi J. Cytokine expression and induction of acinar cell apoptosis after pancreatic duct ligation in mice. J Interferon Cytokine Res. 1999;19:637–644. doi: 10.1089/107999099313785. [DOI] [PubMed] [Google Scholar]
- 14.Adams DH, Lloyd AR. Chemokines: leucocyte recruitment and activation cytokines. Lancet. 1997;349:490–495. doi: 10.1016/s0140-6736(96)07524-1. [DOI] [PubMed] [Google Scholar]
- 15.Murphy PM. The molecular biology of leukocyte chemoattractant receptors. Annu Rev Immunol. 1994;12:593–633. doi: 10.1146/annurev.iy.12.040194.003113. [DOI] [PubMed] [Google Scholar]
- 16.Bhatia M, Ramnath RD, Chevali L, Guglielmotti A. Treatment with bindarit, a blocker of MCP-1 synthesis, protects mice against acute pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2005;288:G1259–G1265. doi: 10.1152/ajpgi.00435.2004. [DOI] [PubMed] [Google Scholar]
- 17.Brady M, Bhatia M, Christmas S, Boyd MT, Neoptolemos JP, Slavin J. Expression of the chemokines MCP-1/JE and cytokine-induced neutrophil chemoattractant in early acute pancreatitis. Pancreas. 2002;25:260–269. doi: 10.1097/00006676-200210000-00008. [DOI] [PubMed] [Google Scholar]
- 18.Ishibashi T, Zhao H, Kawabe K, Oono T, Egashira K, Suzuki K, Nawata H, Takayanagi R, Ito T. Blocking of monocyte chemoattractant protein-1 (MCP-1) activity attenuates the severity of acute pancreatitis in rats. J Gastroenterol. 2008;43:79–85. doi: 10.1007/s00535-007-2126-9. [DOI] [PubMed] [Google Scholar]
- 19.Bhatia M, Hegde A. Treatment with antileukinate, a CXCR2 chemokine receptor antagonist, protects mice against acute pancreatitis and associated lung injury. Regul Pept. 2007;138:40–48. doi: 10.1016/j.regpep.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 20.Grady T, Liang P, Ernst SA, Logsdon CD. Chemokine gene expression in rat pancreatic acinar cells is an early event associated with acute pancreatitis. Gastroenterology. 1997;113:1966–1975. doi: 10.1016/s0016-5085(97)70017-9. [DOI] [PubMed] [Google Scholar]
- 21.Yubero S, Ramudo L, Manso MA, De Dios I. The role of redox status on chemokine expression in acute pancreatitis. Biochim Biophys Acta. 2009;1792:148–154. doi: 10.1016/j.bbadis.2008.12.002. [DOI] [PubMed] [Google Scholar]
- 22.Bhatia M, Brady M, Kang YK, Costello E, Newton DJ, Christmas SE, Neoptolemos JP, Slavin J. MCP-1 but not CINC synthesis is increased in rat pancreatic acini in response to cerulein hyperstimulation. Am J Physiol Gastrointest Liver Physiol. 2002;282:G77–G85. doi: 10.1152/ajpgi.00031x.2002. [DOI] [PubMed] [Google Scholar]
- 23.Tamizhselvi R, Moore PK, Bhatia M. Inhibition of hydrogen sulfide synthesis attenuates chemokine production and protects mice against acute pancreatitis and associated lung injury. Pancreas. 2008;36:e24–e31. doi: 10.1097/MPA.0b013e31816857bb. [DOI] [PubMed] [Google Scholar]
- 24.Sun LK, Reding T, Bain M, Heikenwalder M, Bimmler D, Graf R. Prostaglandin E2 modulates TNF-alpha-induced MCP-1 synthesis in pancreatic acinar cells in a PKA-dependent manner. Am J Physiol Gastrointest Liver Physiol. 2007;293:G1196–G1204. doi: 10.1152/ajpgi.00330.2007. [DOI] [PubMed] [Google Scholar]
- 25.Han B, Logsdon CD. Cholecystokinin induction of mob-1 chemokine expression in pancreatic acinar cells requires NF-kappaB activation. Am J Physiol. 1999;277:C74–C82. doi: 10.1152/ajpcell.1999.277.1.C74. [DOI] [PubMed] [Google Scholar]
- 26.Yang BM, Demaine AG, Kingsnorth A. Chemokines MCP-1 and RANTES in isolated rat pancreatic acinar cells treated with CCK and ethanol in vitro. Pancreas. 2000;21:22–31. doi: 10.1097/00006676-200007000-00048. [DOI] [PubMed] [Google Scholar]
- 27.Xie MJ, Motoo Y, Su SB, Mouri H, Sawabu N. Induction of chemokines in rat pancreatic acinar cell injury. Pancreas. 2002;24:198–204. doi: 10.1097/00006676-200203000-00012. [DOI] [PubMed] [Google Scholar]
- 28.Springer TA. Traffic signals on endothelium for lymphocyte recirculation and leukocyte emigration. Annu Rev Physiol. 1995;57:827–872. doi: 10.1146/annurev.ph.57.030195.004143. [DOI] [PubMed] [Google Scholar]
- 29.Albelda SM, Smith CW, Ward PA. Adhesion molecules and inflammatory injury. FASEB J. 1994;8:504–512. [PubMed] [Google Scholar]
- 30.Zaninovic V, Gukovskaya AS, Gukovsky I, Mouria M, Pandol SJ. Cerulein upregulates ICAM-1 in pancreatic acinar cells, which mediates neutrophil adhesion to these cells. Am J Physiol Gastrointest Liver Physiol. 2000;279:G666–G676. doi: 10.1152/ajpgi.2000.279.4.G666. [DOI] [PubMed] [Google Scholar]
- 31.Ramudo L, De Dios I, Yubero S, Vicente S, Manso MA. ICAM-1 and CD11b/CD18 expression during acute pancreatitis induced by bile-pancreatic duct obstruction: effect of N-acetylcysteine. Exp Biol Med (Maywood) 2007;232:737–743. [PubMed] [Google Scholar]
- 32.Barnes PJ, Karin M. Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. 1997;336:1066–1071. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- 33.Wulczyn FG, Krappmann D, Scheidereit C. The NF-kappa B/Rel and I kappa B gene families: mediators of immune response and inflammation. J Mol Med. 1996;74:749–769. doi: 10.1007/s001090050078. [DOI] [PubMed] [Google Scholar]
- 34.Siebenlist U, Franzoso G, Brown K. Structure, regulation and function of NF-kappa B. Annu Rev Cell Biol. 1994;10:405–455. doi: 10.1146/annurev.cb.10.110194.002201. [DOI] [PubMed] [Google Scholar]
- 35.Yubero S, Ramudo L, Manso MA, De Dios I. Mechanisms of dexamethasone-mediated chemokine down-regulation in mild and severe acute pancreatitis. Biochim Biophys Acta. 2009;1792:1205–1211. doi: 10.1016/j.bbadis.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 36.Samuel I, Yorek MA, Zaheer A, Fisher RA. Bile-pancreatic juice exclusion promotes Akt/NF-kappaB activation and chemokine production in ligation-induced acute pancreatitis. J Gastrointest Surg. 2006;10:950–959. doi: 10.1016/j.gassur.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 37.Satoh A, Gukovskaya AS, Nieto JM, Cheng JH, Gukovsky I, Reeve JR Jr, Shimosegawa T, Pandol SJ. PKC-delta and -epsilon regulate NF-kappaB activation induced by cholecystokinin and TNF-alpha in pancreatic acinar cells. Am J Physiol Gastrointest Liver Physiol. 2004;287:G582–G591. doi: 10.1152/ajpgi.00087.2004. [DOI] [PubMed] [Google Scholar]
- 38.Ramnath RD, Bhatia M. Substance P treatment stimulates chemokine synthesis in pancreatic acinar cells via the activation of NF-kappaB. Am J Physiol Gastrointest Liver Physiol. 2006;291:G1113–G1119. doi: 10.1152/ajpgi.00177.2006. [DOI] [PubMed] [Google Scholar]
- 39.Karin M, Liu Z, Zandi E. AP-1 function and regulation. Curr Opin Cell Biol. 1997;9:240–246. doi: 10.1016/s0955-0674(97)80068-3. [DOI] [PubMed] [Google Scholar]
- 40.Vaquero E, Gukovsky I, Zaninovic V, Gukovskaya AS, Pandol SJ. Localized pancreatic NF-kappaB activation and inflammatory response in taurocholate-induced pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2001;280:G1197–G1208. doi: 10.1152/ajpgi.2001.280.6.G1197. [DOI] [PubMed] [Google Scholar]
- 41.Gukovsky I, Reyes CN, Vaquero EC, Gukovskaya AS, Pandol SJ. Curcumin ameliorates ethanol and nonethanol experimental pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2003;284:G85–G95. doi: 10.1152/ajpgi.00138.2002. [DOI] [PubMed] [Google Scholar]
- 42.Masamune A, Sakai Y, Yoshida M, Satoh A, Satoh K, Shimosegawa T. Lysophosphatidylcholine activates transcription factor NF-kappaB and AP-1 in AR42J cells. Dig Dis Sci. 2001;46:1871–1881. doi: 10.1023/a:1010622828502. [DOI] [PubMed] [Google Scholar]
- 43.Ramnath RD, Sun J, Bhatia M. Role of calcium in substance P-induced chemokine synthesis in mouse pancreatic acinar cells. Br J Pharmacol. 2008;154:1339–1348. doi: 10.1038/bjp.2008.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Darnell JE Jr. STATs and gene regulation. Science. 1997;277:1630–1635. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- 45.Wilks AF, Harpur AG, Kurban RR, Ralph SJ, Zürcher G, Ziemiecki A. Two novel protein-tyrosine kinases, each with a second phosphotransferase-related catalytic domain, define a new class of protein kinase. Mol Cell Biol. 1991;11:2057–2065. doi: 10.1128/mcb.11.4.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chaturvedi P, Reddy MV, Reddy EP. Src kinases and not JAKs activate STATs during IL-3 induced myeloid cell proliferation. Oncogene. 1998;16:1749–1758. doi: 10.1038/sj.onc.1201972. [DOI] [PubMed] [Google Scholar]
- 47.Vona-Davis LC, Frankenberry KA, Waheed U, Peterson E, McFadden DW. Expression of STAT3 and SOCS3 in pancreatic acinar cells. J Surg Res. 2005;127:14–20. doi: 10.1016/j.jss.2005.03.019. [DOI] [PubMed] [Google Scholar]
- 48.Robinson K, Vona-Davis L, Riggs D, Jackson B, McFadden D. Peptide YY attenuates STAT1 and STAT3 activation induced by TNF-alpha in acinar cell line AR42J. J Am Coll Surg. 2006;202:788–796. doi: 10.1016/j.jamcollsurg.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 49.Yu JH, Kim KH, Kim H. Suppression of IL-1beta expression by the Jak 2 inhibitor AG490 in cerulein-stimulated pancreatic acinar cells. Biochem Pharmacol. 2006;72:1555–1562. doi: 10.1016/j.bcp.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 50.Pereda J, Sabater L, Cassinello N, Gómez-Cambronero L, Closa D, Folch-Puy E, Aparisi L, Calvete J, Cerdá M, Lledó S, et al. Effect of simultaneous inhibition of TNF-alpha production and xanthine oxidase in experimental acute pancreatitis: the role of mitogen activated protein kinases. Ann Surg. 2004;240:108–116. doi: 10.1097/01.sla.0000129343.47774.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dabrowski A, Boguslowicz C, Dabrowska M, Tribillo I, Gabryelewicz A. Reactive oxygen species activate mitogen-activated protein kinases in pancreatic acinar cells. Pancreas. 2000;21:376–384. doi: 10.1097/00006676-200011000-00008. [DOI] [PubMed] [Google Scholar]
- 52.Ju KD, Yu JH, Kim H, Kim KH. Role of mitogen-activated protein kinases, NF-kappaB, and AP-1 on cerulein-induced IL-8 expression in pancreatic acinar cells. Ann N Y Acad Sci. 2006;1090:368–374. doi: 10.1196/annals.1378.040. [DOI] [PubMed] [Google Scholar]
- 53.Parekh DB, Ziegler W, Parker PJ. Multiple pathways control protein kinase C phosphorylation. EMBO J. 2000;19:496–503. doi: 10.1093/emboj/19.4.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yuan J, Lugea A, Zheng L, Gukovsky I, Edderkaoui M, Rozengurt E, Pandol SJ. Protein kinase D1 mediates NF-kappaB activation induced by cholecystokinin and cholinergic signaling in pancreatic acinar cells. Am J Physiol Gastrointest Liver Physiol. 2008;295:G1190–G1201. doi: 10.1152/ajpgi.90452.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hunter T. Protein kinases and phosphatases: the yin and yang of protein phosphorylation and signaling. Cell. 1995;80:225–236. doi: 10.1016/0092-8674(95)90405-0. [DOI] [PubMed] [Google Scholar]
- 56.Camps M, Nichols A, Arkinstall S. Dual specificity phosphatases: a gene family for control of MAP kinase function. FASEB J. 2000;14:6–16. [PubMed] [Google Scholar]
- 57.Escobar J, Pereda J, Arduini A, Sandoval J, Sabater L, Aparisi L, López-Rodas G, Sastre J. Cross-talk between oxidative stress and pro-inflammatory cytokines in acute pancreatitis: a key role for protein phosphatases. Curr Pharm Des. 2009;15:3027–3042. doi: 10.2174/138161209789058075. [DOI] [PubMed] [Google Scholar]
- 58.Höfken T, Keller N, Fleischer F, Göke B, Wagner AC. Map kinase phosphatases (MKP's) are early responsive genes during induction of cerulein hyperstimulation pancreatitis. Biochem Biophys Res Commun. 2000;276:680–685. doi: 10.1006/bbrc.2000.3530. [DOI] [PubMed] [Google Scholar]
- 59.Sarmiento N, Sánchez-Bernal C, Ayra M, Pérez N, Hernández-Hernández A, Calvo JJ, Sánchez-Yagüe J. Changes in the expression and dynamics of SHP-1 and SHP-2 during cerulein-induced acute pancreatitis in rats. Biochim Biophys Acta. 2008;1782:271–279. doi: 10.1016/j.bbadis.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 60.Thomas ML. The leukocyte common antigen family. Annu Rev Immunol. 1989;7:339–369. doi: 10.1146/annurev.iy.07.040189.002011. [DOI] [PubMed] [Google Scholar]
- 61.De Dios I, Ramudo L, Alonso JR, Recio JS, Garcia-Montero AC, Manso MA. CD45 expression on rat acinar cells: involvement in pro-inflammatory cytokine production. FEBS Lett. 2005;579:6355–6360. doi: 10.1016/j.febslet.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 62.De Dios I, Ramudo L, García-Montero AC, Manso MA. Redox-sensitive modulation of CD45 expression in pancreatic acinar cells during acute pancreatitis. J Pathol. 2006;210:234–239. doi: 10.1002/path.2037. [DOI] [PubMed] [Google Scholar]
- 63.Yakura H. Phosphatases and kinases in lymphocyte signaling. Immunol Today. 1998;19:198–201. doi: 10.1016/s0167-5699(97)01232-2. [DOI] [PubMed] [Google Scholar]